Abstract
The title compound was isolated as the unexpected reaction product from a reaction attempting to access a glycosyl 1-phosphate. The product was isolated in good yield, as one diastereoisomer, and was characterised by 1H, 13C, and 2D NMR, alongside HRMS analysis.
1. Introduction
Chemically altered variants of native glycosides form part of an important toolkit to study related processes surrounding glycans biological function. One such modification is the replacement of the interglycosidic linkage with sulphur. Switching to this chalcogen imparts improved hydrolytic stability to the linkage, and wider S-glycoside forms of important glycan targets have underpinned advances in glycoscience [1,2,3,4,5]. Replacing sulphur with oxygen provides a similar conformational preference regarding thioglycosidic linkages [6], and this, combined with sulfur’s lower affinity for protons, confers an overall reduced susceptibility of an S-glycan to hydrolysis [7]. As part of a wider programme concerning the synthesis of S-glycosides [8,9,10], we sought access to a mannose 1-phosphate bearing a 2-deoxy-2-thiomodification to then investigate enzymatic construction of α-D-manno-S-linkages.
2. Results
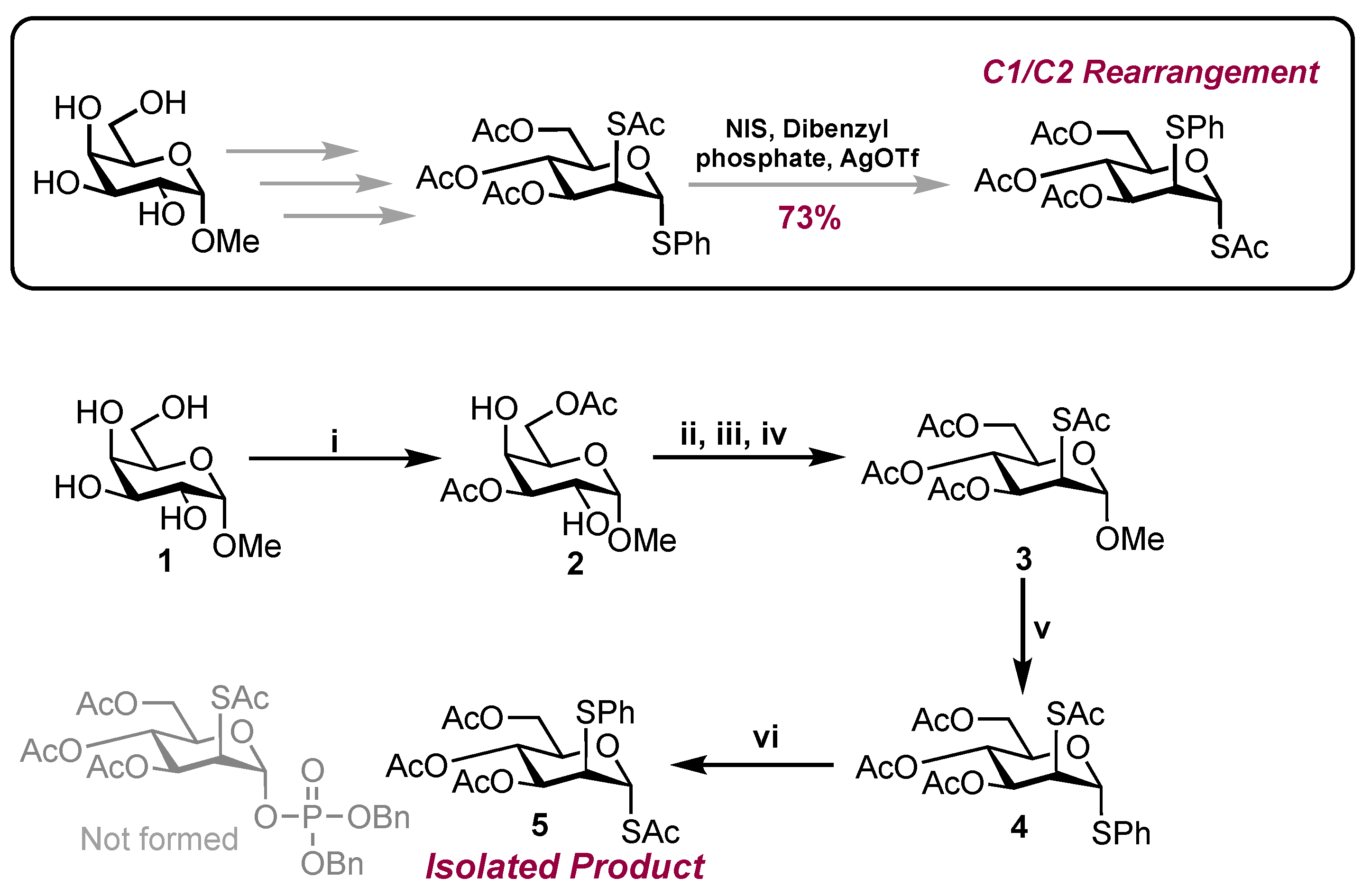
Starting from methyl α-D-galactose 1, the first synthetic step involved regioselective acylation at the 3- and 6-OH positions (Scheme 1) [11]. The reaction delivered diol 2 in 51% yield with characterisation data matching those previously reported [11]. With diol 2 in hand, inversion of configuration was completed at C2 and C4 to install the required mannose configuration. Accordingly, triflation of the free hydroxyl groups was followed by inversion of the 4-O-triflate using ammonium acetate. After 2 h, to the same reaction, potassium thioacetate was added, and after 2 days of stirring, TLC analysis confirmed complete consumption of the material (Rf = 0.54, hexane/EtOAc, 1:1). Purification by column chromatography delivered acetate 3 in 49% yield. The mannose configuration was confirmed through the coupling constant between H-1 and H-2 (3JH1-H2 = 1.2 Hz), matching that previously reported [12]. Glycosylation using thiophenol and BF3·OEt2 then granted access to thioglycoside 4, although this proceeded sluggishly, and after 3 days, the reaction was stopped and, following purification, acetate 4 was isolated in 37% yield alongside recovered starting material 3 (27%).
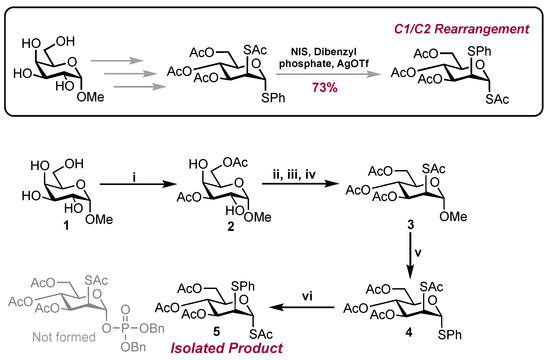
Scheme 1.
Reagents and conditions: i. DIPEA, DMF, MeCN, Ac2O (2.2. equiv.), RT, 51%; ii. Tf2O (5.0 equiv.), DCM, pyridine, −30 °C to 10 °C; iii. NH4OAc (1.0 equiv.), DMF, RT; iv. KSAc (3.0 equiv.), DMF, RT, 49% (3 steps); v. PhSH (16.0 equiv.), BF3·OEt2, (4.0 equiv.) RT, 37% (recovered 3: 27%); vi. NIS, dibenzyl phosphate (1.5 equiv.), AgOTf (0.5 equiv.), DCM, 73%.
Using dibenzyl phosphate, NIS and AgOTf, anomeric phosphorylation of thioglycoside 4 was attempted next. The reaction produced a single new spot by TLC after 1 h (Rf = 0.35, hexane/EtOAc, 6:4). Following workup and purification, 31P NMR analysis revealed no chemical shift for the desired 1-phosphate and 1H NMR lacked additional aromatic region chemical shifts, compared with starting material 4.
Following HMBC and HRMS analysis, the unknown product was indicatively assigned as anomeric S-acetate 5. HMBC analysis revealed a cross-peak between H-1 [δH 5.89 (d, J = 1.9 Hz) ppm] and a carbonyl chemical shift [δc 168.6 (C=O) ppm], suggesting the SAc moiety was located at C1. The chemical shift was also distinctly downfield [δH1 5.89], when compared to H-3 [δH3 5.14], H-4 [δH4 5.29] and H-6 [δH6a 5.89, δH6b 5.89], with it being noted that each had a carbonyl cross-peak. (Figure 1B). The lack of a cross-peak between H-2 and the carbonyl environment indicated the presence of the SPh group at C2. This was further supported by HRMS analysis. Coupled HSQC NMR revealed a 1JC1-H1 value of 168.5 Hz, suggesting that the anomeric SAc substituent was in the α configuration. Coupling constants observed for H-2 were indicative of a mannose configuration as opposed to glucose [1JH1-H2 = 1.9 Hz, 1JH2-H3 = 4.4 Hz].
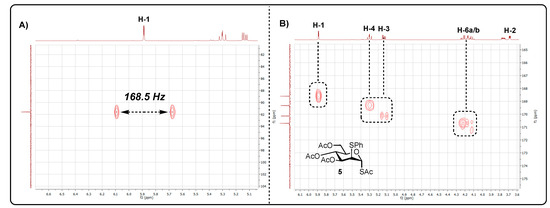
Figure 1.
(A) Coupled HSQC analysis of 5 CDCl3 (400 × 101 MHz); (B) HMBC CDCl3 (400 × 101 MHz) analysis of 5.
In conclusion, an unusual and unexpected C1/C2 rearrangement product was uncovered from a reaction targeting a protected anomeric phosphate. The material was isolated in 73% yield with analytical data indicating the structural assignment. Whilst further mechanistic insight was not gathered here, it is possible that the observed product formed via the migration of thioacetate to C1 with a loss of anomeric thiophenyl, followed by the reintroduction of SPh at C2. Copies of NMR and HRMS data are included in the Supplementary Materials.
3. Materials and Methods
3.1. General
All chemicals were purchased from Acros Organics (Antwerp, Belgium), Biosynth (Staad, Switzerland), Fisher Scientific (Loughborough, UK), Fluorochem (Manchester, UK), and Sigma Aldrich (Gillingham, UK) and were used without further purification unless otherwise stated. NMR spectra were recorded on a Bruker Avance 400 spectrometer. For reactions that required heating, DrySyn heating blocks were used as the heat source. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane, where δ = 0.00 ppm. The number of protons (n) for a given resonance is indicated by nH. The multiplicity of each signal is indicated by s (singlet), bs (broad singlet), as (apparent singlet), ad (apparent doublet), d (doublet), t (triplet), q (quartet), p (pentet), sep (septet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dddd (doublet of doublet of doublet of doublets), dt (doublet of triplets), tt (triplet of triplets), dqd (doublet of quartets of doublets) or m (multiplet). Coupling constants (J) are quoted in Hz and calculated to the nearest 0.1 Hz. Anhydrous DCM, DMF and pyridine were obtained from Sure/SealTM bottles via chemical suppliers. Unless otherwise stated, all reactions were conducted using anhydrous solvents, under an atmosphere of N2 which was passed through a Drierite® drying column. HRMS were recorded on a ThermoScientific LTQ Orbitrap XL at the ESPRC National Mass Spectrometry Facility at Swansea University. Analytical thin-layer chromatography (TLC) was carried out on pre-coated 0.25 mm Merck KgaA 60 F254 silica gel plates. Visualisation was by the adsorption of UV light, or thermal development after dipping in a methanolic solution of sulphuric acid (5% v/v). Automatic flash chromatography was carried out on silica gel (Reveleris® X2 system) under a positive pressure of compressed N2. RT: room temperature. Visualisation was achieved using UV detection at 230 and 270 nm.
3.2. Methyl 3,6-Di-O-acetyl-α-d-galactopyranoside (2)
Methyl α-D-galactose 1 (3.00 g, 15.5 mmol, 1.0 equiv.) was dissolved in MeCN/DMF (10:1, 30 mL) and acetic anhydride (3.22 mL, 34.1 mmol, 2.2 equiv.) and DIPEA (2.36 mL, 13.6 mmol, 0.4 equiv.) added. The reaction mixture was stirred for 12 h at 40 °C. Reaction completion was confirmed by TLC analysis (Rf = 0.29, EtOAc/hexane, 5:1) and the mixture was quenched through the addition of NH4Cl (100 mL). The organic layer was collected, dried (MgSO4), and filtered, and the solvent was removed under reduced pressure. The crude residue was then purified by column chromatography (hexane/EtOAc, 0–100%) yielding the title compound (2.20 g, 7.91 mmol, 51%) as a colourless oil. Rf = 0.29 (EtOAc/hexane, 5:1); 1H NMR (400 MHz, CDCl3) δ 5.07 (dd, J = 10.3, 3.1 Hz, 1H, H-3), 4.85 (d, J = 3.9 Hz, 1H, H-1), 4.34 (dd, J = 11.5, 6.0 Hz, 1H, H-6a), 4.24 (dd, J = 11.5, 6.7 Hz, 1H, H-6b), 4.16–3.94 (m, 3H, H-2, H-4, H-5), 3.46 (s, 3H, OCH3), 2.44 (d, J = 3.8 Hz, 1H, OH), 2.17 (s, 3H, OAc), 2.10–2.08 (m, 4H, OAc, OH); 13C NMR (101 MHz, CDCl3) δ 171.0 (C=O, Ac), 170.9 (C=O, Ac), 99.7 (C1), 73.0 (C3), 68.00, 67.79, 67.14, 62.9 (C6), 55.6 (OCH3), 21.1 (Ac-CH3), 20.8 (Ac-CH3); HRMS m/z (ES+) Found: (M+Na)+ 301.0894, C11H18O8Na, requires M+ 301.8994. Data matched those previously reported [11].
3.3. Methyl 3,4,6-Tri-O-acetyl-2-S-acetyl-2-thio-α-d-mannopyranoside (3)
Methyl 3,6-di-O-acetyl-α-d-glucopyranoside 2 (1.99 g, 7.15 mmol, 1.0 equiv.) was dissolved in DCM (54.0 mL) and pyridine (6.00 mL). The mixture was then cooled to −30 °C and Tf2O (6.00 mL, 10.1 g, 35.8 mmol, 5.0 equiv.) added dropwise. The mixture was warmed to 10 °C and stirred for 4 h, at which point TLC analysis confirmed reaction completion (Rf = 0.78, hexane/EtOAc, 1:1). The resulting mixture was subsequently diluted with DCM (100 mL) and washed sat.aq. NaHCO3 (100 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Crude methyl 3,6-di-O-acetyl-4,6-di-O-triflate-D-glucopyranoside (3.80 g, 7.00 mmol, 1.0 equiv.) was used directly in the next step without further purification. NH4OAc (540 mg, 7.00 mmol, 1.0 equiv.) was added to the crude sugar in DMF (5 mL). After stirring at RT for 2 h, KSAc (2.40 g, 21.0 mmol. 3.0 equiv.) was added to the mixture and stirred at RT for 24 h. TLC analysis confirmed complete consumption of the starting material (Rf = 0.54, hexane/EtOAc, 1:1). The mixture was diluted with EtOAc (100 mL) and washed with NaHCO3 (100 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Purification of the residue was performed by column chromatography (hexane/EtOAc, 0.40%), yielding the title compound (1.32 g, 3.50 mmol, 49%) as a yellow/green oil. Rf = 0.78 (hexane/EtOAc, 1:1); 1H NMR (400 MHz, CDCl3) δ 5.57 (dd, J = 9.9, 4.8 Hz, 1H, H-3), 5.08 (t, J = 10.0 Hz, 1H, H-4), 4.76 (d, J = 1.2 Hz, 1H, H-1), 4.27 (dd, J = 4.7, 1.3 Hz, 1H, H-2), 4.21 (dd, J = 12.2, 4.9 Hz, 1H, H-6a), 4.10 (dd, J = 12.2, 2.4 Hz, 1H, H-6b), 3.97 (ddd, J = 10.0, 4.9, 2.4 Hz, 1H, H-5), 3.41 (s, 3H, OCH3), 2.38 (SAc), 2.11 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.97 (s, 3H, OAc); 13C NMR (101 MHz, CDCl3) δ 193.4 (C=O), 170.7 (C=O), 169.8 (C=O), 169.6 (C=O), 101.0 (C1), 68.6 (C3), 68.5 (C-5), 66.9 (C4), 62.3 (C6), 55.4 (OCH3), 47.1 (C2), 30.5 (SAc-CH3), 20.72 (Ac-CH3), 20.71 (Ac-CH3), 20.6 (Ac-CH3); HRMS m/z (ES+) Found: (M+NH4)+ 396.1310, C15H26NO9S, requires M+ 396.1325. Data matched those previously reported [13].
3.4. Phenyl 3,4,6-Tri-O-acetyl-2-deoxy-2-S-acetyl-1,2-di-thio-α-d-mannopyranoside (4)
To a solution of methyl 3,4,6-tri-O-acetyl-2-deoxy-2-S-acetyl-2-thio-α-d-mannopyranoside (1.06g, 2.77 mmol, 1.0 equiv.) in PhSH (5.0 mL, 49.0 mmol, 16.0 equiv.), BF3·OEt2 (1.37 mL, 11.1 mmol, 4.0 equiv.) was added and the reaction stirred at RT for 3 days. At this point, the reaction mixture was diluted with DCM (50 mL) and washed sat.aq. NaHCO3 (100 mL), and the combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Column chromatography afforded the title compound as a white solid (460 mg, 1.01 mmol, 36%). The starting material was also recovered (286 mg, 0.756 mmol, 27%). Rf = 0.70 (hexane/EtOAc, 1:1); 1H NMR (400 MHz, CDCl3) δ 7.51–7.44 (m, 2H, ArH), 7.35–7.28 (m, 3H, ArH), 5.57–5.50 (m, 2H, H-3, H-1), 5.13 (t, J = 9.9 Hz, 1H, H-4), 4.57 (ddt, J = 9.9, 5.5, 2.3 Hz, 1H, H-5), 4.51 (dd, J = 4.6, 1.3 Hz, 1H, H-2), 4.24 (dd, J = 12.3, 5.5 Hz, 1H, H-6a), 4.09 (dd, J = 12.3, 2.3 Hz, 1H, H-6b), 2.37 (s, 3H, SAc), 2.07 (s, 3H, OAc), 2.06 (s, 3H, OAc), 1.99 (s, 3H, OAc); 13C NMR (101 MHz, CDCl3) δ 193.3 (C=O), 170.6 (C=O), 169.7 (C=O), 169.6 (C=O), 133.1 (Ar-C), 132.1 (Ar-C), 129.2 (Ar-C), 128.1 (Ar-C), 87.7 (C1), 69.7 (C5), 69.1 (C3), 67.1 (C4), 62.3 (C6), 48.6 (C2), 30.5 (SAc-CH3), 20.69 (2 x Ac-CH3), 20.67 (Ac-CH3); HRMS m/z (ES+) Found: (M+NH4)+ 474.1259, C20H28O8S2N, requires M+ 474.1256. Data matched those previously [12].
3.5. Acetyl 3,4,6-Tri-O-acetyl-2-deoxy-2-S-phenyl-1,2-di-thio-α-d-mannopyranoside (5)
To phenyl 3,4,6-tri-O-acetyl-2-deoxy-2-S-acetyl-1,2-di-thio-α-D-mannopyranoside (50.0 mg, 0.110 mmol, 1.0 equiv.) and in DCM (5 mL), dibenzyl phosphate (61.0 mg, 0.220 mmol, 1.5 equiv.) was added. The mixture was cooled to −40 °C; then, AgOTf (14.0 mg, 55.0 µmol, 0.5 equiv.) and NIS (37.0 mg, 0.165 mmol, 1.5 equiv.) were added, and the reaction was warmed to 0 °C and stirred for 1 h. The solution was subsequently diluted with DCM (100 mL), washed with water (100 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The crude residue was purified by column chromatography (hexane/EtOAc, 0–50%). Rf = 0.35 (hexane/EtOAc, 6:4); 1H NMR (400 MHz, CDCl3) δ 7.59–7.53 (m, 2H, ArH), 7.37–7.28 (m, 2H, ArH), 7.28–7.18 (m, 1H, ArH), 5.89 (d, J = 1.9 Hz, 1H, H-1), 5.33–5.26 (m, 1H, H-4), 5.13 (dd, J = 9.5, 4.4 Hz, 1H, H-3), 4.22 (dd, J = 12.4, 4.9 Hz, 1H, H-6a), 4.21–4.12 (m, 1H, H-6b), 3.76 (ddd, J = 9.2, 4.9, 2.7 Hz, 1H, H-5), 3.68 (dd, J = 4.4, 1.9 Hz, 1H, H-2), 2.10 (s, 3H, OAc), 2.06 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.98 (s, 3H, OAc); 13C NMR (101 MHz, CDCl3) δ 170.7 (C=O), 170.1 (C=O), 169.3 (C=O), 168.6 (C=O), 136.9 (Ar-C), 129.0 (Ar-C), 128.6 (Ar-C), 127.5 (Ar-C), 91.6 (C1), 73.5 (C5), 71.8 (C3), 65.5 (C4), 61.9 (C6), 58.5 (C2), 20.7 (2 x Ac-CH3), 20.7 (Ac-CH3), 20.6 (Ac-CH3); HRMS m/z (ES+) Found: (M+NH4)+ 474.1250, C20H28O8S2N, requires M+ 474.1256.
Supplementary Materials
The following pages contain representative spectral characterisation data, including NMR and HRMS for (2) (3), (4) and (5).
Author Contributions
G.J.M. and J.P. conceived and designed the experiments; J.P. performed the experiments and analysed the data; and J.P. and G.J.M. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
G.J.M. thanks UK Research and Innovation (Future Leaders Fellowship, MR/T019522/1) for project grant funding.
Data Availability Statement
The original contributions presented in the study are included in the Supplementary Materials; further inquiries can be directed to the corresponding author.
Acknowledgments
The EPSRC UK National Mass Spectrometry Facility (NMSF) at Swansea University is thanked for MS analyses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Romanò, C.; Jiang, H.; Boos, I.; Clausen, M.H. S-Glycosides: Synthesis of S-Linked Arabinoxylan Oligosaccharides. Org. Biomol. Chem. 2020, 18, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhang, X.; Xia, K.; Green, D.E.; Baytas, S.; Xu, Y.; Pham, T.; Liu, J.; Zhang, F.; Almond, A.; et al. Chemoenzymatic Synthesis of Sulfur-Linked Sugar Polymers as Heparanase Inhibitors. Nat. Commun. 2022, 13, 7438. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Hatton, N.E.; Porter, J.; Vendeville, J.B.; Wheatley, D.E.; Ghirardello, M.; Wahart, A.J.C.; Ahmadipour, S.; Walton, J.; Galan, M.C.; et al. Reverse Thiophosphorylase Activity of a Glycoside Phosphorylase in the Synthesis of an Unnatural Manβ1,4GlcNAc Library. Chem. Sci. 2023, 14, 11638–11646. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.; Colomer, J.P.; Manzano, V.E.; Varela, O. Synthesis of Thiodisaccharides Related to 4-Thiolactose. Specific Structural Modifications Increase the Inhibitory Activity against E. Coli β-Galactosidase. Org. Biomol. Chem. 2023, 21, 2188–2203. [Google Scholar] [CrossRef] [PubMed]
- Driguez, H. Thiooligosaccharides as Tools for Structural Biology. ChemBioChem 2001, 2, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Montero, E.; García-Herrero, A.; Asensio, J.L.; Hirai, K.; Ogawa, S.; Santoyo-González, F.; Cañada, F.J.; Jiménez-Barbero, J. The Conformational Behaviour of Non-Hydrolizable Lactose Analogues: The Thioglycoside, Carbaglycoside, and Carba-Iminoglycoside Cases. Eur. J. Org. Chem. 2000, 2000, 1945–1952. [Google Scholar] [CrossRef]
- Wilson, J.C.; Kiefel, M.J.; Angus, D.I.; Itzstein, M. von Investigation of the Stability of Thiosialosides toward Hydrolysis by Sialidases Using NMR Spectroscopy. Org. Lett. 1999, 1, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Parisi, D.; Miller, T.; Ní Cheallaigh, A.; Miller, G.J. Chemical synthesis of amphiphilic glycoconjugates: Access to amino, fluorinated and sulfhydryl oleyl glucosides. Carbohydr. Res. 2023, 530, 108854. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Lima, M.A.; Pongener, I.; Miller, G.J. Synthesis of 4-Thio-D-Glucopyranose and Interconversion to 4-Thio-D-Glucofuranose. Carbohydr. Res. 2023, 524, 108759. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.; Miller, G.J. Synthesis of S-Glycoside Building Blocks as Mimetics of the Repeating d-GlcN-α-1,4-d-GlcA Heparan Sulfate Disaccharide. Molecules 2024, 29, 5809. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Gan, L.; Zhang, L.; Yan, N.; Dong, H. Diisopropylethylamine-Triggered, Highly Efficient, Self-Catalyzed Regioselective Acylation of Carbohydrates and Diols. Org. Biomol. Chem. 2018, 16, 5591–5597. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Malolanarasimhan, K. Oligomeric Thioglycosides with α-D-Manno-(1′→2) Linkages from a Glycal-1,2-Episulfide. Org. Lett. 1999, 1, 611–613. [Google Scholar] [CrossRef]
- Wu, B.; Ge, J.; Ren, B.; Pei, Z.; Dong, H. Synthesis and Binding Affinity Analysis of Positional Thiol Analogs of Mannopyranose for the Elucidation of Sulfur in Different Position. Tetrahedron 2015, 71, 4023–4030. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).