
Progress in the Correlative Atomic Force Microscopy and Optical Microscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Principles of Atomic Force Microscopy and Optical Microscopy
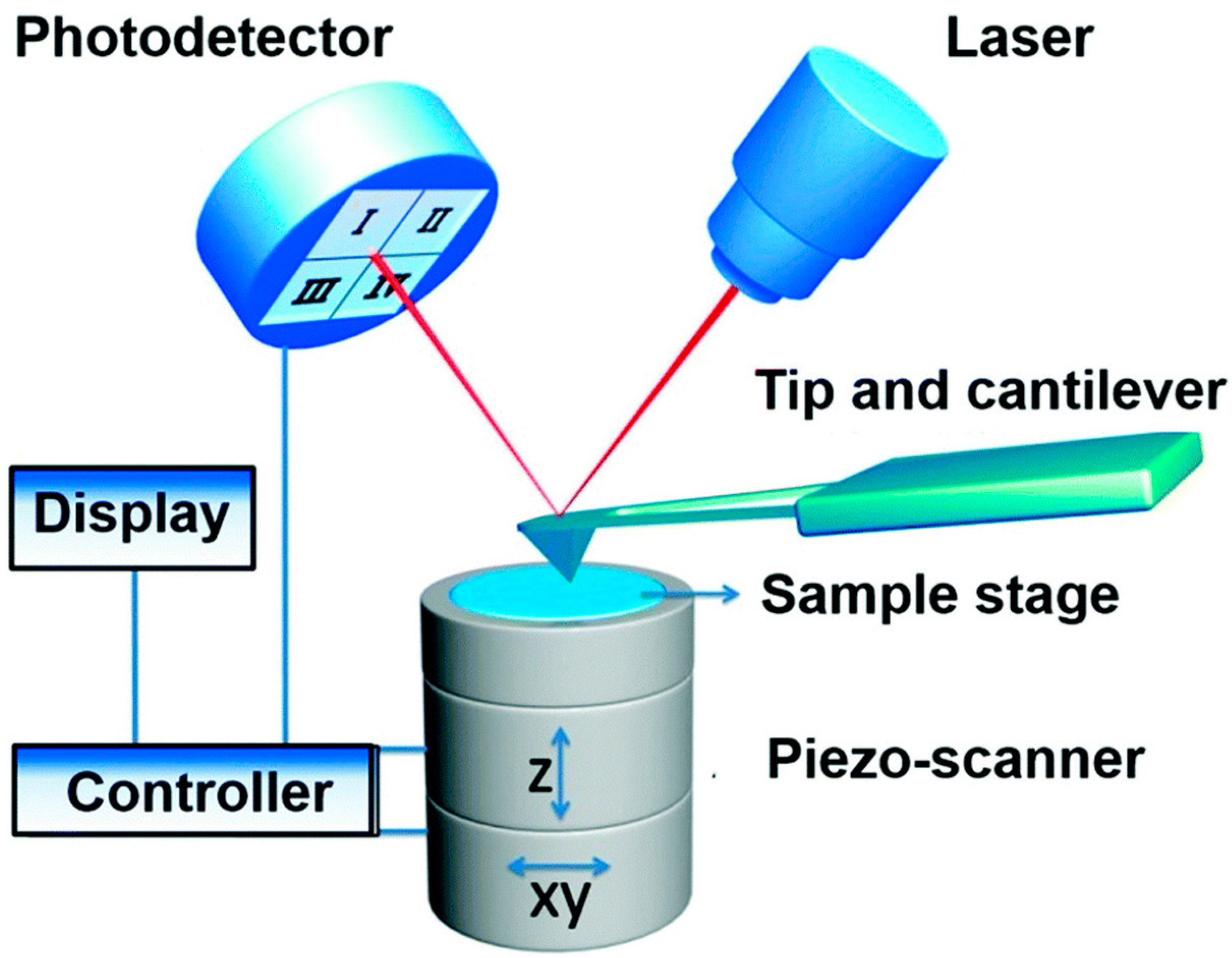
2.1. Atomic Force Microscopy (AFM)
2.2. Conventional Fluorescence Microscopy
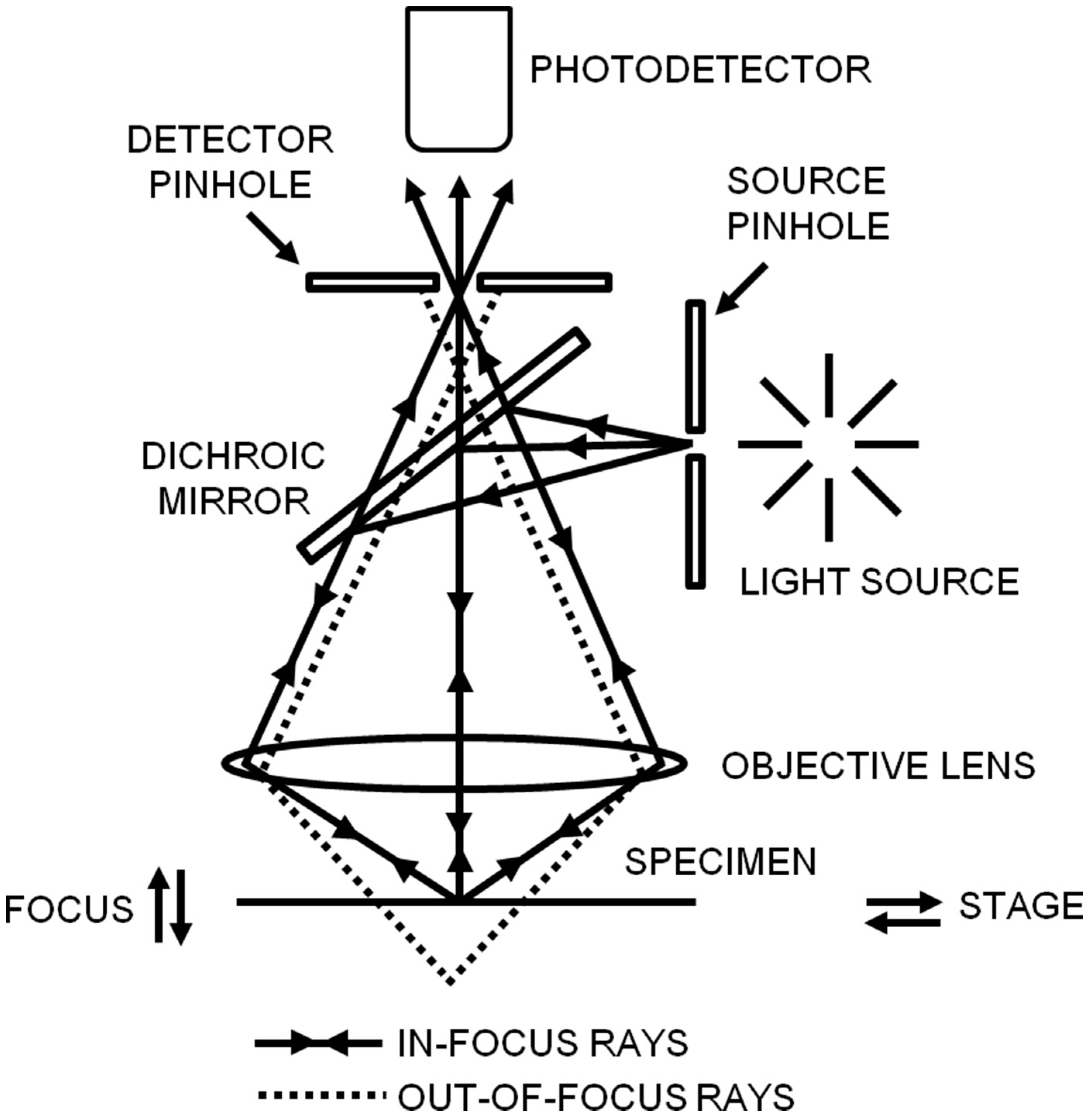
2.2.1. Confocal Laser Scanning Microscopy (CLSM)
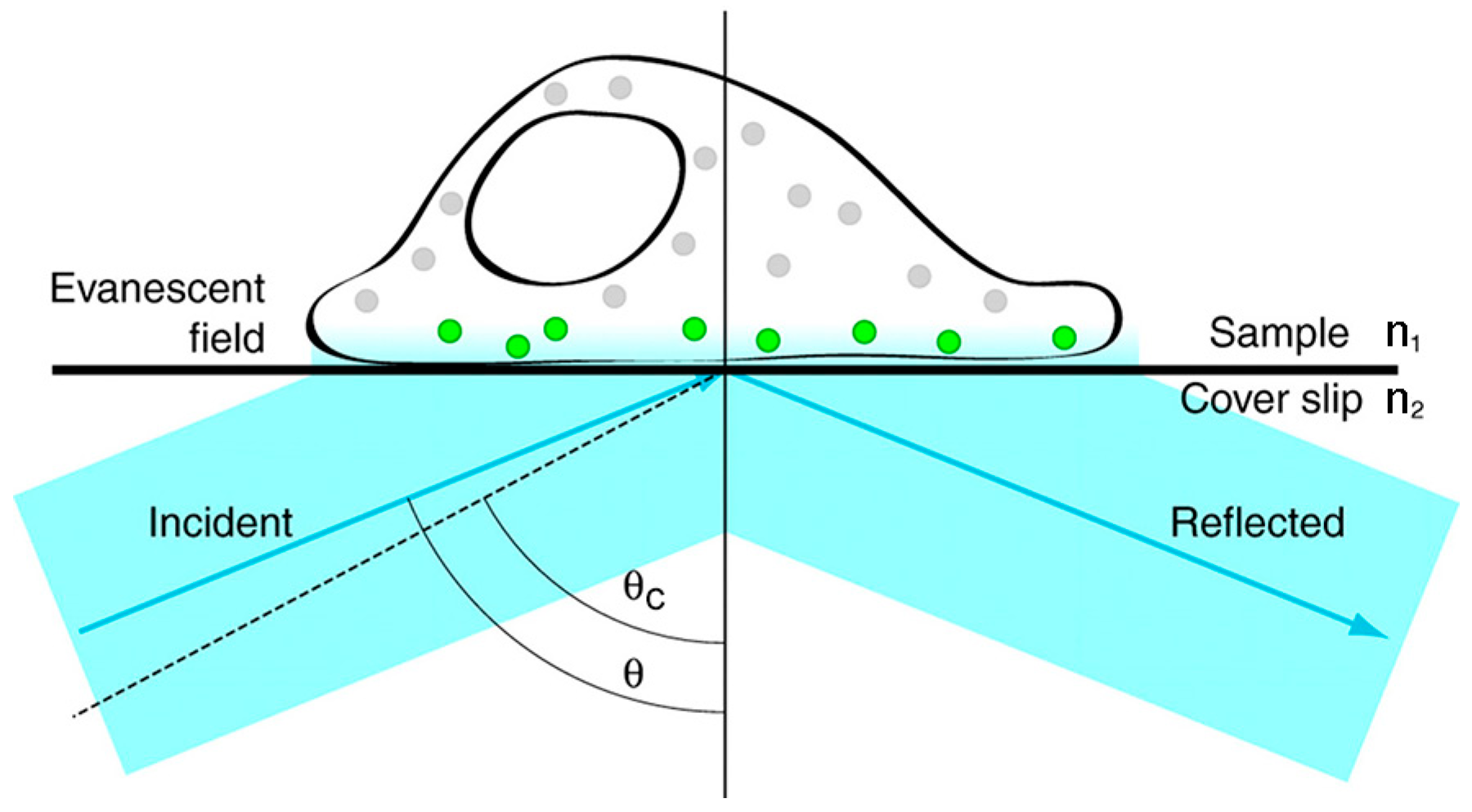
2.2.2. Total Internal Reflection Fluorescence Microscopy (TIRFM)
2.3. Super-Resolution Fluorescence Microscopy
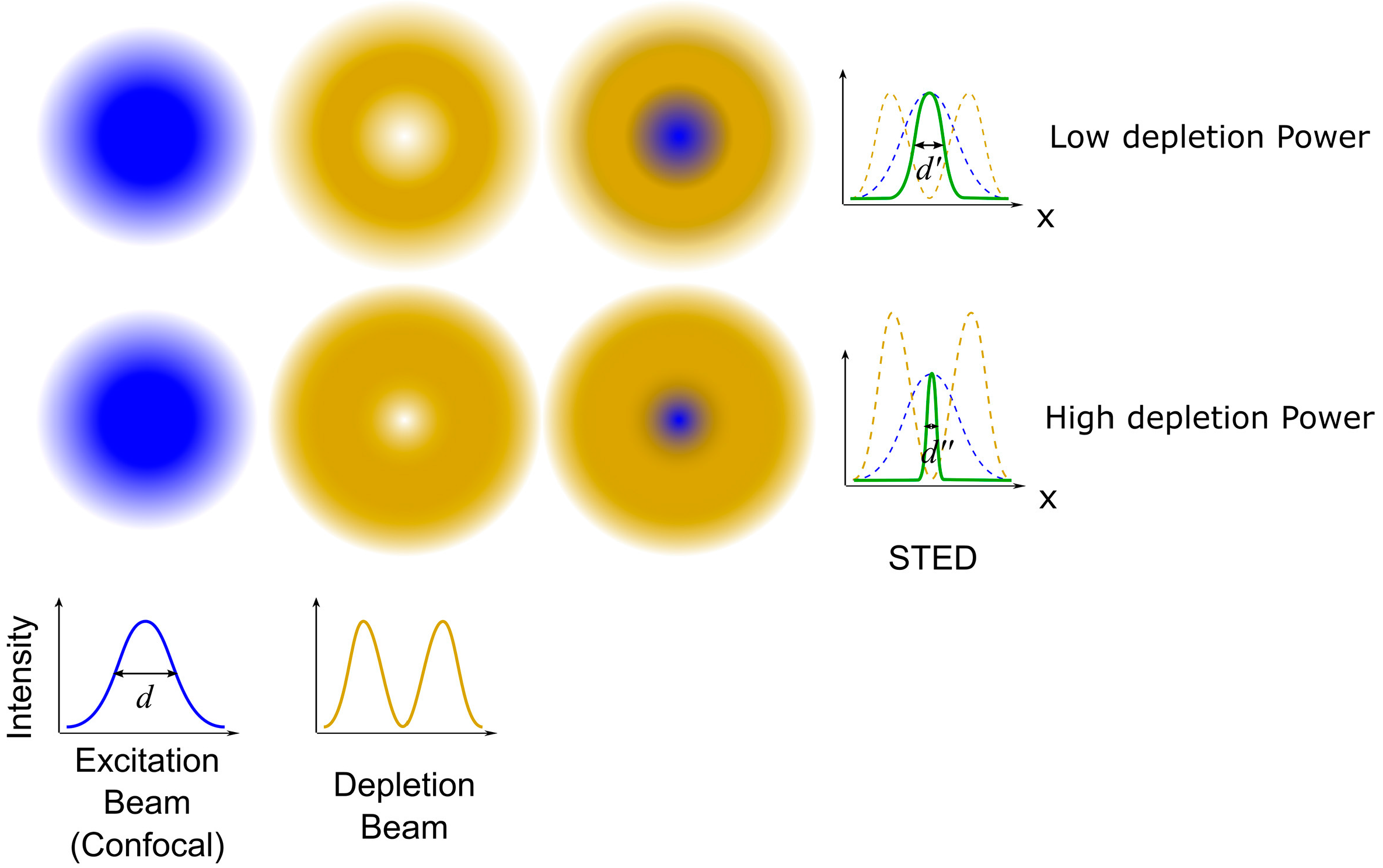
2.3.1. Stimulated Emission Depletion (STED) Microscopy
2.3.2. Single-Molecule Localization Microscopy (SMLM)
3. Correlative Optical Microscopy/Atomic Force Microscopy
3.1. Correlative Conventional Fluorescence Microscopy/Atomic Force Microscopy
3.2. Correlative Super-Resolution Fluorescence Microscopy/Atomic Force Microscopy
4. Challenges and Outlook
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Wang, H. The structure and function of cell membranes examined by atomic force microscopy and single-Molecule force spectroscopy. Chem. Soc. Rev. 2015, 44, 3617–3638. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Dufrêne, Y.F. Atomic force microscopy: A nanoscopic window on the cell surface. Trends Cell Biol. 2011, 21, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Marszalek, P.E.; Dufrêne, Y.F. Stretching single polysaccharides and proteins using atomic force microscopy. Chem. Soc. Rev. 2012, 41, 3523–3534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.G.; Hao, X.; Cai, M.J.; Shan, Y.P.; Shang, X.; Tang, Z.Y.; Wang, H.D. Localization of Na+-K+ ATPases in quasi-Native cell membranes. Nano Lett. 2009, 9, 4489–4493. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.P.; Wang, Z.Y.; Hao, X.A.; Shang, X.; Cai, M.J.; Jiang, J.G.; Fang, X.X.; Wang, H.D.; Tang, Z.Y. Locating the band III protein in quasi-Native cell membranes. Anal. Methods 2010, 2, 805–808. [Google Scholar] [CrossRef]
- Zhao, W.D.; Liu, S.H.; Cai, M.J.; Xu, H.J.; Jiang, J.G.; Wang, H.D. Detection of carbohydrates on the surface of cancer and normal cells by topography and recognition imaging. Chem. Commun. 2013, 49, 2980–2982. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction barrier: Super-Resolution imaging of cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Hein, B.; Willig, K.I.; Wurm, C.A.; Westphal, V.; Jakobs, S.; Hell, S.W. Stimulated emission depletion nanoscopy of living cells using SNAP-Tag fusion proteins. Biophys. J. 2010, 98, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-Resolution microscopy: From single molecules to supramolecular assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef] [PubMed]
- Smith, C. Microscopy: Two microscopes are better than one. Nature 2012, 492, 293–297. [Google Scholar] [CrossRef] [PubMed]
- De Boer, P.; Hoogenboom, J.P.; Giepmans, B.N.G. Correlated light and electron microscopy: Ultrastructure lights up! Nat. Methods 2015, 12, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, P.D.; Shivanandan, A.; Deschout, H.; Jankele, R.; Nievergelt, A.P.; Feletti, L.; Davidson, M.W.; Radenovic, A.; Fantner, G.E. High-Resolution correlative microscopy: Bridging the gap between single molecule localization microscopy and atomic force microscopy. Nano Lett. 2015, 15, 4896–4904. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Gaub, H.E. Structure and mechanics of membrane proteins. Annu. Rev. Biochem. 2008, 77, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Dufrêne, Y.F. Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotechnol. 2008, 3, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Dufrêne, Y.F. Optical and force nanoscopy in microbiology. Nat. Microbiol. 2016, 1, 16186. [Google Scholar] [CrossRef] [PubMed]
- Hansma, P.K.; Cleveland, J.P.; Radmacher, M.; Walters, D.A.; Hillner, P.E.; Bezanilla, M.; Fritz, M.; Vie, D.; Hansma, H.G.; et al. Tapping mode atomic force microscopy in liquids. Appl. Phys. Lett. 1994, 64, 1738–1740. [Google Scholar] [CrossRef]
- Müller, D.J.; Krieg, M.; Alsteens, D.; Dufrêne, Y.F. New frontiers in atomic force microscopy: Analyzing interactions from single-Molecules to cells. Curr. Opin. Biotechnol. 2009, 20, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hinterdorfer, P.; Baumgartner, W.; Gruber, H.J.; Schilcher, K.; Schindler, H. Detection and localization of individual antibody-Antigen recognition events by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 3477–3481. [Google Scholar] [CrossRef] [PubMed]
- Hinterdorfer, P.; Dufrêne, Y.F. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods 2006, 3, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Helenius, J.; Alsteens, D.; Dufrêne, Y.F. Force probing surfaces of living cells to molecular resolution. Nat. Chem. Biol. 2009, 5, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, X.; Xia, T.; Fang, X. Living cell study at the single-Molecule and single-Cell levels by atomic force microscopy. Nanomedicine 2012, 7, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Dufrêne, Y.F.; Martínez-Martín, D.; Medalsy, I.; Alsteens, D.; Müller, D.J. Multiparametric imaging of biological systems by force-Distance curve-Based AFM. Nat. Methods 2013, 10, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, X.; Huang, Y.; Hao, X.; Xu, H.; Cai, M.; Wang, H.; Qin, Q. Entry of a novel marine DNA virus, singapore grouper iridovirus, into host cells occurs via clathrin-Mediated endocytosis and macropinocytosis in a pH-Dependent manner. J. Virol. 2014, 88, 13047–13063. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xu, H.; Wang, S.; Cai, M.; Shi, Y.; Yang, G.; Wang, H.; Shan, Y. Studying the dynamic mechanism of transporting a single drug carrier-Polyamidoamine dendrimer through cell membranes by force tracing. Nanoscale 2016, 8, 18027–18031. [Google Scholar] [CrossRef] [PubMed]
- Paddock, S.W. Principles and practices of laser scanning confocal microscopy. Mol. Biotechnol. 2000, 16, 127–149. [Google Scholar] [CrossRef]
- Furrer, P.; Gurny, R. Recent advances in confocal microscopy for studying drug delivery to the eye: Concepts and pharmaceutical applications. Eur. J. Pharm. Biopharm. 2010, 74, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Toomre, D.; Pawley, J.B. Disk-Scanning confocal microscopy. In Handbook of Biological Confocal Microscopy, 3rd ed.; Pawley, J.B., Ed.; Springer: Boston, MA, USA, 2006; pp. 221–238. [Google Scholar]
- Ustione, A.; Piston, D.W. A simple introduction to multiphoton microscopy. J. Microsc. 2011, 243, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Schmoranzer, J.; Simon, S.M. Role of microtubules in fusion of post-Golgi vesicles to the plasma membrane. Mol. Biol. Cell 2003, 14, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, J.Z. Focusing on clathrin-Mediated endocytosis. Biochem. J. 2008, 412, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Mattheyses, A.L.; Simon, S.M.; Rappoport, J.Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 2010, 123, 3621–3628. [Google Scholar] [CrossRef] [PubMed]
- Toomre, D. Cellular imaging using total internal reflection fluorescence microscopy: Theory and instrumentation. Cold Spring Harb. Protoc. 2012, 2012, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Merino, D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J. Neurochem. 2015, 135, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Toward fluorescence nanoscopy. Nat. Biotechnol. 2003, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Dyba, M.; Jakobs, S. Concepts for nanoscale resolution in fluorescence microscopy. Curr. Opin. Neurobiol. 2004, 14, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-High resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Rossy, J.; Owen, D.M.; Williamson, D.J.; Yang, Z.M.; Gaus, K. Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat. Immunol. 2013, 14, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, J.; Guo, X.; Tong, T.; Shi, X.; Li, L.; Qi, M.; Wang, Y.; Cai, M.; Jiang, J. Regulation of EGFR nanocluster formation by ionic protein-Lipid interaction. Cell Res. 2014, 24, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Oddone, A.; Vilanova, I.V.; Tam, J.; Lakadamyali, M. Super-Resolution imaging with stochastic single-Molecule localization: Concepts, technical developments, and biological applications. Microsc. Res. Tech. 2014, 77, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Van Engelenburg, S.B.; Lippincott-Schwartz, J. Superresolution imaging of biological systems using photoactivated localization microscopy. Chem. Rev. 2014, 114, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.; Huang, B.; Dempsey, G.T.; Zhuang, X.W. Multicolor super-Resolution imaging with photo-Switchable fluorescent probes. Science 2007, 317, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, U.; Malkusch, S.; Flottmann, B.; Mondry, J.; Liguzinski, P.; Verveer, P.J.; Heilemann, M. Chemically induced photoswitching of fluorescent probes-A general concept for super-Resolution microscopy. Molecules 2011, 16, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.R. Domain structure in model membrane bilayers investigated by simultaneous atomic force microscopy and fluorescence imaging. Langmuir 2003, 19, 8358–8363. [Google Scholar] [CrossRef]
- Frankel, D.J.; Pfeiffer, J.R.; Surviladze, Z.; Johnson, A.E.; Oliver, J.M.; Wilson, B.S.; Burns, A.R. Revealing the topography of cellular membrane domains by combined atomic force microscopy/fluorescence imaging. Biophys. J. 2006, 90, 2404–2413. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.M.; Müller, D.J. Analyzing focal adhesion structure by atomic force microscopy. J. Cell Sci. 2005, 118, 5315–5323. [Google Scholar] [CrossRef] [PubMed]
- Haga, H.; Sasaki, S.; Kawabata, K.; Ito, E.; Ushiki, T.; Sambongi, T. Elasticity mapping of living fibroblasts by AFM and immunofluorescence observation of the cytoskeleton. Ultramicroscopy 2000, 82, 253–258. [Google Scholar] [CrossRef]
- Charras, G.T.; Horton, M.A. Single cell mechanotransduction and its modulation analyzed by atomic force microscope indentation. Biophys. J. 2002, 82, 2970–2981. [Google Scholar] [CrossRef]
- Hecht, E.; Knittel, P.; Felder, E.; Dietl, P.; Mizaikoff, B.; Kranz, C. Combining atomic force-Fluorescence microscopy with a stretching device for analyzing mechanotransduction processes in living cells. Analyst 2012, 137, 5208–5214. [Google Scholar] [CrossRef] [PubMed]
- Alsteens, D.; Newton, R.; Schubert, R.; Martinez-Martin, D.; Delguste, M.; Roska, B.; Müller, D.J. Nanomechanical mapping of first binding steps of a virus to animal cells. Nat. Nanotechnol. 2017, 12, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Delcea, M.; Schmidt, S.; Palankar, R.; Fernandes, P.A.L.; Fery, A.; Moehwald, H.; Skirtach, A.G. Mechanobiology: Correlation between mechanical stability of microcapsules studied by AFM and impact of cell-Induced stresses. Small 2010, 6, 2858–2862. [Google Scholar] [CrossRef] [PubMed]
- Palankar, R.; Pinchasik, B.-E.; Schmidt, S.; De Geest, B.G.; Fery, A.; Moehwald, H.; Skirtach, A.G.; Delcea, M. Mechanical strength and intracellular uptake of CaCO3-Templated LbL capsules composed of biodegradable polyelectrolytes: The influence of the number of layers. J. Mater. Chem. B 2013, 1, 1175–1181. [Google Scholar] [CrossRef]
- Brown, A.E.X.; Hategan, A.; Safer, D.; Goldman, Y.E.; Discher, D.E. Cross-Correlated TIRF/AFM reveals asymmetric distribution of force-Generating heads along self-Assembled, “synthetic” myosin filaments. Biophys. J. 2009, 96, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Gudzenko, T.; Franz, C.M. Studying early stages of fibronectin fibrillogenesis in living cells by atomic force microscopy. Mol. Biol. Cell 2015, 26, 3190–3204. [Google Scholar] [CrossRef] [PubMed]
- Oreopoulos, J.; Yip, C.M. Probing membrane order and topography in supported lipid bilayers by combined polarized total internal reflection fluorescence-Atomic force microscopy. Biophys. J. 2009, 96, 1970–1984. [Google Scholar] [CrossRef] [PubMed]
- Oreopoulos, J.; Epand, R.F.; Epand, R.M.; Yip, C.M. Peptide-Induced domain formation in supported lipid bilayers: Direct evidence by combined atomic force and polarized total internal reflection fluorescence microscopy. Biophys. J. 2010, 98, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.B.; Truskey, G.A.; Monty Reichert, W. Atomic force and total internal reflection fluorescence microscopy for the study of force transmission in endothelial cells. Biophys. J. 2000, 78, 1725–1735. [Google Scholar] [CrossRef]
- Trache, A.; Lim, S.-M. Live cell response to mechanical stimulation studied by integrated optical and atomic force microscopy. J. Vis. Exp. 2010, 44, 2072. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-M.; Trzeciakowski, J.P.; Sreenivasappa, H.; Dangott, L.J.; Trache, A. RhoA-Induced cytoskeletal tension controls adaptive cellular remodeling to mechanical signaling. Integr. Biol. 2012, 4, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Harke, B.; Chacko, J.; Haschke, H.; Canale, C.; Diaspro, A. A novel nanoscopic tool by combining AFM with STED microscopy. Opt. Nanoscopy 2012, 1, 1–6. [Google Scholar] [CrossRef]
- Chacko, J.V.; Canale, C.; Harke, B.; Diaspro, A. Sub-Diffraction nano manipulation using STED AFM. PLoS ONE 2013, 8, e66608. [Google Scholar] [CrossRef] [PubMed]
- Chacko, J.V.; Harke, B.; Canale, C.; Diaspro, A. Cellular level nanomanipulation using atomic force microscope aided with superresolution imaging. J. Biomed. Opt. 2014, 19, 105003. [Google Scholar] [CrossRef] [PubMed]
- Chacko, J.V.; Zanacchi, F.C.; Diaspro, A. Probing cytoskeletal structures by coupling optical superresolution and afm techniques for a correlative approach. Cytoskeleton 2013, 70, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Monserrate, A.; Casado, S.; Flors, C. Correlative atomic force microscopy and localization-Based super-Resolution microscopy: Revealing labelling and image reconstruction artefacts. ChemPhysChem 2014, 15, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Opazo, F.; Levy, M.; Byrom, M.; Schaefer, C.; Geisler, C.; Groemer, T.W.; Ellington, A.D.; Rizzoli, S.O. Aptamers as potential tools for super-Resolution microscopy. Nat. Methods 2012, 9, 938–939. [Google Scholar] [CrossRef] [PubMed]
- Leenheer, D.; ten Dijke, P.; Hipolito, C.J. A current perspective on applications of macrocyclic-Peptide-Based high-Affinity ligands. Pept. Sci. 2016, 106, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Shim, S.-H.; He, J.; Zhuang, X. Fast, three-Dimensional super-Resolution imaging of live cells. Nat. Methods 2011, 8, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Chmyrov, A.; Keller, J.; Grotjohann, T.; Ratz, M.; d’Este, E.; Jakobs, S.; Eggeling, C.; Hell, S.W. Nanoscopy with more than 100,000 ‘doughnuts’. Nat. Methods 2013, 10, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Toshio, A. High-Speed atomic force microscopy coming of age. Nanotechnology 2012, 23, 062001. [Google Scholar]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Cai, M.; Tong, T.; Wang, H. Progress in the Correlative Atomic Force Microscopy and Optical Microscopy. Sensors 2017, 17, 938. https://doi.org/10.3390/s17040938
Zhou L, Cai M, Tong T, Wang H. Progress in the Correlative Atomic Force Microscopy and Optical Microscopy. Sensors. 2017; 17(4):938. https://doi.org/10.3390/s17040938
Chicago/Turabian StyleZhou, Lulu, Mingjun Cai, Ti Tong, and Hongda Wang. 2017. "Progress in the Correlative Atomic Force Microscopy and Optical Microscopy" Sensors 17, no. 4: 938. https://doi.org/10.3390/s17040938
APA StyleZhou, L., Cai, M., Tong, T., & Wang, H. (2017). Progress in the Correlative Atomic Force Microscopy and Optical Microscopy. Sensors, 17(4), 938. https://doi.org/10.3390/s17040938