Deregulation of Hepatic Mek1/2–Erk1/2 Signaling Module in Iron Overload Conditions


 , and
, and 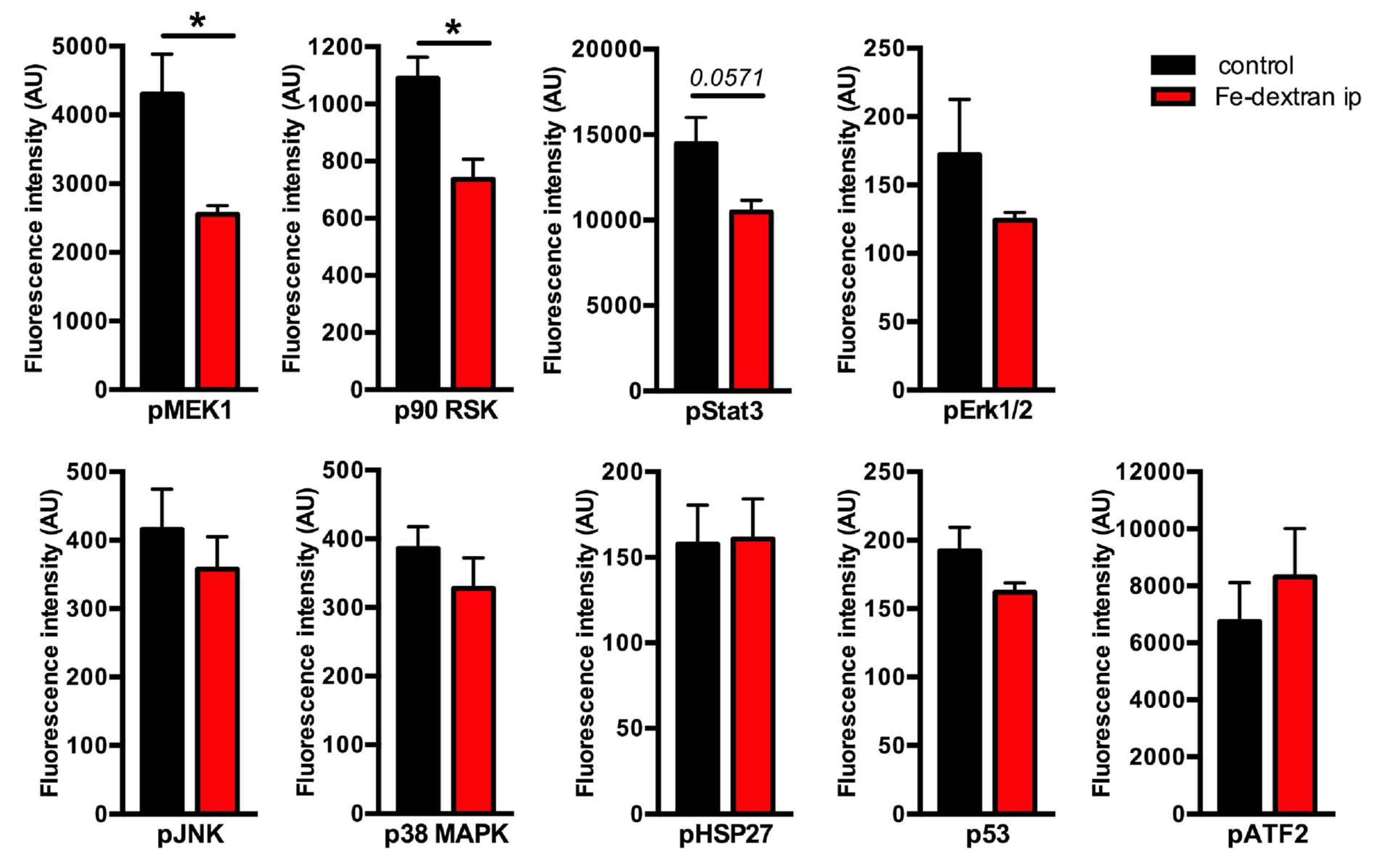{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
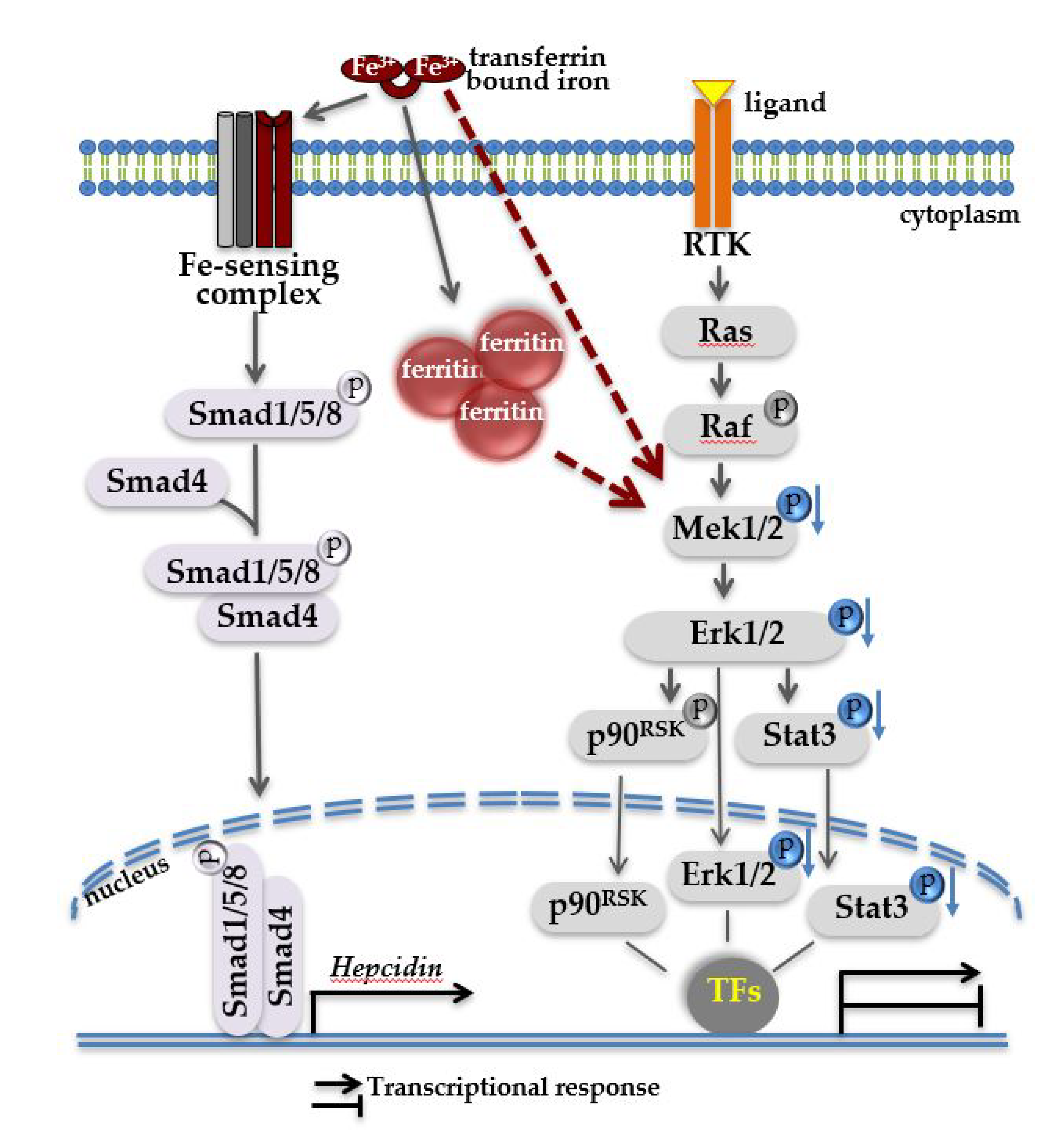
2. Results and Discussion
2.1. Classical and Stress-Induced MAPKs Activation in Iron-Loaded Livers
2.2. Association of Mitogen-activated Protein Kinases (MAPK) Activity with Hepatic Iron Overload
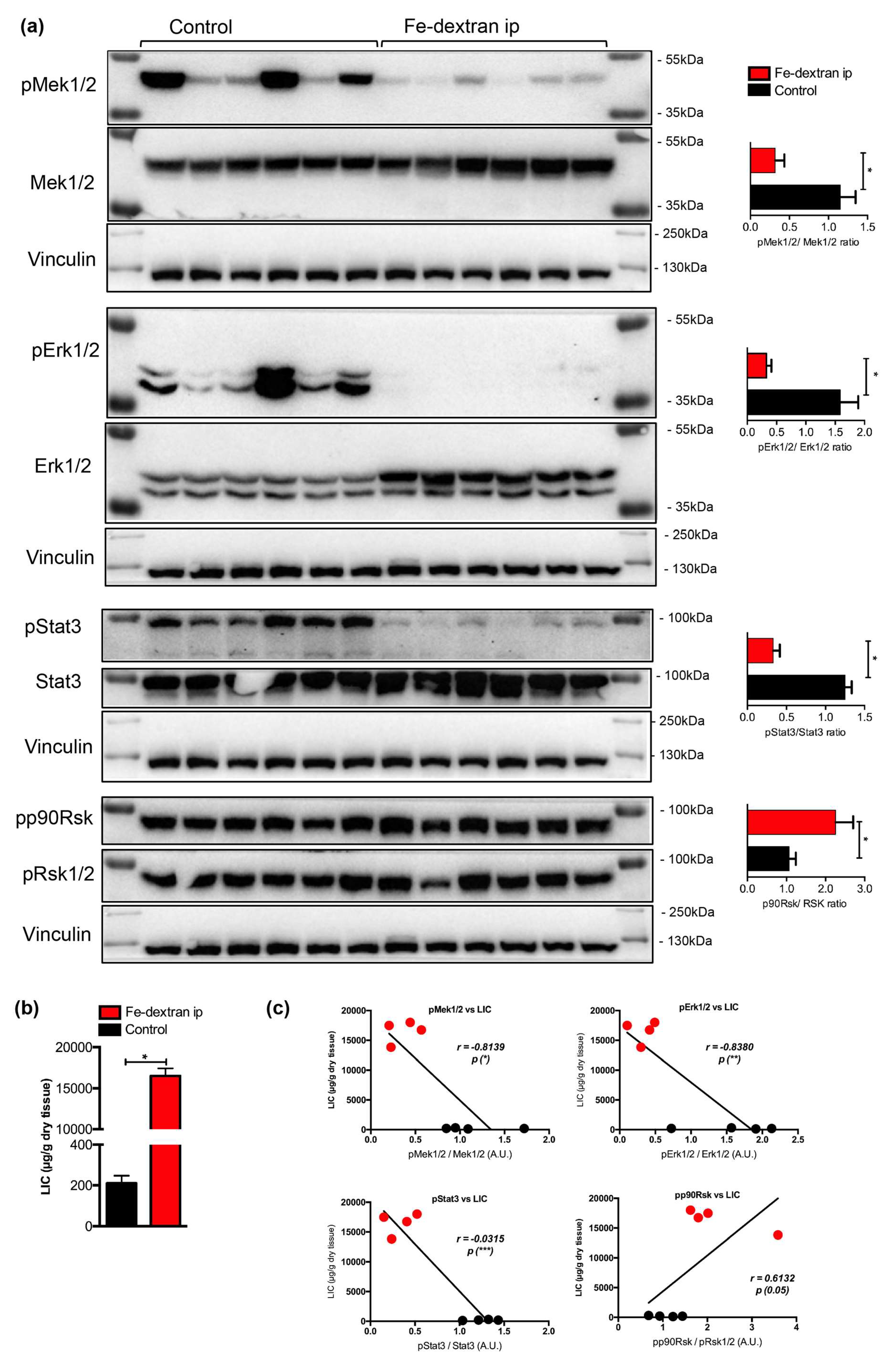
2.3. Nutritional Iron Overload is Characterized by Low Hepatic Mek1/2-Erk1/2-Stat3 Signaling
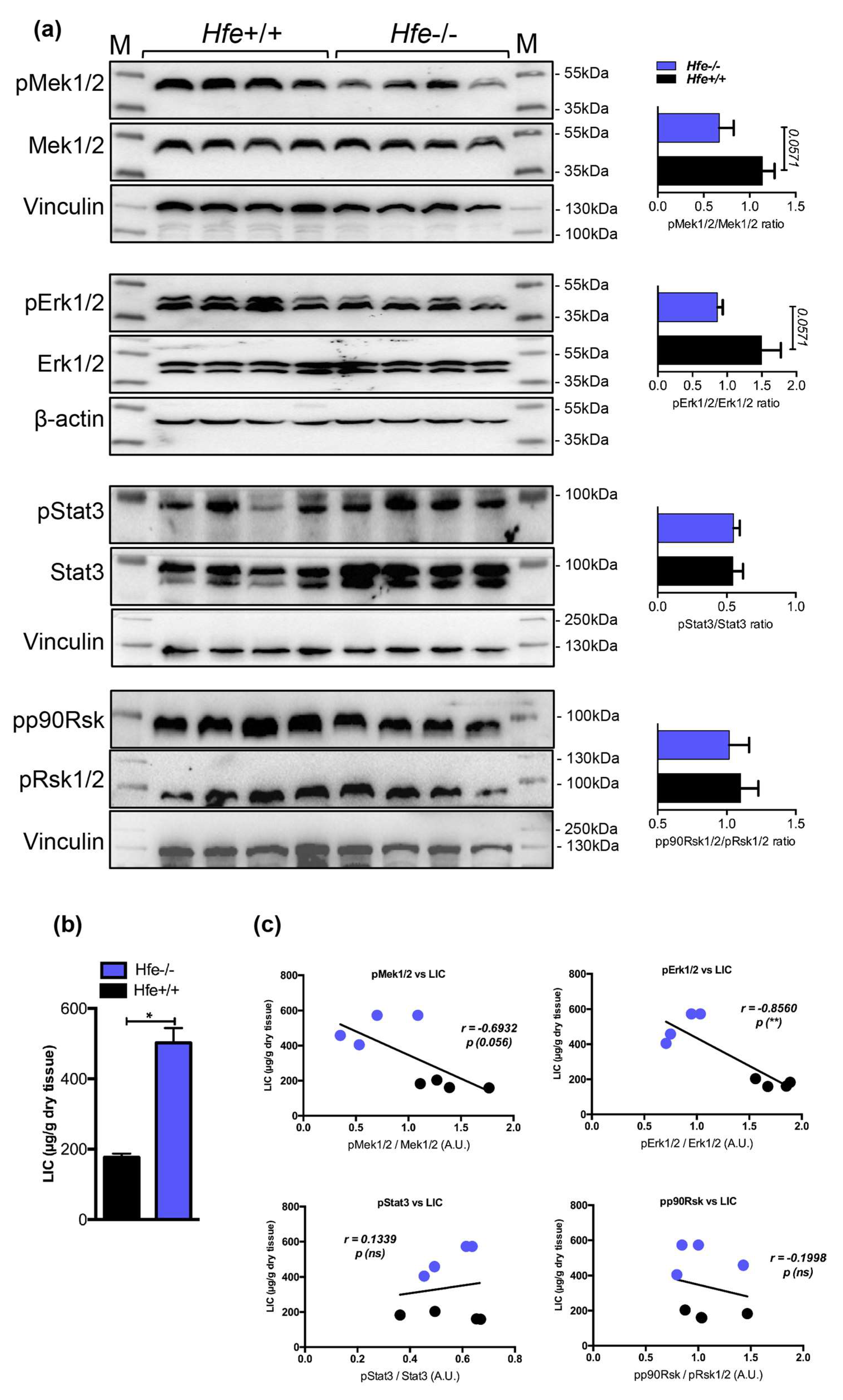
2.4. Low Hepatic Mek1/2-Erk1/2 Signaling is Present in Hfe-/- Mice in Spite of Low Bmp-Smad Signaling
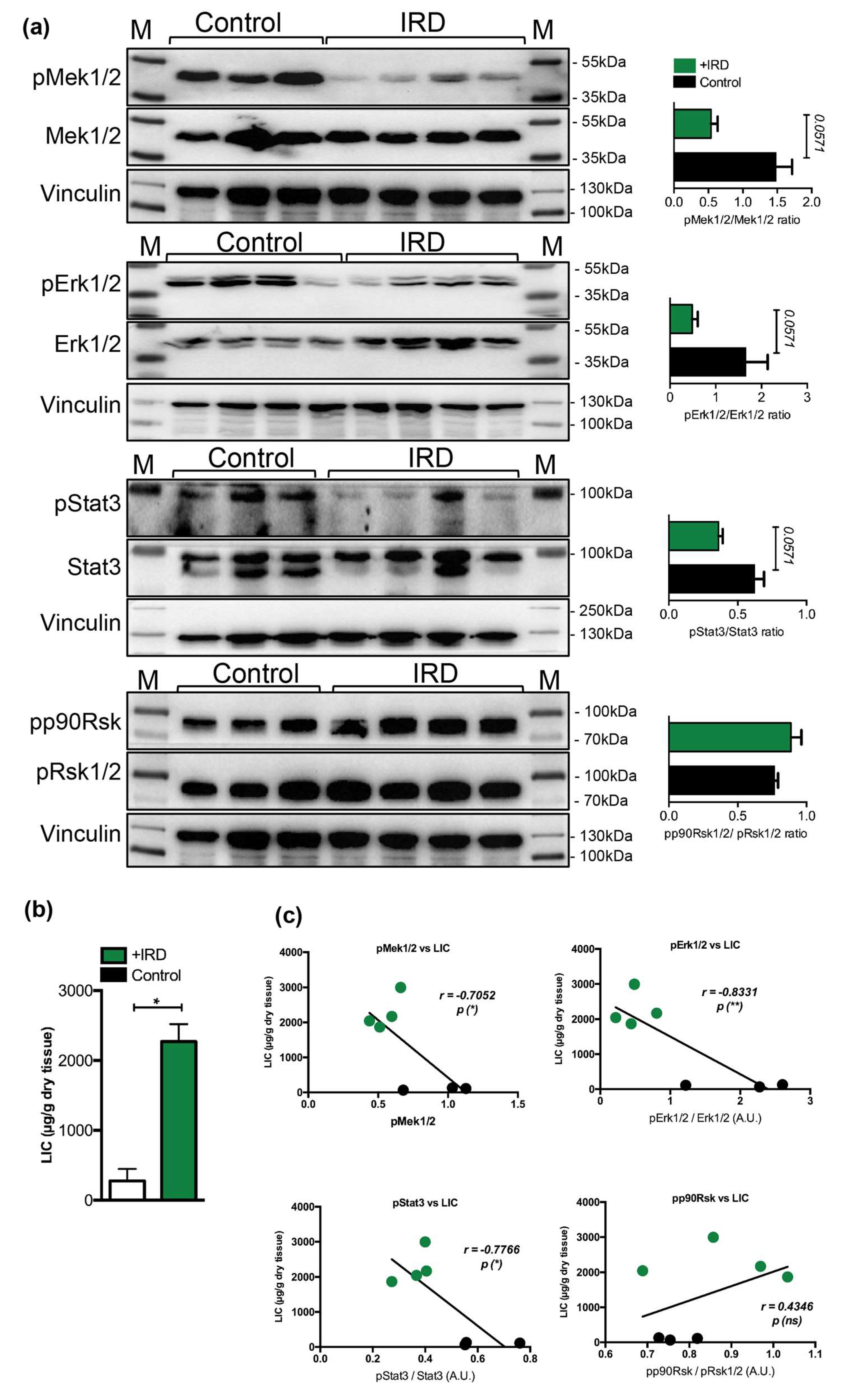
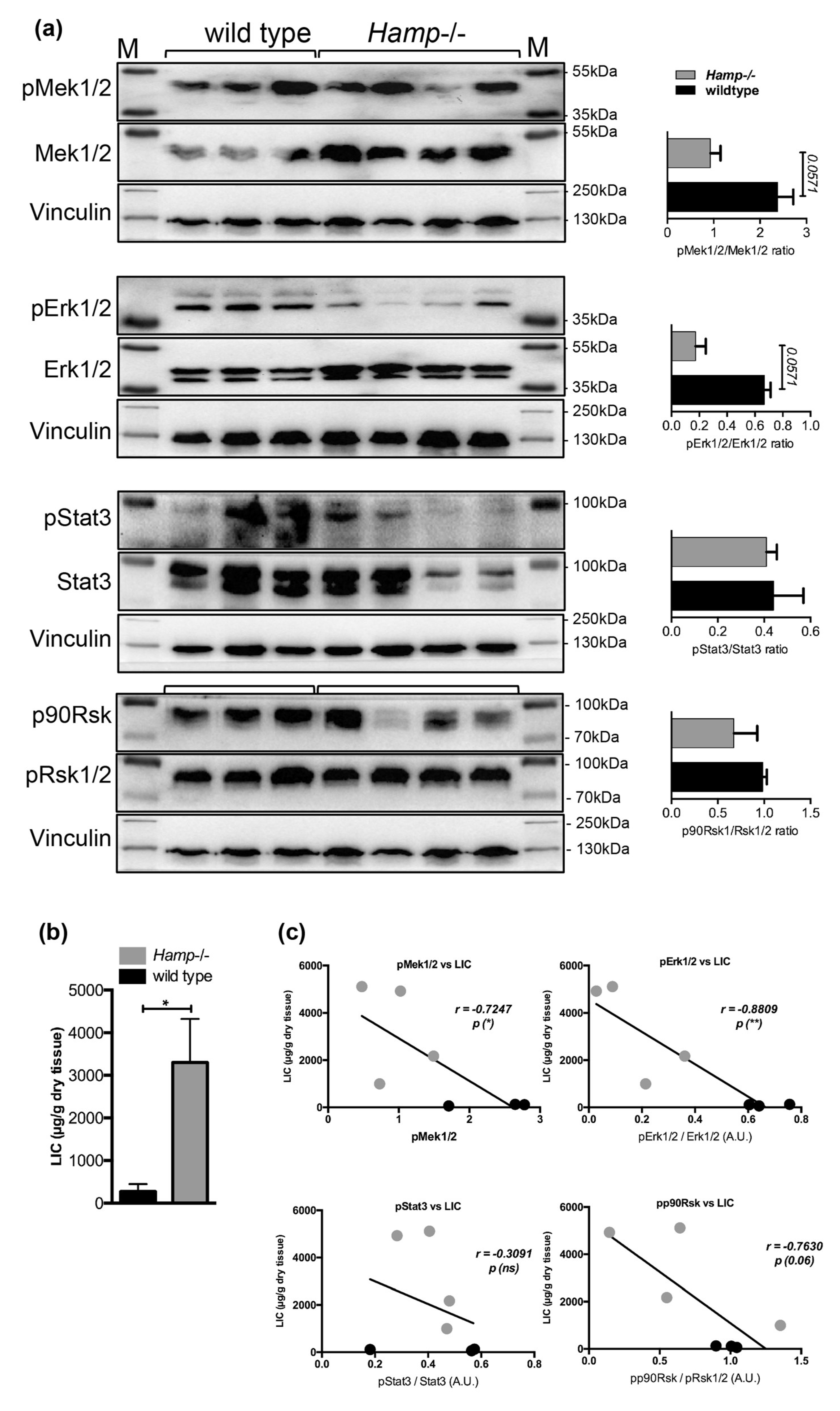
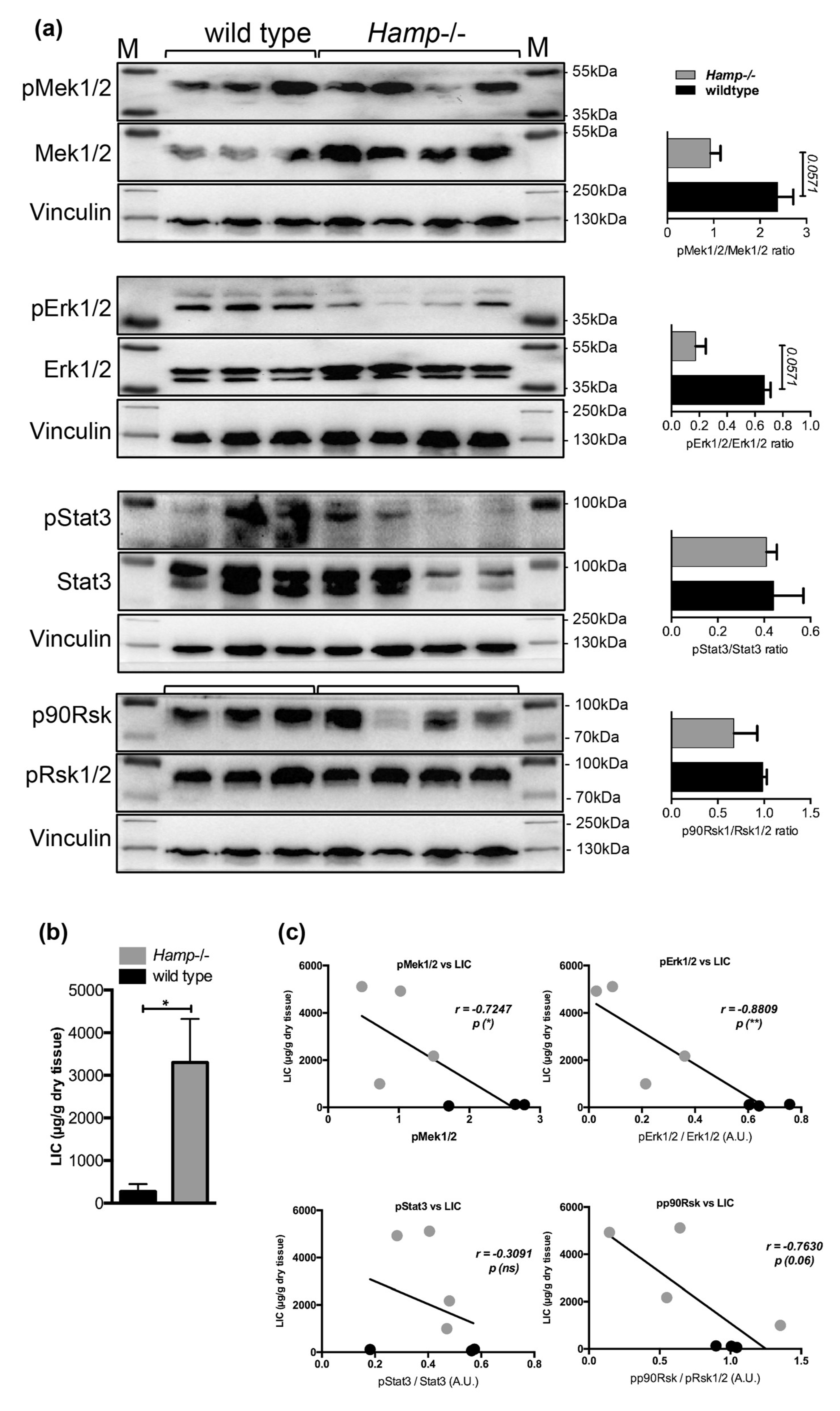
2.5. Low Hepatic Mek1/2-Erk1/2 Signaling is Present in Hepcidin-Deficient Mice and is Further Aggravated by Iron Excess
3. Materials and Methods
3.1. Mice and Treatments
3.2. Phosphoprotein Analysis in Liver Lysates
3.3. Protein Isolation and Immunoblot Analysis
3.4. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lesbordes-Brion, J.C.; Viatte, L.; Bennoun, M.; Lou, D.Q.; Ramey, G.; Houbron, C.; Hamard, G.; Kahn, A.; Vaulont, S. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 2006, 108, 1402–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deugnier, Y.; Turlin, B. Pathology of hepatic iron overload. World J. Gastroenterol. 2007, 13, 4755–4760. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron Overload in Patients with Chronic Liver Disease. Gastroenterol. Hepatol. 2016, 12, 695–698. [Google Scholar]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Vujic Spasic, M.; Kiss, J.; Herrmann, T.; Galy, B.; Martinache, S.; Stolte, J.; Grone, H.J.; Stremmel, W.; Hentze, M.W.; Muckenthaler, M.U. Hfe acts in hepatocytes to prevent hemochromatosis. Cell Metab. 2008, 7, 173–178. [Google Scholar] [CrossRef]
- Wallace, D.F.; Summerville, L.; Subramaniam, V.N. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 2007, 132, 301–310. [Google Scholar] [CrossRef]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef]
- Roetto, A.; Daraio, F.; Alberti, F.; Porporato, P.; Cali, A.; De Gobbi, M.; Camaschella, C. Hemochromatosis due to mutations in transferrin receptor 2. Blood Cells Mol. Dis. 2002, 29, 465–470. [Google Scholar] [CrossRef]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Investig. 2005, 115, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Steinbicker, A.U.; Bartnikas, T.B.; Lohmeyer, L.K.; Leyton, P.; Mayeur, C.; Kao, S.M.; Pappas, A.E.; Peterson, R.T.; Bloch, D.B.; Yu, P.B.; et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 2011, 118, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.D.; Ryan, E.; Fabre, A.; Lawless, M.W.; Crowe, J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology 2010, 52, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Vujic Spasic, M.; Sparla, R.; Mleczko-Sanecka, K.; Migas, M.C.; Breitkopf-Heinlein, K.; Dooley, S.; Vaulont, S.; Fleming, R.E.; Muckenthaler, M.U. Smad6 and Smad7 are co-regulated with hepcidin in mouse models of iron overload. Biochim. Biophys. Acta 2013, 1832, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzolari, A.; Raggi, C.; Deaglio, S.; Sposi, N.M.; Stafsnes, M.; Fecchi, K.; Parolini, I.; Malavasi, F.; Peschle, C.; Sargiacomo, M.; et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 2006, 119, 4486–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, M.; Luscieti, S.; Gandini, V.; Maccarinelli, F.; Finazzi, D.; Silvestri, L.; Roetto, A.; Arosio, P. Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation. Haematologica 2010, 95, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramey, G.; Deschemin, J.C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Boil. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Bennett, A.M. Mitogen-Activated Protein Kinase Regulation in Hepatic Metabolism. Trends Endocrinol. Metab. 2017, 28, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Browning, A.C.; Garner, A.N.; Kelly, D.P. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: A potential role in the cardiac metabolic stress response. J. Boil. Chem. 2001, 276, 44495–44501. [Google Scholar] [CrossRef]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Aouadi, M.; Caron, L.; Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Subramaniam, S.; Dramane, G.; Khelifi, D.; Khan, N.A. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie 2017, 137, 78–87. [Google Scholar] [CrossRef]
- Kujiraoka, T.; Satoh, Y.; Ayaori, M.; Shiraishi, Y.; Arai-Nakaya, Y.; Hakuno, D.; Yada, H.; Kuwada, N.; Endo, S.; Isoda, K.; et al. Hepatic extracellular signal-regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J. Am. Heart Assoc. 2013, 2, e000361. [Google Scholar] [CrossRef]
- Yu, Y.; Richardson, D.R. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J. Boil. Chem. 2011, 286, 15413–15427. [Google Scholar] [CrossRef]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Boil. 1997, 17, 6508–6516. [Google Scholar] [CrossRef]
- Lunova, M.; Goehring, C.; Kuscuoglu, D.; Mueller, K.; Chen, Y.; Walther, P.; Deschemin, J.C.; Vaulont, S.; Haybaeck, J.; Lackner, C.; et al. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J. Hepatol. 2014, 61, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Zerrad-Saadi, A.; Lambert-Blot, M.; Mitchell, C.; Bretes, H.; Collin de l’Hortet, A.; Baud, V.; Chereau, F.; Sotiropoulos, A.; Kopchick, J.J.; Liao, L.; et al. GH receptor plays a major role in liver regeneration through the control of EGFR and ERK1/2 activation. Endocrinology 2011, 152, 2731–2741. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life 2000, 50, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Horne, W.; Kamimura, S.; Niemela, O.; Parkkila, S.; Yla-Herttuala, S.; Brittenham, G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Investig. 1995, 96, 620–630. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Vega, M.I.; Chatterjee, D.; Goodglick, L.; Bonavida, B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res. 2004, 64, 7117–7126. [Google Scholar] [CrossRef]
- Rouault, T.A. Hepatic iron overload in alcoholic liver disease: Why does it occur and what is its role in pathogenesis? Alcohol 2003, 30, 103–106. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Dominitz, J.A.; Weiss, N.S.; Heagerty, P.J.; Kowdley, K.V. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 2004, 126, 1293–1301. [Google Scholar] [CrossRef]
- Harrison-Findik, D.D.; Klein, E.; Crist, C.; Evans, J.; Timchenko, N.; Gollan, J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979–1985. [Google Scholar] [CrossRef] [Green Version]
- Aroor, A.R.; Shukla, S.D. MAP kinase signaling in diverse effects of ethanol. Life Sci. 2004, 74, 2339–2364. [Google Scholar] [CrossRef] [PubMed]
- Sampey, B.P.; Stewart, B.J.; Petersen, D.R. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J. Boil. Chem. 2007, 282, 1925–1937. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ishac, E.J.; Dent, P.; Kunos, G.; Gao, B. Effects of ethanol on mitogen-activated protein kinase and stress-activated protein kinase cascades in normal and regenerating liver. Biochem. J. 1998, 334 Pt 3, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Marmur, J.; Beshara, S.; Eggertsen, G.; Onelov, L.; Albiin, N.; Danielsson, O.; Hultcrantz, R.; Stal, P. Hepcidin levels correlate to liver iron content, but not steatohepatitis, in non-alcoholic fatty liver disease. BMC Gastroenterol. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Datz, C.; Felder, T.K.; Niederseer, D.; Aigner, E. Iron homeostasis in the metabolic syndrome. Eur. J. Clin. Investig. 2013, 43, 215–224. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; Network, N.C.R. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron metabolism in Nonalcoholic Fatty Liver Disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef]
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Q.; Greten, T.F. Nonalcoholic fatty liver disease promotes hepatocellular carcinoma through direct and indirect effects on hepatocytes. FEBS J. 2018, 285, 752–762. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Vanni, E.; Grieco, A.; Miele, L.; Consonni, D.; Fatta, E.; Lombardi, R.; Marchesini, G.; et al. Risk of nonalcoholic steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease and low visceral adiposity. J. Hepatol. 2011, 54, 1244–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver Int. 2011, 31, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Sugimoto, R.; Takeo, M.; Urawa, N.; Mifuji, R.; Tanaka, H.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Adachi, Y.; et al. Hepcidin expression in the liver: Relatively low level in patients with chronic hepatitis C. Mol. Med. 2007, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Nishina, S.; Hino, K.; Korenaga, M.; Vecchi, C.; Pietrangelo, A.; Mizukami, Y.; Furutani, T.; Sakai, A.; Okuda, M.; Hidaka, I.; et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 2008, 134, 226–238. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangudu, N.K.; Buth, N.; Strnad, P.; Cirstea, I.C.; Spasić, M.V. Deregulation of Hepatic Mek1/2–Erk1/2 Signaling Module in Iron Overload Conditions. Pharmaceuticals 2019, 12, 70. https://doi.org/10.3390/ph12020070
Tangudu NK, Buth N, Strnad P, Cirstea IC, Spasić MV. Deregulation of Hepatic Mek1/2–Erk1/2 Signaling Module in Iron Overload Conditions. Pharmaceuticals. 2019; 12(2):70. https://doi.org/10.3390/ph12020070
Chicago/Turabian StyleTangudu, Naveen Kumar, Nils Buth, Pavel Strnad, Ion C. Cirstea, and Maja Vujić Spasić. 2019. "Deregulation of Hepatic Mek1/2–Erk1/2 Signaling Module in Iron Overload Conditions" Pharmaceuticals 12, no. 2: 70. https://doi.org/10.3390/ph12020070
APA StyleTangudu, N. K., Buth, N., Strnad, P., Cirstea, I. C., & Spasić, M. V. (2019). Deregulation of Hepatic Mek1/2–Erk1/2 Signaling Module in Iron Overload Conditions. Pharmaceuticals, 12(2), 70. https://doi.org/10.3390/ph12020070