3. Lectures
3.1. Radio-Iodination Using a C-H Activation Approach (L1)
Thomas Cailly 1,2
1
Normandie Univ, UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), 14000 Caen, France
2
Department of Nuclear Medicine, CHU Côte de Nacre, 14000 Caen, France
Radio-iodinated molecules are used in a wide range of scientific fields, from life sciences to human health applications. These radiotracers are prepared from pre-functionalized chemical precursors, requiring important and time-consuming synthetic efforts. The development of general and easy to implement methods providing access to complex radio-iodinated molecules is of paramount importance to accelerate the discovery process in these fields. Recently transition metal mediated C-H activation has allowed late stage functionalization of complex substrates while using chelating groups already present on these structures. We herein report the palladium mediated C-H radio-iodination of arenes using sodium iodide as the primary isotopic source. This reaction can be performed without chemical know-how in 30 min and has been applied to the synthesis of complex radio-iodinated compounds of biological importance.
3.2. Late-Stage C-H arylation of thiazolo[5,4-f]quinazolin-9(8H)-One Backbone: Synthesis of an Array of Potential Kinase Inhibitors (L2)
Corinne Fruit
Normandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA UMR 6014, F-76000 Rouen, France
Our research group was focused on the synthesis of a novel library of thiazolo[5,4-f]quinazolin-(on)es as potential kinase inhibitors involved to some extent in Alzheimer’s disease. We previously reported that 4-N-protected thiazolo[5,4-f]quinazolines displayed single-digit nanomolar IC50 values and are among the most potent DYRK1A/1B inhibitors disclosed to date (Nguyen, T. L., et al. Expert Opin. Ther. Pat. 2017, 11, 1183–1199; Courtadeur, S., et al. J. Neurochem. 2015, 133, 440–451). Driven by the design of an efficient route allowing a direct transformation of thiazoloquinazolin-4-one derivatives identified as kinase inhibitors (Hédou, D., et al. Molecules 2016, 21, 578; Hédou, D., et al. Molecules 2016, 21, 794), the synthesis of an array of C2 and/or C7 arylated compounds was further envisioned. In this context, transition-metal-catalyzed C-C coupling of heteroarene through C-H arylation represents an extremely attractive approach (Rossi, R., et al. Tetrahedron 2016, 72, 1795–1837; Segawa, Y., et al. Angew. Chem. Int. Ed. 2015, 54, 66–81; Ackermann, L. Chem. Rev. 2011, 111, 1315–1345). This methodology has emerged as an important tool for incorporating structural diversity into complex nitrogen containing heterocycles (Aziz, J., et al. Synthesis 2017, 49, 4562–4585; Fruit, C. Science 2016, 352, 1277–1278; Wencel-Delord, J., et al. Nat. Chem. 2013, 5, 369–375). Following our previous study on C-H arylation of quinazolinone skeleton (Laclef, S., et al. Org. Lett. 2015, 17, 1700–1703; Godeau, J., et al. Eur. J. Org. Chem. 2015, 7705–7717), a selective palladium-catalyzed and copper-assisted direct C-H (hetero)-arylation of thiazolo[5,4-f]quinazolin-9(8H)-one has been developed with aryl halides as coupling partners under microwave irradiation(Besson, T., et al. Synthesis 2016, 48, 3879–3889). Electron-deficient heteroarenes are also readily introduced, a notable feature with respect to medicinal agent synthesis (Couly, F., et al. Synthesis 2017, 49, 4615–4622; Harari, M., et al. Org. Lett. 2016, 18, 3282–3285).
Differently substituted N8-protected-aryl-thiazoloquinazolin-9(8H)-ones were thereby obtained in a facile manner. This late-stage functionalization of drug candidates allows a streamlined route to compounds with increased novelty, essential in drug discovery.
3.3. Disrupting the Metabolic Adaptation of Cancer Cells with Small Molecules as a Promising Anticancer Strategy (L3)
Raphaël Frédérick
Université Catholique de Louvain (UCL), Medicinal Chemistry Research Group, Louvain Drug Research Institute (LDRI), 73 avenue Mounier, B-1200 Bruxelles, Belgium
Our current research efforts aims at providing necessary pharmacological or diagnostic tools to detail important evolutionary metabolic strategies allowing cancer cells to cope with fluctuating resource availability and grow. In very recent years, we have been interested in three different axes: the design and study of serine synthetic pathway (SSP) inhibitors, the development of novel original diagnostic tools to study the exchange of lactate between hypoxic/glycolytic cancer cells, and more recently we have started to develop first-in-class lactate dehydrogenase B (LDHB) tetramerization inhibitors. Our last results in this field will be presented (Van Hée, V.F., et al. Oncotarget 2017, 8, 24415–24428; Ravez, S. et al. J. Med. Chem. 2017, 60, 1591–1597; Ravez, S. et al. J. Med. Chem. 2017, 60, 1227–1237; Brisson, L. et al. Cancer Cell 2016, 30, 418–431).
3.4. Benzoxepin-Type Selective Estrogen Receptor (ER) Modulators and Downregulators with Subtype-Specific ERα and ERβ Activity (L4)
Niamh M. O’Boyle
School of Pharmacy and Pharmaceutical Sciences, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St, Dublin 2 D02 R590, Ireland
The two nuclear estrogen receptors (ERα and ERβ) mediate the biological effects of the estrogen hormones and ERα is an attractive therapeutic target for diseases including breast cancer and osteoporosis. Estrogens are known to have tissue selective effects, and there is considerable interest in the therapeutic use of selective estrogen receptor modulators (SERMs). ERα is an important target for drugs such as tamoxifen and fulvestrant. There is ongoing debate about the role of ERβ in cancer.
Three series of ER-ligands based on the benzoxepin scaffold structure were synthesized—series I containing an acrylic acid, series II with an acrylamide and series III with a saturated carboxylic acid substituent. These compounds were shown to be high affinity ligands for the ER with nanomolar IC50 binding values. Series I acrylic acid ligands were generally ERα selective. In particular, compound 13e featuring a phenylpenta-2,4-dienoic acid substituent was shown to be antiproliferative and downregulated ERα and ERβ expression in MCF-7 breast cancer cells. Interestingly, from series III, the phenoxybutyric acid derivative compound 22 was not antiproliferative and selectively downregulated ERβ. A docking study of the benzoxepin ligands was undertaken. Compound 13e is a promising lead for development as a clinically relevant SERD, whilst compound 22 will be a useful experimental probe for helping to elucidate the role of ERβ in cancer cells.
3.5. Combining Organofluorine Chemistry and C-H Activation: New Tools to Fluorinated Molecules (L5)
Tatiana Besset
Normandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA UMR 6014, F-76000 Rouen, France
Over the last years, the organofluorine research field has known a fast expansion (Liu, H. et al. Adv. Synth. Catal. 2013, 355, 617–626; Furuya, T., et al. Nature 2011, 473, 470–477; Besset, T., et al. Angew. Chem. Int. Ed. 2012, 51, 5048–5050; Wu, X.-F., et al. Chem. Asian J. 2012, 7, 1744–1754; Landelle, G., et al. Beilstein J. Org. Chem. 2013, 9, 2476–2536; Besset, T., et al. Chem. Eur. J. 2014, 20, 16830–16845; Besset, T., et al. Eur. J. Org. Chem. 2015, 2765–2789), which could be explained by the plethora of pharmaceuticals and agrochemicals containing at least one fluorine atom (Wang, J., et al. Chem. Rev. 2014, 114, 2432–2506; Ilardi, E. A., et al. J. Med. Chem. 2014, 57, 2832–2842; Gillis, E. P., et al. J. Med. Chem. 2015, 58, 8315–8359; Purser, S., et al. Chem. Soc. Rev. 2008, 37, 320–330; Landelle, G., et al. Curr. Top. Med. Chem. 2014, 14, 941–951). Consequently, a large panel of fluorinated groups (F, CF3, SCF3, CF2R…) is available and their introduction onto molecules can be realized by means of various transformations. Besides, transition metal catalyzed direct C-H bond functionalization has witnessed tremendous progress over the last decade allowing new retrosynthetic disconnections and innovative approaches. The combination of organofluorine chemistry and the transition metal catalyzed C-H bond functionalization is really appealing and is considered as a powerful synthetic tool. However, the introduction of fluorine-containing groups on versatile alkenes as well as aliphatic derivatives by direct C-H bond functionalization is still scarce. To take up this challenge, we have developed innovative methodologies to access these important fluorinated building blocks. Transition metal catalyzed trifluoromethylation (Besset, T., et al. J. Org. Chem. 2014, 79, 413–418) and trifluoromethylthiolation (Xiong, H.-Y., et al. J. Org. Chem. 2015, 80, 4204–4212; Zhao, Q., et al. Org. Lett. 2017, 19, 5106–5109) of vinylic and aliphatic amides were particularly studied. Besides, a special attention was paid to the development of modern strategies in organofluorine chemistry with a special focus on emergent fluorinated groups and the design of original electrophilic reagents (Xiong, H.-Y., et al. Org. Chem. Front. 2016, 3, 620–624; Xiong, H.-Y., et al. Angew. Chem. Int. Ed. 2016, 55, 13490–13494).
![Pharmaceuticals 12 00073 i005]()
3.6. Molecular Interrogation of Multiple Signalling States of the β1-Adrenoceptor (L9)
Shailesh N. Mistry 1,*
and
Jillian G. Baker 2
1
School of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, Nottingham NG7 2RD, UK
2
Cell Signalling, The University of Nottingham Medical School, Queen’s Medical Centre, Nottingham NG7 2UH, UK
The β1-adrenoceptor (β1-AR) is a predominantly cardiac G protein-coupled receptor (GPCR) and regulates both the rate and force of heartbeat. β1-AR blockade is important in managing symptoms and prolonging life in heart failure and ischaemic heart disease, by blocking the actions of endogenous catecholamine agonists—epinephrine and norepinephrine. Conventionally it was believed that β1-AR activation occurred solely through agonist binding to the catecholamine binding site and stabilisation of an active receptor conformation, however β-blockers (β1-AR antagonists) such as CGP12177 have been found to exhibit an unusual behaviour (Kaumann, A. J., et al. Pharmacol. Therapeut. 2008, 118, 303–336). It is now well established that the β1-AR exists in at least two agonist signalling states. A ‘primary conformation’, which is activated by endogenous catecholamines and competitively blocked by β-blockers with high binding affinity and, a ‘secondary conformation’, through which some ligands (e.g., CGP12177, pindolol, alprenolol) can generate agonist responses that are relatively resistant to β-blocker antagonism (Baker, J. G., et al. Mol. Pharmacol. 2003, 63, 1312–1321).
![Pharmaceuticals 12 00073 i006]()
Of concern, some β-blockers that exhibit this activity (e.g., carvedilol) and are used in the clinic reach plasma concentrations in patients that are sufficient to enable secondary conformation-mediated signalling. In order to understand this phenomenon further it is essential to elucidate the specific ligand-protein interactions and ligand structural features that are responsible for this behaviour. This presentation will summarise our findings to date of an ongoing ligand-based SAR study which started with two pharmacologically unusual β-blockers – CGP12177 and alprenolol.
3.7. Epigenetic Reactivation: HDAC and LSD1 Inhibitors as Anticancer Agents (L10)
A. Ganesan
School of Pharmacy, University of East Anglia, Norwich Research Park, Norwich NR4 7TJ, UK
The activation or silencing of genes in nucleosomes is regulated by structural modifications of the DNA double helix and the N-terminal tails of histone proteins. Two of the major histone modifications involve methylation or acetylation of lysine residues which are then removed by deacetylase (HDAC) and demethylase (KDM) enzymes. Both the HDACs and KDMs are important drug discovery targets, with five recently approved agents for the treatment of haematological cancers and multiple other candidates in clinical trials. The presentation will review the state of the art for HDAC and KDM inhibitors with illustrative examples based on natural products and synthetic scaffolds from our group (Lecointre, B., et al. Phil. Trans. R. Soc. B 2018, 373, 20170364; Conforti, F. et al. Oncotarget 2017, 8, 48737–48754; Borrello, M. T. et al. Bioorg. Med. Chem. Lett. 2017, 27, 2099–2101; Liu, Q. et al. RSC Adv. 2015, 5, 1109–1112; Tortorici, M., et al. ACS Chem. Biol. 2013, 8, 1677–1682).
3.8. New Azole Antifungals with Fused Pyrimidinone and Triazinone Derivatives (L11)
Cédric Logé 1,*,
David Montoir 1,
Rémi Guillon 1,
Alain Tonnerre 1,
Carine Picot 2,
Sophie Gazzola 2,
Fabrice Pagniez 2
and
Patrice Le Pape 2
1
Department of Medicinal Chemistry, University of Nantes, F-44200 Nantes, France
2
Department of Parasitology and Medical Mycology, University of Nantes, F-44200 Nantes, France
Azoles (fluconazole, itraconazole, voriconazole, posaconazole) are important drugs for the treatment of invasive fungal infections (IFIs). Unfortunately, their use as first intention has led to resistance. Besides azole antifungal agents, alternative classes include polyenes such as amphotericine B and its lipidic forms, as well as the echinocandines. However, these alternative antifungal agents are generally costly and not suitable for oral administration. Thus, there is a continued interest in developing azole antifungals in particular those able to circumvent resistance phenomenon’s which can be attributed either to mutations in ERG11 gene encoding the cytochrome P450 14α-demethylase (CYP51), or to an overexpression of genes encoding membrane transport proteins (CDR1, CDR2, and MDR1). Recently, isavuconazole, a new extended-spectrum triazole was also approved for the treatment of invasive aspergillosis and mucormycosis, an order of molds that most antifungals (voriconazole included) do not treat.
We have already reported the synthesis of a novel antifungal agent bearing a thiazoloquinazolinone scaffold (Guillon, R. et al. ACS Med. Chem. Lett. 2013, 4, 288–292). Taking into account that this compound has showed in vivo antifungal efficacy in a mice model of systemic candidiasis, we decided to synthesize new analogues with a more easily accessible pyrrolotriazinone or imidazopyrimidinone scaffolds (Logé, C., et al. PCT WO 2017/021178 A1). These compounds exhibit in vitro activity against pathogenic Candida species (fluconazole-susceptible and fluconazole-resistant) and promising activity against both Aspergillus fumigatus strains and a Rhizopus oryzae strain, the most prevalent agent of mucormycosis. Our lead compounds displayed also in vivo efficacy against three lethal systemic infections caused by C. albicans.
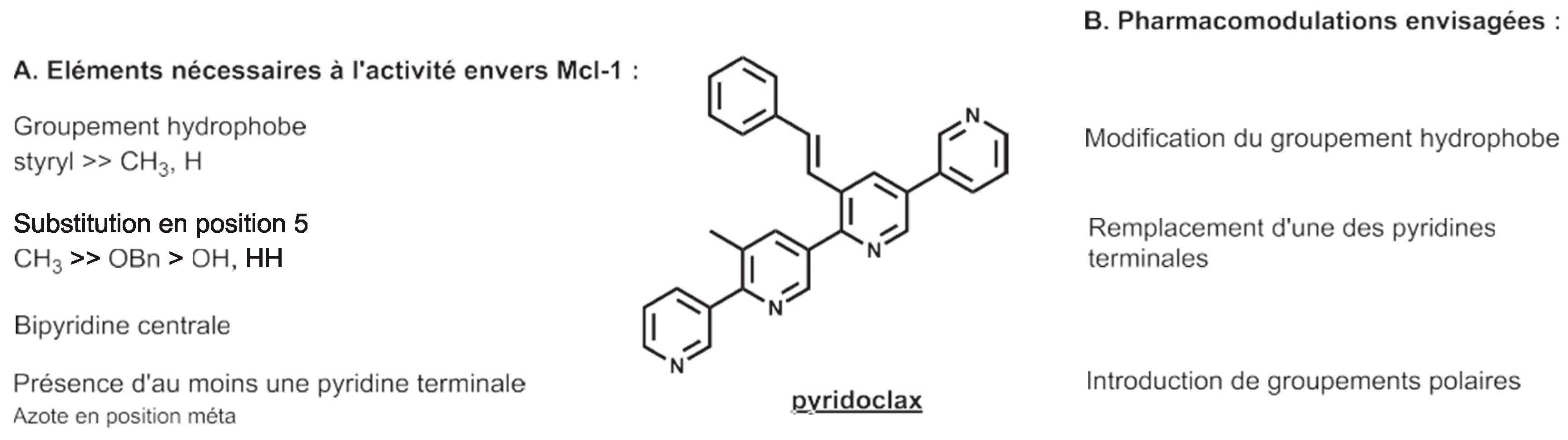
3.9. Pyridoclax and Its Derivatives Directly Inhibit Mcl-1 and Exert Potent Antitumor Effects on Ovarian Cancers In Vitro and In Vivo (L13)
Anne Sophie Voisin-Chiret
Normandie Univ, UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), 14000 Caen, France
Protein-protein interactions (PPIs) are attractive targets because they control numerous cellular processes. In oncology, apoptosis regulating Bcl-2 family proteins are of particular interest. Apoptotic cell death is controlled via PPIs between the anti-apoptotic proteins hydrophobic groove and the pro-apoptotic proteins BH3 domain. In ovarian carcinoma, it has been previously demonstrated that Bcl-xL and Mcl-1 cooperate to protect tumor cells against apoptosis.
If clinically relevant pharmacologic inhibition of Bcl-xL is available using ABT-263 (Navitoclax), selective direct inhibition of Mcl-1 remains problematic. In this context, our teams have designed and synthesized small compounds based on a pyridyl scaffold, named oligopyridines, which potentially target the Mcl-1 hydrophobic binding pocket. We demonstrated that the lead of the first generation of oligopyridines, named Pyridoclax, interacts directly with Mcl-1, releases its pro-apoptotic partners Bim and Bak and induces massive apoptosis at 25 µM concentration in combination with anti-Bcl-xL strategies in chemoresistant ovarian cancer cell lines.
In order to improve its biological activity, we evaluated the cytotoxic effects of a second generation of oligopyridines derived from the Pyridoclax. This allowed us to identify the MR31367, one of the most potent oligopyridines that shows a stronger pro-apoptotic activity in association with to Bcl-xL-targeting strategies in ovarian cancer cell lines. Further characterization showed that this derivative binds Mcl-1 and releases Bim and Bak from it, leading to Bak-mediated apoptosis.
Overall, these results open up interesting perspectives for the clinical use of Mcl-1 inhibitors as single agent or in combination with anticancer drugs to improve the clinical management of ovarian cancers.
5. Flash Communications
5.1. Phenotype Characterization and Mechanism Determination of TMEM45A Protein (FC01)
Thomas Dal Maso 1,*,
Sebastien Marx 1,
Carine Michiels 2,
Johan Wouters 1
and
Benjamin Le Calvé 2
1
Department of Chemistry, University of Namur, 5000 Namur, Belgium
2
Department of Biology, University of Namur, 5000 Namur, Belgium
More and more cancers display resistance to chemotherapeutic drugs and are linked to well- known factors such as tumor microenvironment. For example, hypoxia has been shown to favour tumor growth and to be involved in the drug resistance of cancer cells. During hypoxia, cell metabolism switches for adapting to environment modifications. Flamant et al. highlighted a new transmembrane protein, TMEM45A, which could be involved in the resistance of cancer cell in hypoxic conditions (Flamant, L. et al. BMC Cancer 2012, 12, 391). It is known that the silencing of TMEM45A in human breast cancer cells (MDA-MB-231) incubated in the presence of etoposide or taxol under hypoxia become sensitive to cell death induced by these chemotherapeutic molecules. Sun et al. have already shown that the knockdown of this protein has an impact on invasion, migration and proliferation of glioma cells (T98G) (Rebucci, M. et al. Biochem. Pharmacol, 2013, 85, 1219–1226; Sun, W. et al. Int. J. Clin. Exp. Pathol. 2015, 8, 12657–12667).
Until now, various phenotypes were observed and associated to TMEM45A expression. However, the structure, function and biological pathway of this protein are unknown. This work aims to unravel the mechanism of actions and functions by determining partners of TMEM45A using immunoprecipitation assays coupling to mass spectrometry. Moreover, the phenotype was studied using shRNA technology in MDA-MB-231 and T98G cells.
Acknowledgments: Thomas Dal Maso is grateful for financial support from the Fonds pour la Recherche Scientifique (FNRS). This work is supported by FRIA Grant.
5.2. Conception and Synthesis of Novel MT5-MMP Inhibitors for Alzheimer’s Disease (FC02)
Pauline Zipfel 1,*,
Julien Lalut 1,
Johanna Giovannini 1,
Peggy Suzanne 1,
Alban Lepailleur 1,
Ronan Bureau 1,
Kevin Baranger 2,
Michel Khrestchatisky 2,
Santiago Rivera 2,
Christophe Rochais 1
and
Patrick Dallemagne 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
2
Institute of Neuropathophysiology (INP), UMR7051, CNRS, Aix Marseille Université, 13331 Marseille Cedex 03, France
In 2017, over 46 million people were living with dementia worldwide and Alzheimer’s disease (AD) is the most common form of dementia in France, with about 850,000 cases. AD is a neurodegenerative and incurable brain disorder; only treatments for symptoms are available at this time. Because of the heavy economic and societal impact, there is an urgent need to find new treatments that target the molecular causes of neuronal cell death.
Two abnormal structures in the brain called β-amyloid (Aβ)-containing plaques and neurofibrillary tangles are considered as two of the main features of AD. In this context, several studies support the hypothesis that alterations in the processing of β-amyloid precursor protein (APP), resulting in the accumulation of Aβ and other proteolytic products contributes to AD pathogenesis. Thus, current research focuses on the enzymes involved in the amyloid cascade such as α-, β-, and γ-secretase. However, recent studies have revealed the existence of another physiological APP processing pathway, mediated by a novel AD-related enzyme, membrane-type 5-matrix metalloproteinase (MT5-MMP), that can process APP and promote Aβ and CTFβ accumulation, as well as the inflammatory process in AD transgenic mice (Baranger, K. et al. Cell. Mol. Life Sci. 2016, 73, 217–236; Baranger, K. et al. J. Neuroinflammation 2016, 13, 167; Baranger et al. Front. Mol. Neurosci. 2017, 9, 1–17). Moreover, MT5-MMP can cleave APP upstream from the β-secretase cleavage site (the so called η-cleavage site) (Ahmad, M. et al. J. Biochem. 2006, 139, 517–526) and release a N-terminally elongated Aβ fragment (Aη-α), which appears to be synaptotoxic (Willem, M. et al. Nature 2015, 526, 443–447).
We aim to design and synthesize the first MT5-MMP inhibitors through an interdisciplinary approach including molecular modelling, medicinal chemistry and biology. Starting from a hit compound identified by screening of CERMN’s chemical library, we are now investigating the pharmacomodulations on that scaffold to gain in affinity and selectivity for MT5-MMP.
5.3. Multicomponent Ugi-Synthesis and Biological Evaluation of New Multitarget Directed Small Molecules for Alzheimer’s Disease Therapy (FC04)
Irène Pachón-Angona *
and
Lhassane Ismaili
Neuroscience EA481, Université de Bourgogne Franche-Comté, 19 rue Ambroise Paré, 25000 Besançon, France
Alzheimer’s disease (AD) is a multifactorial and fatal neurodegenerative disorder characterized by decline of cholinergic function, deregulation of other neurotransmitter systems, beta-amyloid (Aβ) fibril aggregation and neurofibrillary tangles deposition (León, R., et al. Med. Res. Rev. 2013, 33, 139–189). Due to this complex nature of AD and the involvement of a number of relevant biological systems in AD progression, multitarget directed ligands (MTDL) may enable therapeutic efficacy. Accordingly, compounds possessing, besides anticholinergic activity and Aβ-aggregation inhibition properties, biometal chelating, nitric oxide releasing properties, antioxidant capacity, beta-secretase and monoamine oxidase inhibition power, as well as serotonin and sigma receptor modulation capacities have been developed.
In this context, we describe here new MTDL synthesized by the multicomponent Ugi reaction (Pachón-Angona I., et al., J. Enzyme Inhib. Med. Chem. 2018, 34, 479–489). The new hybrids result from the juxtaposition of different pharmacophoric groups, such as N-benzylpiperidine, a well-known functional motif present in ChE inhibitors, melatonin, a natural product showing broad-spectrum antioxidant and free radical scavenger activity, and a chromone moiety, as an antioxidant and MAO inhibitor.
Accordingly, the new MTDL showed a strong antioxidant power, EeAChE, eqBuChE and hMAO-A/B inhibitory activities, and neuroprotective capacity in SH-SY5Y neuronal cells against toxics insults, such as Oligomycin/Rotenone, Aβ1-35 and okadaic acid.
5.4. Synthesis of Piperazine-Based Siderophores to Design Broad Spectrum Antibiotics (FC06)
Pauline Loupias 1,*,
Alexandra Dassonville-Klimpt 1,
Elodie Lohou 1,
Nicolas Taudon 2
and
Pascal Sonnet 1
1
AGIR, UFR de pharmacie, Université de Picardie Jules Verne, 1 rue des Louvels, 80000 Amiens, France
2
Unité de Toxicologie Analytique, Institut de Recherche Biomédicales des Armées, 91223 Brétigny-sur-Orge, France
Resistance to antibiotics is an emerging phenomenon, fast becoming a major medical problem. The resistance of Gram-negative bacteria such as Pseudomonas aeruginosa and the Burkholderia group to conventional antibiotics leads to therapeutic failure and requires new antibiotic therapies. The use of iron transport systems is a promising strategy to overcome this phenomenon. These TonB-dependent receptors, essential for the survival of microorganisms, allow specific recognition of ferric siderophore complexes in order to transport iron within bacteria (Miethke, M. et al., Microbiol. Mol. Biol. Rev. 2007, 71, 413–451). Bacteria, according to their kind, express different types of receptors that allow them to recognize their endogenous siderophores but also xenosiderophores. Pseudomonas aeruginosa and Burkholderia pseudomallei in particular possess FptA receptors allowing the recognition of pyochelin (Butt A.T., et al. Front. Cell. Infect. Microbiol. 2017, 7).
These specific systems may allow the introduction of antibacterial agents by forming antibiotic-siderophore conjugates or toxic complexes such as gallium complexes, in the bacteria to kill it. Siderophores have three types of chelating function: catechols, hydroxamates and hydroxy-carboxylates. Previous work in the laboratory has shown that piperazine 1,4-dicatechol structures (MPPS0225) could be recognized by Pseudomonas aeruginosa strains. In order to further investigate this piperazine platform, we have synthesized iron chelators bearing 3-hydroxypyridin-4-ones and 1,3-dihydroxypyridin-4-one ligands. At the same time, we were interested in the synthesis of a more complex 2,5-dioxopiperazine platform, part of the rhodotorulic acid (RA), a natural siderophore produced by Rhodotorula pilimanae showing an interesting iron affinity (pFe = 21,8). Two RA synthesis strategies will be developed as well as the corresponding 3,6-disubstituted analogs. Through the synthesis of these chelators, we would like to study the influence, on the complexation of iron, the nitrogenous platform (piperazine or dioxopiperazine), the presence of stereogenic centers (3,6-disubstituted dioxopiperazine vs piperazines 1,4 -disubstituted) and the nature of the iron ligands (hydroxypyridinone vs catechol). An evaluation of the siderophore-like potential and a measurement of the complexing force of these will be carried out.
![Pharmaceuticals 12 00073 i014]()
Acknowledgments: This work was funded by the DGA and the Haut de France region.
5.5. Docking Studies of Melatonergic and Serotonergic Multi-Target Directed Ligands of Potential Interest for Alzheimer’s Disease (FC07)
Sangeetha-Laura Thirumaran *,
Feng Xiao,
Stéphane Lemaître,
Ronan Bureau,
Sylvain Rault,
Alban Lepailleur
and
Christophe Rochais
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
Alzheimer’s disease (AD) is the most common form of dementia, affecting millions of patients worldwide, for which the current treatments produce only symptomatic benefits. Among the biological targets implied in the physiopathology, and especially among the G-protein coupled receptors (GPCRs), melatonergic MT1 and MT2 and serotonergic 5-HT2c receptors present a growing interest. Modulations of these receptors have been proved to promote the non-amyloidogenic cleavage of Amyloid Protein Precursor (APP) and to ameliorate the symptoms through several actions such as anti-oxidant effect and regulation of the transmission of other neurotransmitters (Shukla, M., et al., Curr. Neuropharmacol. 2017, 15, 1010–1031; Švob Štrac, D., et al., Transl. Neurosci. 2016, 7, 35–49).
As AD is a multifactorial disorder, a simultaneous action on these two types of receptors with Multi-Target Directed Ligands (MTDLs) could represent a novel therapeutic approach. To this aim, we screened our chemical library in order to identify both potent MT1 and MT2 receptors agonists and 5-HT2c receptors antagonists. We performed docking studies of the selected molecules into homology models of MT1 and MT2 receptors and into the crystal structure of 5-HT2c receptors (Peng, Y., et al. Cell. 2018, 172, 719–730). The resulting ligand-receptor interactions are in agreement with the literature and allow to understand the polypharmacological profile of this promising new series of compounds (Pala, D., et al., Int. J. Mol. Sci. 2013, 14, 8093–8121; Renault, N. in Di Giovanni, G.; Esposito, E.; Di Matteo V. (Editors), 5HT2C Receptors in the Pathophysiology of CNS Disease, 2011, 97–127).
5.6. Versatile Approach to 5,6-Member Ring Cephalosporin Derivatives as Promising P2X7R Ligands via Morin Rearrangement (FC08)
Fanny Danton *,
Mohamed Othman,
Ata Martin Lawson
and
Adam Daich
Normandie Univ, UNILEHAVRE, URCOM, CNRS FR-3038, Le Havre, France Université Le Havre Normandie, 25 rue Philippe Lebon, BP. 1123, 76063 Le Havre, France
The high P2X7R receptor expression in immune cells suggests its considerable role in numerous diseases such as cancer and neurodegenerative diseases. Previous studies have already proved the anti-inflammatory, analgesic and in some cases anticancer properties of selective P2X7R antagonists (Baudelet, D., et al. Curr. Med. Chem. 2015, 22, 713–729).
Among the different chemical series developed by the major pharmaceutical companies, some lead compounds underwent to clinical trials mainly for the treatment of inflammatory diseases.1 GSK products of types 1 and 2 (Chambers, L. J. et al., WO 2008003697, 2008, Chem. Abstr. 2008, 148, 145026) mentioned in the Figure are allosteric modulator of P2X7R receptor. The latter were proposed for the first time in 2008 by GlaxoSmithKline for the treatment of chronic inflammatory.
The aim of our work is to elaborate new molecules able to interact with P2X7R receptors. The design of our compound series was done on the basis of the GSK lead compounds 1 and 2. Indeed, the 2-amido-lactam “-C-CO-NH-C-” sequence found in compounds 1 and 2 was considered as the key fragment responsible of such hypothetic activity. Therefore, we envisioned to insert such moiety in our series. As highlighted in the Figure, a skeleton containing a polycyclic N,X-acetal backbone (I, II; X = S, O, N), fused 1,4-thiazine III and 1,4-diazine IV bearing the expected 2-amidolactam fragment were selected to ensure a structure-activity relationship (SAR) study pivotal for further biological activities.
![Pharmaceuticals 12 00073 i016]()
In this line, the expected N,X-acetals I, II were synthetized by using a modified standard Meyer’s cyclocondensation (Groaning, M. D.; Meyers, A. I. Tetrahedron 2000, 56, 9843–9873). Then an oxidation step followed by a Morin rearrangement provided the compounds III starting from I and II. During these investigations, the experimental conditions for the sulfoxides intermediates as well as the Morin rearrangement were investigated and optimized.
5.7. Design, Synthesis and Biological Evaluation of Dibenzofuran Derivatives Inspired from Cercosporamide as Kinase Inhibitors (FC09)
Viet Hung Dao 1,
Isabelle Ourliac-Garnier 2,
Marc-Antoine Bazin 1,
Blandine Baratte 3,
Sandrine Ruchaud 3,
Stéphane Bach 3,
Patrice Le Pape 2
and
Pascal Marchand 1,*
1
Department of Medicinal Chemistry, University of Nantes, 44200 Nantes, France
2
Department of Parasitology and Medical Mycology, IICiMed—EA1155, IRS2, University of Nantes, 44200 Nantes, France
3
Sorbonne Universities, UPMC Paris 06, CNRS USR3151 Protein Phosphorylation and Human Diseases, Plateforme de criblage KISSf (Kinase Inhibitor Specialized Screening facility), Station Biologique, 29688 Roscoff, France
(–)-Cercosporamide is a natural product isolated from the phytopathogen fungus Cercosporidium henningsii (Sugawara, F., et al. J. Org. Chem. 1991, 56, 909–910). It was identified as a broad-spectrum antifungal agent displaying an in vitro mean MIC value of 89 µg/mL (Conover, M.A., et al., Phytochemistry 1992, 31, 2999–3001) and of 10 µg/mL (Sussman, A., et al., Eukaryot. Cell 2004, 3, 932–943) against C. albicans. Interestingly, it appeared to act as a potent CaPkc1 ATP-competitive inhibitor with an IC50 of 44 nM (Sussman, A., et al., Eukaryot. Cell 2004, 3, 932–943). Furthermore, cercosporamide inhibited human PKCα (IC50 = 1 µM) and PKCβ (IC50 = 0.3 µM) (Konicek, B.W., et al., Cancer Res. 2011, 71, 1849–1857) and was later shown to inhibit other human kinases, including Mnk1/2, Jak3, GSK3β, ALK4 and Pim1, from nanomolar to low micromolar ranges (Konicek, B.W., et al., Cancer Res. 2011, 71, 1849–1857; Hou, J., et al., Oncotarget 2012, 3, 118–131).
Polyoxygenated dibenzofurans are important heteroaromatic compounds which display a wide variety of biological activities (Love, B.E. Eur. J. Med. Chem. 2015, 97, 377–387). Consequently, in continuation of our successful attempts in the search of biologically active cercosporamide inspired derivatives (Bazin, M.-A., et al. Eur. J. Med. Chem. 2013, 69, 823–832), we report here the synthesis and biological evaluation of dibenzo[b,d]furans targeting protein kinases. In addition, cercosporamide was found to recognize ATP-binding site of Mnk2 kinase through hydrogen bond network due to the 3-OH and the 4-CONH2 of the phenyl portion, justifying the strategy of keeping dihydroxybenzofuran-carboxamide part of the natural product model for the design of new tricyclic compounds (Hou, J., et al. Oncotarget 2012, 3, 118–131).
5.8. Fluorinated Indazole Derivatives: Promising Structures for 5-HT4 Receptors PET Radiotracers (FC10)
Reynald Mangeant 1,*,
Silvia Stiebing 1,
Thomas Cailly 1,2,
Audrey Davis 1,
Christine Fossey 1,
Frédéric Fabis 1
and
Valérie Collot 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
2
Department of Nuclear Medicine, CHU Côte de Nacre, 14000 Caen, France
Since its discovery in 1988, the serotonin 4 receptor subtype (5-HT4R) has emerged as a promising target for drug discovery and development resulting from their implications in cognition, learning, memory processes and many neuropsychiatric disorders such as Alzheimer’s disease, anxiety, depression or anorexia nervosa (Bockaert, J., et al. A. Neuropharmacology 2008, 55, 922–931.). Thus, discovery of active 5-HT4R agonists and antagonists remains a continuing interest in clinical research. To this end, positron emission tomography (PET) (Marner, L., et al. Neuroimage 2010, 50, 855–861.) (Caillé, F., et al. Bioorg. Med. Chem. Lett. 2013, 6243–6247.) coupled with effective radioligands constitutes a valuable tool, both in clinical studies and drug discovery’s program. Based on previous works in CERMN (Lam, B.V., et al. Chem. Eur. J. 2016, 22, 4440–4446.), we aimed to develop new fluorinated indazole derivatives as potential brain 5-HT4R PET tracers.
![Pharmaceuticals 12 00073 i018]()
A convergent synthesis pathway to obtain fluorinated analogues has been established. A methodology allowing selective functionalization at position 3 leading to polyfunctional indazoles within a minimum of step has been developed. New pharmacomodulations studies were realized in order to increase receptor affinity, decrease lipophilicity and increase metabolic resistance.
5.9. Conception and Synthesis of New Pleiotropic Compounds that Both Display 5-HT4R Agonist Activity and Antioxidant Property in Alzheimer’s Disease (FC12)
Caroline Lanthier *,
Cédric Lecoutey,
Julien Lalut,
François-Xavier Toublet,
Patrick Dallemagne
and
Christophe Rochais
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
In a world where life expectancy is increasing, Alzheimer disease (AD) is the main cause of dementia, and touch approximatively 17% of people who are more than 75 years in France. This is a progressive neurodegenerative disorder characterized by memory loss and cognitive decline. Despite the fact that the physiopathology of AD is not entirely known at the time, some molecular causes were found such as the β-amyloid peptides aggregation, tau-dependent neurofibrillary tangles, as well as oxidative stress. Currently, treatments available for patients are mainly Acetylcholine esterase (AChE) inhibitor, which only have symptomatic benefits and do not cure AD. Then there is still a strong medical need in the AD population.
In this context, the concept of Multi-Target Directed Ligands (MTDLs) was applied to design a drug with several therapeutic targets to treat a disease. The MTDL should be able in first hand, to limit the development of β-amyloid plaques obtained by the aggregation of β-amyloïd peptides (Aß). Our compounds should be able to promote the cleavage of amyloid protein precursor (APP) by a—secretase in order to produce a neuroprotective and soluble peptide sAPPa. This is the role of the 5HT4R agonist (blue part—fig) which is already studied in the CERMN in other MTDL projects and led to the discovery of Donecopride (Rochais, C. et al., J. Med. Chem. 2015, 58, 3172–3187). In another hand, it appears that the oxidative stress has a central role in AD (Rosini, M. et al., J. Med. Chem. 2014, 57, 2821–2831). Adding antioxidant moiety such as polyphenol or ubiquinone (orange part—fig) could trap free radicals or reactive oxygen species (ROS). To that end, different compounds will be designed and synthetized in order to evaluate their in vitro/in vivo properties regarding their agonist activity on 5-HT4R and antioxidant property.
![Pharmaceuticals 12 00073 i019]()
5.10. Antiplasmodial Activity of Polyphenolic Derivatives (FC13)
Gilles Degotte 1,2,*,
Sylvain G. Alson 1,3,4,
Aurore Hans 2,
Olivia Jansen 1,
Ewa Cieckiewicz 1,
Hajatiana Rakotoarimanana 3,
Henintsoa Rafatro 3,
Fanantenanirainy Randimbivololona 4,
Pierre Francotte 2
and
Michel Frederich 1
1
Laboratoire de Pharmacognosie, Centre Interdisciplinaire de Recherches sur le Médicament (CIRM), ULiège, Liège, Belgium
2
Laboratoire de Chimie Pharmaceutique, Centre Interdisciplinaire de Recherches sur le Médicament (CIRM), ULiège, Liège, Belgium
3
Laboratoire d’Evaluation Pharmaco-Clinique (LEPC), Institut Malgaches de Recherches Appliquées (IMRA) BP, Antananarivo, Madagascar
4
Laboratoire de Pharmacologie Générale, de Pharmacocinétique et de Cosmétologie (LPGPC), Faculté des sciences, Université d’Antananarivo BP, Antananarivo, Madagascar
Despite the progress in the struggle against malaria, this parasitic disease remains a major public health problem with 216 million cases in 2016. The last advance was the development of artemisinin and its derivatives, near 15 years ago. These compounds are employed with success in combination with other antimalarial drugs and are now the recommended treatment by the World Health Organization. However, resistance of Plasmodium to these molecules appears and spreads over in Asia and Africa. Thus, the design of new antiplasmodial derivatives is imperative to hope the eradication of this infection (World Health Organisation, 2017).
Polyphenolics compounds are well known to have multiple pharmacological activities such as antioxidant (Wright, J. S. et al., J. Am. Chem. Soc. 2001, 123, 1173–1183), antitumor (Okuda, T. in Food Factors for Cancer Prevention 1997, 280–285), antimicrobial (Bisignano, G. et al., J. Pharm. Pharmacol. 1999, 51, 971–974) and antiplasmodial (Köhler, I. et al., Z. Naturforsch. 2002, 57c, 277–281.). Regarding this last effect, the screening of caffeic acid (1) and its derivatives was performed in vitro and in vivo. These evaluations permitted to select ethyl caffeate (2) as the most potent derivatives against a 3D7 strain in culture. More interestingly, this ester was found active in mice with a stage specificity on the young trophozoites. The major interest of this molecule is that caffeic acid is widely distributed in plants and is considered non-toxic.
Structures of caffeic acid (1), ethyl caffeate (2) and ellagic acid (3).
Considering the results obtained with this first series of polyphenolic analogs and based on the known antimalarial activity of ellagic acid (3) in vitro and in vivo (Soh, P. N. et al., Antimicrob. Agents Chemother. 2009, 53, 1100–1106.), we currently investigate this widely distributed polyphenol as a scaffold for further pharmacomodulation. The new structures will be screened to determine their antiplasmodial effect.
Acknowledgments: This work was financed by FNRS.
5.11. Chemical Modifications of Mycobacterium Tuberculosis MabA Inhibitors (FC16)
Léo Faïon 1,*,
Catalin Pintiala 1,
Martin Moune 2,
Rosangela Frita 2,
Kamel Djaout 2,
Catherine Piveteau 1,
Benoit Deprez 1,
Alain Baulard 2,
Nicolas Willand 1
and
Marion Flipo 1
1
Univ. Lille, Inserm, Institut Pasteur de Lille, U1177—Drugs and Molecules for living Systems, F-59000 Lille, France
2
Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, U1019—UMR 8204—CIIL—Center for Infection and Immunity of Lille, F-59000 Lille, France
This infection, caused by
Mycobacterium tuberculosis, kills each year 1.7 million people (World Health Organization, Global Tuberculosis Report, 2017,
http://www.who.int/tb/publications). A treatment, involving a combination of drugs for a minimum of six months, is available but the rapid emergence of multidrug resistant (MDR) strains of
M. tuberculosis stresses the need for alternative therapies. The enzyme MabA (FabG1) is involved in the biosynthesis of mycolic acids (Marrakchi, H, et al.
A. Microbiology 2002,
148, 951), which are very long-chain fatty acids playing a major role in the bacterial cell wall. The gene coding for this enzyme has been shown to be essential for M. tuberculosis (Parish, T.
J. Bacteriol. 2007,
189, 3721), therefore our goal is to find drug-like inhibitors of MabA that prevent mycobacterial growth. The screening of a 1280 fragment-library on MabA using a new enzymatic assay allowed the identification of several families of inhibitors. One of this series was selected for further optimization and structure-activity relationships were established. This led to the discovery of the first low-molecular-weight MabA inhibitors. The synthesis of these compounds and the biological results will be presented.
![Pharmaceuticals 12 00073 i021]()
5.12. Mcl-1 Selective Inhibitor Modulation: Discovery of Mcl-1/Bcl-xL Dual Inhibitors, for the Treatment of Chemoresistant Ovarian Cancers (FC17)
Martina De Pascale 1,*,
Camille Denis 1,
Emilie Brotin 2,
Laurent Poulain 2
and
Anne-Sophie Voisin-Chiret 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 1400 Caen, France
2
Plate-forme ImpedanCELL, Inserm U1086 ANTICIPE, axe BIOTICLA, Centre de Lutte Contre le Cancer François Baclesse, 14000 Caen, France
Protein-protein interactions (PPIs) are attractive targets because they control numerous cellular processes. In particular, in oncology, our laboratory is interested in Bcl-2 family proteins, apoptosis regulators. This family is composed by pro-apoptotic (Bax, Bak, Noxa…) and anti-apoptotic members (Bcl-2, Bcl-xL, Mcl-1…) that interact via PPIs to regulate the cellular death thanks to BH domains (BH1, BH2, BH3). In solid human tumors, the anti-apoptotic members are over expressed and their inhibition represents a novel and promising strategy for new anticancer drugs. In ovarian carcinoma, it has been previously demonstrated that Bcl-xL and Mcl-1 cooperate to protect tumor cells against apoptosis and their concomitant inhibition leads to massive apoptosis even in absence of chemotherapy. In this context, we have designed and synthesized an oligopyridine BH3-mimetic, named pyridoclax (Gloaguen, C., et al. J. Med. Chem. 2015, 58, 1644–1668) that interacts selectively with Mcl-1 hydrophobic pocket.1 Know that Bcl-xL inhibition is also accessible using BH3-mimetics, we would design a novel class of BH3-mimetics able to inhibit simultaneously Bcl-xL and Mcl-1 (Zhang, Z., et al. Arch. Pharm. Chem. Life Sci. 2015, 348, 1–11). We will present our approach to design such dual inhibitors from pyridoclax scaffold taking into account the structure-activity relationship.
![Pharmaceuticals 12 00073 i022]()
5.13. Benzoylthiosemicarbazides as Promising Antimicrobial Agents Targeting D-alanine-D- alanine Ligase in bacterio (F18)
Alice Ameryckx 1,*,
Léopold Thabault 1,
Lionel Pochet 2,
Françoise Van Bambeke 1
and
Raphael Frédérick 1
1
Medicinal Chemistry Research Group/Cellular and Molecular Pharmacology, Louvain Drug Research Institute (LDRI), Université catholique de Louvain, Av. E Mounier 74, 1200 Woluwé-Saint-Lambert, Belgium
2
Department of Pharmacy, Namur Medicine & Drug Innovation Center (NAMEDIC), Namur Research Institute forLife Sciences (NARILIS), University of Namur, Namur, Belgium
As the phenomenon of antibiotic resistance is dramatically increasing these days, searching for new therapeutic targets less vulnerable to this resistance issue appears as a real need (Cassell, G. H., et al., JAMA 2001, 285, 601–605). The cell wall of bacteria and the enzymes that are involved in its synthesis are promising targets for many antibiotics, which inhibit the late stages of peptidoglycan biosynthetic pathway (Fan, C., et al., Science 1994, 266, 439–443). But the resistance phenomena have revealed the high flexibility in this assembly pathway, and the need to target other enzymes acting on earlier steps of peptidoglycan synthesis (Tytgat, I., et al., Curr. Med. Chem. 2009, 16, 2566–2580) D-alanyl-D-alanine ligase (Ddl) is of particular interest as it utilizes a substrate (D-alanine) which is specific for bacterial peptidoglycan biosynthesis and essential for bacterial growth (Zawadzke, L. E., et al., Biochemistry 1991, 30, 1673–1682; Gholizadeh, Y., et al., Protein Sci. 2001, 10, 836–844).
In this work, a series of 37 benzoylthiosemicarbazides inhibitors of D-Ala-D-Ala ligase (Ddl) were designed and synthesized in order to target resistant strains of bacteria. Among these, the 4-(3,4-dichlorophenyl)-1-(2-hydroxybenzoyl)-3-thiosemicarbazide (1) was identified as a very potent Ddl inhibitor with an activity in the micromolar range. This compound, possessing strong antimicrobial activities including against multidrug resistant strains, was proved to act through a bactericidal mechanism and demonstrated very low cytotoxicity on THP-1 human monocytic cell line.
![Pharmaceuticals 12 00073 i023]()
Further structure-activity relationships (SARs) studies provided evidence that the hydroxyl substituent in the 2-position (R1) of the benzoylthiosemicarbazide scaffold is essential for the enzymatic inhibition. The in vivo biochemical mechanism was then determined by UPLC-MS dosage (S. Putty, A., et al., Chem. Biol. Drug. Des. 2011, 78, 757–763) of intracellular L-Ala, D-Ala and D-Ala-D-Ala levels in response to 4-(3,4-dichlorophenyl)-1-(2-hydroxybenzoyl)-3- thiosemicarbazide (1). Finally, these encouraging results prompted us to investigate the selectivity of this compound towards various enzymes and receptors. This work thus highlights the thiosemicarbazide motif as very promising for the development of novel antibacterial compounds acting through an interesting mechanism of action, good selectivity and low cytotoxicity.
6. Posters
6.1. PfRab6 and PfPyrK1: New Putative Plasmodial Targets for Designing Novel Antimalarials (P01)
Dyhia Amrane 1,
Nicolas Primas 1,*,
Gilles Lanzada 1,
Sébastien Hutter 2,
Nadine Azas 2,
Pierre Verhaeghe 3
and
Patrice Vanelle 1
1
Aix-Marseille Univ, CNRS, ICR UMR 7273, Pharmaco-Chimie Radicalaire, Faculté de Pharmacie, 13385 Marseille, France
2
Aix-Marseille Univ, IHU Méditerranée Infection, UMR VITROME, 13005 Marseille, France
3
Université Paul Sabatier, CNRS UPR 8241, Laboratoire de Chimie de Coordination, 31077 Toulouse, France
Malaria is still responsible for a major burden on human health and is becoming increasingly difficult to treat due to the emergence of drug resistance toward Artemisinin-based Combination Therapy (WHO, World Malaria Report, 2017). In order to discover new antimalarials with an original mechanism of action, our laboratory previously described the synthesis and the biological activities of a 2-trichloromethylquinazoline hit active against the multiresistant K1 P. falciparum strain (Verhaeghe, P., et al. Bioorg. Med. Chem. 2009, 17, 4313–4322). A chemoproteomics approach was used in order to elucidate its mechanism of action as it was different from the ones of commercial antimalarials. Two putative plasmodial targets were identified—PfRab6 and PfPyrK1—which are essential for the parasite. The confirmation of the mechanism of action implicating these two proteins is in progress and pharmacomodulation refinement is still required.
![Pharmaceuticals 12 00073 i024]()
In previous work, we report that the 4-sulfonamidoquinazoline derivatives were unactive (Primas, N., et al., Molecules 2012, 17, 8105–8117). Isosteric replacement with a carboxamide substituent showed promising activity. Thus, we prepared a new library of aliphatic, aryl or heteroaryl carboxamide derivatives. Moreover, a scaffold hopping strategy previously showed that the replacement of the quinazoline moiety by a quinoxaline one improved the cytotoxicity profile (Desroches, J., et al., Eur J. Med. Chem. 2017, 125, 68–86). Thus, we synthetized a new series of 2-trichloromethylquinoxalines bearing either arylether, arylthioether or aniline substituents at position 3. We report herein this new pharmacomodulation work: the synthesis of the corresponding derivatives and the biological results will be detailed in the poster.
Acknowledgments: This work was supported by the ANR NINTARMAL project, grant ANR-17-CE11-0017 of the French Agence Nationale de la Recherche.
6.2. Synthesis and Antiparasitic Evaluation of New 2,9-bis[(substituted-aminomethyl)phenyl]-1,10-phenanthroline Derivatives (P05)
Jean Guillon 1,*,
Anita Cohen 2,
Rabindra Nath Das 1,
Clotilde Boudot 3,
Stéphane Moreau 1,
Luisa Ronga 1,
Solène Savrimoutou 1,
Lilian Elmi 1,
Sandra Rubio 1,
Alexandra Dassonville-Klimpt 4,
Nadine Azas 2,
Bertrand Courtioux 3,
Jean-Louis Mergny 1,
Catherine Mullié 4
and
Pascal Sonnet 4
1
Université de Bordeaux, INSERM U1212, UMR CNRS 5320, ARNA Laboratory, UFR des Sciences Pharmaceutiques, Bordeaux, France
2
Aix-Marseille Univ, IRD, AP-HM, SSA, VITROME, Marseille, France
3
INSERM U1094, Tropical Neuroepidemiology, Université de Limoges, Institute of Neuroepidemiology and Tropical Neurology, Limoges, France
4
Université de Picardie Jules Verne, UFR de Pharmacie, AGIR—EA 4294 Amiens, France
Malaria represents one of the most prevalent parasitic infections worldwide. In 2017, an estimated 219 million cases of malaria occurred all over the world and there were an estimated 435,000 deaths from malaria globally (World Health Organization, World Malaria Report 2018). The increase in drug resistance in the malaria endemic areas has significantly reduced the potency of most currently used antimalarial compounds. Thus, new antimalarial drugs with new potential mechanisms of action are now required to overcome this emerging resistance and also to control an ever-increasing number of epidemics due to the parasites (Biot, C. et al., Infect. Disorders-Drug Targets 2006, 6, 173–204; Barnett, D.S., et al., Chem. Rev. 2014, 114, 11221–11241). Following this strategy, a series of new 2,9-bis[(substituted-aminomethyl)phenyl]-1,10-phenanthroline derivatives 1a-s was synthesized, and these corresponding derivatives were biologically evaluated in vitro against three protozoan parasites (Plasmodium falciparum, Leishmania donovani and Trypanosoma brucei brucei). Obtained results showed antiparasitic activity with IC50 values in the μM range. In order to assess the effective selectivity of this series, the in vitro cytotoxicity of these molecules was also evaluated against human HepG2 cells; for some derivatives, cytotoxicity was observed at significantly higher concentrations than antiparasitic activity, revealing an interesting selectivity index (Guillon, J. et al., Chem. Biol. Drug Des. 2018, 91, 974–995).
![Pharmaceuticals 12 00073 i026]()
The 2,9-bis[(substituted-aminomethyl)phenyl]-1,10-phenanthroline 1h was identified as the most potent antimalarial candidate, with cytotoxic to antiparasitic activity ratios of 107 and 39 against a chloroquine-sensitive and a chloroquine-resistant strain of P. falciparum, respectively. As the telomeres of the parasite P. falciparum are the likely target of this compound, we also investigated stabilization of the Plasmodium telomeric G-quadruplexes by our phenanthroline derivatives through a FRET melting assay. Consequently, the ligands 1f and 1m were noticed to be more specific for FPf8T with higher stabilization for FPf8T than for the human F21T sequence.
Acknowledgments: We thank the DGA and ANR (projects ANR-12-ASTR-003, OligoSwitch [ANR-12-IS07-0001], “Quarpdiems” [ANR-12-BSV8-0008-01], and “VIBBnano” [ANR-10-NANO-04-03]) for financial support of this study.
6.3. Development of a Binding Assay Using Fluorescence Polarization (P06)
Marie Jouanne 1,*,
Camille Denis 1,
Martina De Pascale 1,
Charline Kieffer 1,
Louis-Bastien Weiswald 2,
Jana Sopková-de Oliveira Santos 1,
Laurent Poulain 2
and
Anne Sophie Voisin-Chiret 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie Normandie Univ, UNICAEN, CERMN, 14000 Caen, France
2
Inserm U1086 « ANTICIPE » Unité de Recherche Interdisciplinaire pour la Prévention et le Traitement des Cancers. Axe 2: Biologie et Thérapies Innovantes des Cancers Localement Agressifs (BioTICLA), Université de Caen Normandie, UFR Santé et SF 4206 ICORE, 14000 Caen, France
For few years, our laboratory is interested in design and synthesis of small molecules as potential modulators of Protein Protein Interactions (PPIs). Particularly, we are interested in proteins involved in apoptosis process such as anti-apoptotic Bcl-2 family proteins or more recently the X-linked inhibitor of apoptosis protein (XIAP).
Based on its expertise in molecular modelling and in medicinal chemistry, our team has developed a large library of abiotic foldamers able to mimic the secondary structures of proteins (Voisin-Chiret, A.S., et al., Tetrahedron 2012, 68, 4381–4389; Voisin-Chiret, A.S., et al., Pure Appl. Chem. 2012, 84, 2467–2478; Sopkova-de Oliveira Santos, J., et al., J. Chem. Inf. Model. 2012, 52, 429–439; Jouanne, M., et al., Eur J. Org. Chem. 2016, 34, 5686–5696). The biological evaluation of these compounds has been realized with our partner (binding affinity by SPR, cellular tests…) and some of them have been shown very interesting anti-proliferative activities against Mcl-1 protein (Gloaguen, C., et al., J. Med. Chem. 2015, 58, 1644–1668). New molecules are still synthesized in order to disturb targeted proteins. In front of the amount of synthesized compounds, we decided recently to implement screening binding affinities by fluorescence polarization to obtain a first result to select best compounds before to perform cellular and in vivo tests and to improve structure-activity relationship.
![Pharmaceuticals 12 00073 i027]()
Acknowledgments: The authors are grateful for financial support provided by Regional council of Normandy, FEDER, the University of Caen and the Ministry of Research.
6.4. C-H (Radio-)iodination of N-acylsulfonamides, Access to New Iodinated Radiotracers in Cancerology (P07)
Alexandra Hebert 1,*,
Florian Benoist 1,
Emmanuelle Dubost 1,
Frédéric Fabis 1
and
Thomas Cailly 1,2
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie CERMN UNICAEN EA 4258 FR CNRS 3038 INC3M SF 4206 ICORE Boulevard Becquerel, University of Caen Normandie, 14032 Caen, France
2
Department of Nuclear Medicine, CHU Cote de Nacre, 14000 Caen, France
Labelling of (bio)molecules with radioactive isotopes is of high interest to the scientific community, as it strongly impacts the discovery process in life science and nuclear medicine. Radiolabelled molecules have been extensively used to assess biochemical reactions, to measure in vivo distribution of a substance or to perform RadioImmunoAssay (RIA). In nuclear medicine, radio-therapeutics for RadioIsotope Therapy (RIT) and radio-tracers for molecular imaging experiments such as Positron Emission Tomography (PET), Single Photon Emission Computed Tomography (SPECT) or scintigraphy have been described. Several useful isotopes of iodine can be used for both diagnosis and therapy: 123I for SPECT imaging, 124I for PET imaging, 125I for biological assays and nuclear medicine and 131I for radio-therapy and scintigraphy. Our group has recently developed a method to radio-iodinate N-acylsulfonamides through a room temperature palladium mediated C-H radio-iodination. This original strategy allows radiolabelling in very mild conditions without the use of chemical precursors.
![Pharmaceuticals 12 00073 i028]()
In this context, we are currently enlarging the scope of this methodology toward the radio-iodination of antitumoral agents containing a N-acylsulfonamide group. Thus, an antiproliferative agent LY32262 (Nathubhai, A. et al., J. Med. Chem. 2016, 60, 814–820), a Bcl-xL/Mcl1 dual inhibitor (Cominetti, M.D.D. et al., Org. Biomol. Chem. 2016, 14, 10161–10164) and ABT-737 (Broughton, L.J. et al. J. Photochem. Photobiol. B 2016, 163, 374–384) have been selected and progress toward the (radio-)iodination of these molecules will be presented.
6.5. New Multifunctional Diamine Drugs to Prevent Oxidative and Carbonyl Stress Extension in Alzheimer’s Disease (P08)
Elodie Lohou 1,*,
N. André Sasaki 1,
Agnès Boullier 2,3,4
and
Pascal Sonnet 1
1
Laboratoire AGIR, UFR de Pharmacie, Université de Picardie Jules Verne, F-80037 Amiens, France
2
UFR de Médecine, Université de Picardie Jules Verne, F-80037 Amiens, France
3
MP3CV, Centre Universitaire de Recherche en Santé (CURS), F-80054 Amiens (Salouël), France
4
CHU Amiens Picardie, F-80054 Amiens (Salouël), France
Carbonyl stress is now considered to play an important role in Alzheimer’s disease (AD) pathogenesis. Indeed, extensive Advanced Glycation Endproducts (AGEs) and Advanced Lipid peroxidation Endproducts (ALEs) accumulation linked to enhanced reactive carbonyl species (RCS) level has been reported in amyloid β (Aβ) plaques and tau-associated neurofibrillary tangles (NFT). In AD, Aβ-oligomers induce oxidative stress whereas transition metals (Zn2+, Cu2+ and Fe3+) stimulate Aβ aggregation and amyloid precursor protein (APP) processing. RCS such as methylglyoxal (MGO) or malondialdehyde (MDA) are endogenously formed during the sugar glycoxidation and lipid peroxidation of polyunsaturated fatty acids induced by oxidative stress exacerbation. Their condensation with amino groups of tissue proteins gives AGE and ALE. Consequently, carbonyl stress takes part in the vicious downward redox amyloid spiral leading to neurodegeneration as toxic mediator and oxidative stress promoter (Butterfield, D.A., et al. Trends Mol. Med. 2001, 7, 548–554; Tiiman, A., et al., Neurochem. Int. 2013, 62, 367–378). Glycated Aβ cross-linking promotion accelerates its deposition and its protease resistance. Moreover, AGE promote via their receptors RAGE oxidative stress and inflammation as well as cell apoptosis (Krautwald, M. et al., Exp. Gerontol. 2010, 45, 744–751).
Considering the multifactorial pathogenesis of AD, we designed new multifunctional diamine drugs that are simultaneously able to trap RCS (primary vicinal diamine function) as well as ROS and biometals (phenolic acid or hydroxypyridinone (HOPO) moiety) (Lohou, E. et al., Eur. J. Med. Chem. 2016, 122, 702–722). In the poster, synthesis of these new promising hybrid AGE/ALE inhibitors, but also their physicochemical and biological evaluation in terms of their carbonyl trapping capacity, antioxidant activity, Cu2+-chelating capacity as well as their protective properties against in vitro MGO-induced apoptosis in the model AD cell-line PC12 are reported.
Acknowledgments: We thank “SATT Nord” for financial support of this study.
6.6. From the Design to the In Vitro Evaluation of New Iodinated Ligands as Potential 5-HT4 SPECT Radiotracers (P09)
Victor Babin 1,*,
Benjamin Tournier 2,
Audrey Davis 1,
Philippe Millet 2,
Thomas Cailly 1,
Jean-Philippe Bouillon 3
and
Frédéric Fabis 1
1
Normandie University, UNICAEN, CERMN, 14000 Caen, France
2
Hôpitaux Universitaires de Genève, Département de Santé Mentale et de Psychiatrie, Service de Psychiatrie Générale, Unité des Biomarqueurs de Vulnérabilité, Chemin du Petit-Bel-Air, 2 CH-1225 Genève, Suisse
3
Normandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA UMR 6014, F-76000 Rouen, France
Serotonin (5-hydroxytryptamine 5-HT) is a neurotransmitter acting on the central and peripheral nervous system through a large variety of receptors subdivided in seven families. The serotonin receptor subtype-4 (5-HT4R) is known to be involved in physiological and pathological disorders such as Alzheimer disease (Lezoualc’h, F., Exp. Neurol. 2007, 205, 325–329). The development of radiotracer is essential for the evaluation of new drugs targeting 5-HT4R and also for investigation of the receptor involvement in a variety neurodegenerative and neuropsychiatric disorders.
Since a few years, our group has been involved in the development of new Single Photon Emission Computed Tomography (SPECT) radiotracers targeting the 5-HT4R and many iodinated ligands based on phenanthridine (Dubost, E., et al. J. Med. Chem. 2012, 55, 9693–9707) and diazaphenanthridine (Fresneau, N. et al. Eur. J. Med. Chem. 2015, 94, 386–396) scaffolds have been designed. An efficient synthetic access to these compounds has been established and many potent ligands have been synthetized. Biological evaluation has led to a “hit” compound and this ligand and some analogues have been labeled with 125-iodine for SPECT imaging but to date none of these radioligands have been able to image the 5-HT4R specifically. In order to obtain an efficient 5-HT4R radiotracer, our group has decided to synthesize a series of new iodinated ligands with reduced lipophilicity to promote in vivo specificity.
6.7. Fragment-Based Screening Approach Leading to the Inhibition of Matrix Metalloproteinase MT5-MMP (P10)
Julien Lalut 1,*,
Johanna Giovannini 1,
Peggy Suzanne 1,
Alban Lepailleur 1,
Ronan Bureau 1,
Kevin Baranger 2,
Santiago Rivera 2,
Patrick Dallemagne 1,
Jean-François Briere 3
and
Christophe Rochais 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
2
Institute of Neuropathophysiology (INP), UMR7051, CNRS, Aix Marseille Université, Marseille, France
3
Normandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA UMR 6014, F-76000 Rouen, France
Fragment-based drug design is a promising and modern concept in drug discovery which allows to obtain affine and selective ligands for many biological targets from small molecules chosen as starting points. With less complex structures, fragments reach inaccessible binding sites and have more points of interaction contrary to the larger lead candidates developed in traditional medicinal chemistry or generated from HTS. However, fragments generally have less affinity for their targets, that’s why it is necessary to grow the molecules in order to obtain the desired affinity with favorable selectivity and ADME properties (Wermuth, C.G., et al., The Practice of Medicinal Chemistry, 4th Edition, Academic Press: London, UK 2015).
![Pharmaceuticals 12 00073 i032]()
While the majority of screened chemical libraries are essentially linear or planar fragments, we have decided to focus our attention on the screening of three-dimensional geometry molecules from different research programs with scaffolds able to distribute substituents spatially (Mahé, O., et al., Ang. Chem. Int. Ed. 2010, 49, 7072–7075; Gembus, V., et al., Org. Biomol. Chem. 2010, 8, 3287–3293). Several common techniques (NMR, cristallography…) can be used for the identification of a hit from a library of fragments. Our original chemical library has been screened in silico and in vitro to identify fragments interacting towards MT5-MMP, a matrix metalloprotease recently identified as a potential relevant target for the treatment of Alzheimer’s disease (Baranger, K., et al., Cell. Mol. Life Sci. 2016, 73, 217–236).
A 3,4-substituted pyrazoline derivative was identified from screenings as a first hit, especially with a selectivity towards MT5 vs. MT1. Molecular docking seems to indicate a better score for the S enantiomer. Their separation as well as other pharmacomodulations are underway to increase the affinity of this first fragment.
6.8. Anti-Trypanosomatid Lead Optimization in 3-nitroimidazo[1,2-a]pyridine series (P11)
Cyril Fersing 1,
Clotilde Boudot 2,
Julien Pedron 3,
Sébastien Hutter 4,
Romain Paoli-Lombardo 1,
Nicolas Primas 1,*,
Caroline Castera-Ducros 1,
Sandra Bourgeade-Delmas 5,
Alix Sournia-Saquet 3,
Susan Wyllie 6,
Alan Fairlamb 6,
Alexis Valentin 5,
Pascal Rathelot 1,
Nadine Azas 2,
Bertrand Courtioux 4,
Pierre Verhaeghe 3
and
Patrice Vanelle 1
1
Aix-Marseille Univ, CNRS, ICR UMR 7273, Pharmaco-Chimie Radicalaire, Faculté de Pharmacie, 13385 Marseille, France
2
Université de Limoges, UMR Inserm 1094, Neuroépidémiologie Tropicale, 87025 Limoges, France
3
Université Paul Sabatier, CNRS UPR 8241, Laboratoire de Chimie de Coordination, 31077 Toulouse, France
4
Aix-Marseille Univ, IHU Méditerranée Infection, UMR VITROME, 13005 Marseille, France
5
Université Paul Sabatier, IRD UMR 152, PHARMA-DEV, 31400 Toulouse, France
6
Division of Biological Chemistry and Drug Discovery, Wellcome Trust Building, School of Life Sciences, University of Dundee, Dundee, Scotland, UK
Among the drugs currently developed to treat parasitic diseases caused by trypanosomatids such as Leishmania and Trypanosoma, the nitrodrug fexinidazole exerts its mechanism of action via a reductive bioactivation step, leading to cytotoxic metabolites. This reaction is catalyzed by nitroreductases (NTR) enzymes expressed by these protozoa (Wyllie, S., et al. Antimicrob. Agents Chemother. 2013, 57, 901–906; Wyllie, S., et al., PLoS Pathog. 2016, 12, e1005971).
In order to design new NTR substrates, we synthesized original 3-nitroimidazo[1,2-a]pyridines and highlighted a hit molecule (Castera-Ducros, C., et al. Bioorg. Med. Chem. 2013, 21, 7155–7164). A pharmacomodulation work afforded an antileishmanial lead compound, functionalized at the C8 position by a p-chlorophenylsulfane substituent and displaying both a very good activity against three species of Leishmania and a low cytotoxicity toward the HepG2 and THP1 cell line. This molecule is selectively activated by the type 1 NTR of Leishmania, and shows neither mutagenic effect in the Ames test nor genotoxic properties in the comet assay. It has a poor microsomal stability, with minimal influence on the antiparasitic potential as the probable metabolites (sulfoxide and sulfone, similarly to fexinidazole) remain active. More recently, an antitrypanosomal Lead molecule bearing a 4-hydroxybutyn-1-yl substituent at C8 was discovered: also activated by a trypanosomal NTR, this compound displayed an improved microsomal stability, along with a better hydrosolubility. Therefore, this molecule showed suitability for an in vivo evaluation, currently undertaken on a Trypanosoma brucei infected mouse model. Further pharmacomodulation works are also conducted in the aim of optimizing both the activity and the PK properties of these anti-trypanosomatid 3-nitroimidazo[1,2-a]pyridines.
![Pharmaceuticals 12 00073 i033]()
6.9. Development and Characterization of a Versatile Nanoemulsion of Interest for Pre-Clinical Assays (P12)
Line Séguy *,
Anne-Claire Groo,
Etienne Demissy
and
Aurélie Malzert-Fréon
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UniCaen, 14000 Caen, France
About half of new chemical entities entering drug development pipelines are poorly water soluble. To overcome this serious hurdle and to limit the number of lead compounds or drug candidates to be eliminated from discovery pipelines, formulation strategies can be envisaged as soon as possible in drug discovery to enhance the solubility, and in fine the bioavailability of compounds. Among formulations to envisage, nanoemulsions refer to nanosized (typically below 300 nm) droplets of oil dispersed in water. They can be considered as a perfect delivery system for high lipophilic drugs since they permit high drug loading. In the same time, nanoemulsions assure protection of the drug, reduce its intrinsic toxicity, and even promote a prolonged drug release and an increased bioavailability. In regard of these various advantages, formulation of drug-loaded nanoemulsions appears particularly appealing from the early drug discovery to permit early animal experiments.
Nanoemulsions that we want to develop must be administrable by different routes (oral or parenteral routes), and hence must be safe, easily sterilisable, with adapted granulometric properties (monodisperse diameter of about 100 nm), with neutral surface charge, with pH and osmolarity compatible with parenteral adminstration, and transposable to many drugs with various physico-chemical properties.
In regard of these objectives, nanoemulsions were prepared with GRAS excipients by spontaneous emulsification method, a low-energy process requiring no sophisticated equipment, and without using any organic solvent. In order to define appropriate excipients, an in vitro test was set up in the laboratory to define hemolytic properties on human blood cells of excipients used alone or formulated. Among the twenty excipients tested, a medium chain triglyceride and two surfactants were selected for formulation assays. The feasibility of nanoemulsions with appropriate properties was determined by varying process parameters and the relative proportion of excipients. Based on a ternary phase diagram, the best formulation with optimal granulometric properties was defined. Encapsulation assays of a model drug were performed, and the stability of the formulation was determined in time, and also after dilution in various complex biomimetic media.
From the results, it appears that the developed nanoemulsions present appropriate properties for oral and parenteral administration routes. Thus, they would provide a valuable option as formulation strategy for drug discovery.
6.10. Conception of Pleiotropic (pro)Drugs MAO-B Inhibitor/AChE Inhibitor for Alzheimer’s Disease (P14)
Benjamin Guieu *,
Cédric Lecoutey,
Francois-Xavier Toublet,
Rochais Christophe
and
Dallemagne Patrick
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder, leading to the most common (Anand, R., et al. Neuropharmacology 2014, 76, 27–50) form of dementia in the elderly. Given AD’s multifactorial causes, the classical pharmacological approach consisting in interacting very selectively with a single target has shown clinical limitations, failing to restore such a complex biological system. As a result, more and more examples illustrate the concept of Multi-Target Directed Ligands (MTDLs), molecules which display several activities by interacting with different biological in order to obtain a synergy of action (Cavalli, A., et al. J. Med. Chem. 2008, 51, 347–372). For example, our group described the donecopride, first MTDL inhibiting acetylcholinesterase (AChE) and activating 5-HT4 serotoninergic receptors, and now in preclinical evaluation (Lecoutey, C., et al., Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830).
Ladostigil is a novel type of MTDL currently in phase II clinical trials. It is the first compound able to release an active compound, hydroxyrasagiline, a MonoAmine Oxidase B (MAO-B) inhibitor, through the interaction with a first target, the acetylcholinesterase (AChE), which is temporarily inhibited during the process (Youdim, M. B. Curr. Alzheimer Res. 2006, 3, 541–550).
The objective of this project is to illustrate the concept the concept of molecules which are both drugs and prodrugs: the action on a first target will release a second agent acting on another target. Our objective remains original by leading to the design of (pro)drugs interacting with MAO-B to inhibit it and release another anti-AD drug. Using AChE inhibitors, we may then obtain an action similar to ladostigil but in an opposite mechanistic way.
6.11. Enhancing the Potency of Aminoquinolinealcohols toward Gram-Negative Bacteria with Conjugated Peptides (P15)
Pierre Laumaillé,
Alexandra Dassonville-Klimpt *,
Sophie Da-Nascimento,
François Peltier,
Catherine Mullié,
Sandrine Castelain
and
Pascal Sonnet
AGIR (AGents antiInfectieux Résistances et chimiothérapie), Université de Picardie Jules Verne, 80037 Amiens, France
The development of bacterial resistance to antibiotics (ATBs) constitutes a major threat to public health. The ESKAPEE pathogens (E. faecium, S. aureus, K. pneumonia, A. baumannii, P. aeruginosa, Enterobacter spp., E. coli) and Mycobacterium tuberculosis are a major problem in hospitals and in particular for immunocompromised patients. Moreover, very few new classes of ATBs have emerged over the last decades (Fischback, M.A., et al., Science 2009, 325, 1089–1093). Consequently, there is urgency to set up new strategies to prevent and control the emergence and spread of antimicrobial resistant micro-organisms. The range of basic mechanisms of resistance is surprisingly limited, among them diminution of ATB concentration into the bacterial cell through a reduction of cell permeability is a common way used by Gram-negative bacteria and Mycobacterium tuberculosis.
One promising approaches to overcome these barriers is the disruption of the bacterial membrane by natural or short synthetic antimicrobial peptides (AMPs) (Schmidt, N.W., et al., Curr. Opin. Solid State Mater. Sci. 2013, 17, 151–163). Attachment of such peptides to ATBs should improve outer membrane penetration and antibacterial efficacy ((a) Arnusch, C.J., et al., PLoS ONE 2012 7, e39768; (b) Chen, H., et al., Mol. Pharm. 2015, 12, 2505–2516; (c) Schmidt, N.W., et al., ACS Nano 2014, 8, 8786–8793).
Aminoquinolinemethanols (AQMs), synthesized in our laboratory, possess a very higher antiplasmodial activity at close to nanomolar concentrations ((a) Jonet, A., et al., EP11154229.6-2101 2011; (b) Jonet, A., et al., Tetrahedron:Asymmetry 2011, 22, 138–148; (c) Jonet, A., et al., J. Antibiotics 2013, 66, 683–686; (d) Bentzinger, G., et al., Tetrahedron:Asymmetry 2016, 27, 1–11). During an antibacterial screening performed in our laboratory, these AQMs showed good activities against Gram-positive bacteria (MIC = 4–16 µg/mL) and low against Gram-negative bacteria (MIC often more than 128 µg/mL). AQM-AMP conjugates with short cationic AMP sequences (4 and 6 AAs) were prepared to broaden the antibacterial spectrum of the AQM core and to delay the appearance of resistance phenomena (Yeaman, M.R., et al. Pharmacol. Rev. 2003, 55, 27–55). The synthesis and the promising antimicrobial activity of these compounds will be presented.
Acknowledgments: We would like to thank the Haut de France region for their financial support.
6.12. From Estrothiazine to Estrothiazine Sulfone: Deciphering Transcription from Cell Proliferation in Breast Cancer Cells (P16)
Yves Jacquot 1,*,
Danny Spaggiari 2,
Julie Schappler 2,
Anne Lykkesfeldt 3,
Eric Lesniewska 4,
Serge Rudaz 2
and
Guy Leclercq 5
1
Laboratoire des biomolécules, Université Pierre et Marie Curie—Paris 6, École normale supérieure—Paris, Centre National de la Recherche Scientifique, UMR7203 Paris, France
2
School of Pharmaceutical Sciences, University of Geneva & University of Lausanne, Geneva, Switzerland
3
Institute of Cancer Biology, Copenhagen, Denmark
4
Laboratoire Interdisciplinaire Carnot de Bourgogne, Université de Technologie de Belfort-Montbeliard, Université de Bourgogne, Centre National de la Recherche Scientifique, UMR-CNRS 6303 Dijon, France
5
Institut Jules Bordet, Brussels, Belgium
Menopause is characterized by a panel of crippling effects that are characteristic of the decrease of the amount of the endogenous female hormone 17β-estradiol in blood. To address a better quality of life and health, hormone replacement therapy (HRT) is currently used to counteract such deleterious effects. However, HRT significantly enhances the emergence of breast tumors by ~5% in the case of estrogen-only HRT and by ~50% for combined HRT, after 10 years of treatment. This effect is principally due to the fact that drugs used in HRT activate estrogen-dependent transcription and cell proliferation. Thus, alternative strategies are required to control the carcinogenic effects associated with HRT.
Recently, we have synthesized a sulfonylated coumarin with structural analogies with coumestrol and we have tested its effects in breast carcinoma MCF-7 cells. Remarkably, this compound, which interacts directly with the estrogen receptor α (ERα) but independently from coactivators, promotes the dimerization of the receptor and activates transcription but not cell proliferation. Moreover, it preserves the turnover of the protein, revealing that this phenomenon is exclusively dependent on the ability of a molecule to induce cell proliferation. This effect, which requires ERα, depends of the presence of the sulfone function, which replaces the classical methoxy or phenolic pharmacophore of estrogens. According to in vitro binding studies and modelling calculations, this compound interacts within the binding site of 17β-estradiol, in such a way that the sulfone is engulfed within a small pocket defined by the residues Leu-346, Leu-349, Ala-350 and Leu-384.
In conclusion, our work opens new avenues for the development of estrogenic compounds that exert estrogen-dependent transcription but not cell proliferation. Such molecules could be widely used for the management of menopause and diseases that require a supplementation of estrogens. Future transcriptomic studies will be very informative to decipher the mechanism of action of this sulfone.
6.13. Conception of Multi-Target Directed Ligands Prodrugs to Treat Alzheimer’s Disease (P17)
François-Xavier Toublet 1,
Julien Lalut 1,
Cédric Lecoutey 1,
Bérénice Hatat 1,2,
Benjamin Guieu 1,
Caroline Lanthier 1,
Christophe Rochais 1
and
Patrick Dallemagne 1
1
Normandie Univ, UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), 14000 Caen, France
2
CNRS, UMR-5203, Institut Génomique Fonctionnelle, F-34000 Montpellier, France
In 1906, Alois Alzheimer described the disease for the first time: the patient was suffering from particular memory disorders and present brain pathologic deposits. As a result of multiple research, the disease has proved to be more and more complex: amyloid plaque formation is due to hyperactivation of þ-secretase resulting in the formation of the amyloid peptide (Aþ). In addition, hyperphosphorylation of the tau protein was observed, causing disaggregation of microtubules, a neuronal death and a decreased cholinergic transmission (Yun, H., et al. Exp. Neurobiol. 2011, 20, 159–168).
In order to treat Alzheimer’s disease, a multitude of molecules have been synthesized but only 4 are currently marketed (Cummings, J., et al., Res. Clin. Interv. 2017, 3, 367–384): they are mostly acetylcholinesterase inhibitors (AChE), as for example rivastigmine a covalent pseudo-irreversible inhibitor, and a NMDA inhibitor, memantine.
Facing the complexity of the disease and the lack of effectiveness of the current molecules, we have developed multi-target directed ligands (MTDLs) a new strategy based on a drug with several therapeutic targets of interest to treat a disease. Thus, our laboratory synthesized donecopride (Lecoutey, C., et al., Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830), a molecule inhibiting AChE and simultaneously activating 5-HT4 serotoninergic receptors.
My project consists in the development, the synthesis and the biological evaluation of new molecules with novel mechanism: prodrug MTDLs. Normally a prodrug is an inactive molecule activated by an enzyme (Rautio, J., et al. Nat. Rev. Drug Discov. 2008, 7, 255–270). In this context, the first target is inactivated by prodrug, which finds herself cut in half, releasing the second active drug.
This poster will detail the various prodrugs and explain in more details this new notion of MTDL prodrugs.
6.14. Structure-Guided Design of Pyridoclax Derivatives with Noxa-like Mcl-1 Binding Profile (P19)
Jade Fogha 1,
Siham Hedir 2,3,
Marcella De Giorgi 1,
Fabien Gautier 4,5,
Philippe Juin 4,5,
Laurent Poulain 2,3,
Anne Sophie Voisin-Chiret 1
and
Jana Sopková-de Oliveira Santos 1,*
1
Normandie Univ, UNICAEN, EA 4258 CERMN—FR CNRS INC3M, Caen, France
2
Normandie Univ, UNICAEN, Inserm U1086 ANTICIPE, Biology and Innovative Therapeutics for Ovarian Cancers group (BioTICLA), Centre de Lutte Contre le Cancer F. Baclesse, 3 avenue du Général Harris, 14076 Caen, France
3
UNICANCER, Centre de Lutte Contre le Cancer F. Baclesse, 3 avenue du Général Harris, 14076 Caen, France
4
Team 8 “Stress adaptation and tumor escape”, CRCINA, UMR 1232 INSERM, Université de Nantes, Université d’Angers, Institut de Recherche en Santé-Université de Nantes, Nantes, France
5
ICO site René Gauducheau, Boulevard Jacques Monod, Saint Herblain 44805, France
Protein-protein interactions are attractive targets because they control numerous cellular processes. In oncology, apoptosis regulating Bcl-2 family proteins are of particular interest. Bcl-2 proteins are crucial regulators of the intrinsic mitochondrial pathway of apoptosis and comprise both pro-apoptotic and anti-apoptotic proteins (Cory, S., et al. Nat. Rev. Cancer 2002, 2, 647–656). Apoptotic cell death is controlled via PPIs between the anti-apoptotic proteins hydrophobic groove and the pro-apoptotic proteins BH3 domain. Mcl-1 (an anti-apoptotic Bcl-2 member) is a key regulator of cancer cell survival and a known resistance factor to Bcl-2ƒBcl-xL pharmacological inhibitors making it an attractive therapeutic target. As interaction among Bcl-2 family proteins occurs through a-helices, our laboratory has developed a new family of compounds able to mimic a-helix side chain distribution (abiotic foldamers) using as structural chemical units pyridine andƒor phenyl (Sopková-de Oliveira Santos et al. J. Chem. Inf. Model. 2012, 52, 429–439). The designed and synthesized oligopyridines were evaluated by their capacity to inhibit Mcl-1 in live cells and to sensitize ovarian carcinoma cells to Bcl-xL-targeting strategies and Pyridoclax was emerged a lead (Gloaguen, et al., J. Med. Chem., 2015, 58, 1644–1668; Poulain et al. M Patent EP14305309.8. 2014). Here, using a structure-guided design from the Pyridoclax, we identified a novel selective Noxa-like Mcl-1 inhibitor, MR31367 that selectively binds to Mcl-1 hydrophobic groove and releases Bak and Bim from Mcl-1.
6.15. Alzheimer’s Disease: Development of a Multistep Procedure to Characterize Modulators of Amyloid Peptide Aggregation (P20)
Willy Smeralda 1,*,
Marc Since 1,
Michael Boisserie 2,
Sophie Corvaisier 1,
Julien Cardin 2
and
Aurélie Malzert-Fréon 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
2
NIMPH Team, CIMAP CNRS UMR6252, EnsiCaen-UNICAEN-CEA, 14050 Caen Cedex, France
Alzheimer’s disease (AD) is the most widespread form of senile dementia worldwide and represents the leading socioeconomic problem in healthcare. The onset and the progression of this neurodegenerative disease is associated with the aggregation of the amyloid-þ peptide (Aþ) through a fibrillization process. Hence, an attractive therapeutic strategy against this disorder is the development of molecules able to interfere at specific steps of this fibrillization pathway (Habchi, J., et al. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208), i.e., able to inhibit the formation of the soluble toxic oligomeric Aþ aggregates. To identify such promising therapeutic molecules, experimental methods are required to precisely follow and characterize the state (monomer ƒ oligomers ƒfibrils) and the conformation of Aþ peptide during its fibrillization process. In the same time, these methods must be simple enough to remain compatible with the High Throughput Screening (HTS) of new compounds with potential interest in AD treatment.
In the present study, we propose to combine experimental methods to permit a multiparameter characterization of potential modulators of Aþ1-42 fibrillization. These kinetic studies are carried out in presence of liposomes as biomimetic neuronal membranes. Indeed, it has been established that lipids of the neuronal membrane are an important factor in the cascade of events that leads to the formation of the amyloid fibers.
Thus, fluorescence of Thioflavin T, a specific dye for amyloid species (Biancalana, M., et al., Biochim. Biophys. Acta BBA—Proteins Proteomics 2010, 1804, 1405–1412), was kinetically monitored to follow the Aþ fibrillization pathway. In parallel, considering that the toxic fibers formed by Aþ are mainly organized into þ-sheets, assays were correlated with the secondary structure analysis of the peptide using ATR-FTIR spectroscopy (Sarroukh, R., et al. Biochim. Biophys. Acta BBA—Biomembr. 2013, 1828, 2328–2338). Moreover, a liposome leakage assay supported by dynamic light scattering (DLS) studies was carried out to evaluate the impact of the interactions between the peptide and membranes to predict any destabilization effect.
From results obtained to date, it appears that the tested modulators present a range of effects that can affect defined stages of the aggregation of the Aþ1-42 peptide, organized in defined secondary structure. This study highlights also some advantages of the use of liposomes to characterize and discriminate newly Aþ aggregation modulators synthetized by chemists and could even be expanded to other amyloid diseases.
6.16. Synthesis of Guanylated Hydroxyethylene Peptide Isosteres for Inhibition of FXIIa (P21)
François Simon,
Steffy Pierard,
Lionel Pochet
and
Steve Lanners *
Namur Medicine & Drug Innovation Center (NAMEDIC), Namur Research Institute in Life Science (NARILIS), University of Namur, B-5000 Namur, Belgium
A great challenge in anticoagulation therapy is to limit thrombosis without causing increased risk of bleeding in patients. Oral anticoagulants have come closer to this goal but hemorrhage is still reported. Coagulation factors are further investigated as potential targets to inhibit. Among them, FXIIa, a serine protease, appears to be promising according to numerous animal studies (Weitz, I., et al., J. Front. Med. 2017, 4, 19). Up to now, FXIIa inhibitors under development include proteins, peptides and RNA-based macromolecules. (Larsson, M., et al., Sci. Transl. Med. 2014, 6, 222ra17). On the contrary, low-molecular-weight inhibitors are overlooked. Our goal is to synthesize and assess small molecules as potential FXIIa inhibitors. Based on a 3D model of FXIIa developed in our laboratory, we decided to investigate two series of molecules, including chiral compounds 1.
![Pharmaceuticals 12 00073 i036]()
Each fragment was selected to fit one of the binding pockets around the active site of FXIIa. The chiral hydroxyethylene group was added to mimic the tetrahedral intermediate involved in serine protease enzymes. In this work, the synthesis of model compounds is discussed.
Acknowledgments: This work was supported by a research fellow grant from the F.R.S.-FNRS.
6.17. Computational ‘Microscopy’ of Pyridoclax—Mcl-1 Interaction (P22)
Mohammed Benabderrahmane *,
Camille Denis,
Anne-Sophie Voisin-Chiret,
Ronan Bureau
and
Jana Sopkova-de Oliveira Santos
Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Normandie Univ, UNICAEN, 14000 Caen, France
Myeloid cell leukemia-1 (Mcl-1) is an anti-apoptotic member of the BCL-2 family of proteins. It is often over-expressed in human cancers. Pyridoclax (Poulain, L., et al. EP14305309, 04/03/201) is an original oligopyridine lead, very promising in treatment of chemoresistant cancers. Using different biological and biophysical tests (Gloaguen, C., et al., J. Med. Chem. 2015, 58, 1644–1668), it was identified as a selective Mcl-1 inhibitor.
Computational ‘microscopy’ refers to the use of computational resources to simulate the dynamics of a molecular system. This computational ‘microscopy’ technique is able to capture the interplay between a protein and a ligand at a spatio-temporal resolution that is unmatched by other methods. In this work, the mechanism of the interaction between pyridoclax and Mcl-1 key-residues is unraveled using a combined molecular docking/dynamics investigation.
(Left) Pyridoclax (light brown & licorice representation) interacting within the binding groove of Mcl-1 (cartoon representation). (Right): detailed view of Mcl-1 interacting residues: H-bond (solid red) & hyrdrophobic interactions (dashed green) represent the main contribution.
6.18. Synthesis and Biological Evaluation of Functionalized 4,5-dihydropyridazinones as Anti-Inflammatory Agents (P23)
Ingrid Allart-Simon 1,
Aurélie Moniot 2,
Chantal Barberot 1,
Laurette Malleret 3,
Christine Guillaume 2,
Azzaq Bentaher 3,
Sophie Gangloff 2,
Eric Hénon 1,
Frédéric Velard 2,
Janos Sapi 1
and
Stéphane Gérard 1,*
1
Université de Reims Champagne-Ardenne, Institut de Chimie Moléculaire de Reims (ICMR), UMR CNRS 7312, UFR Sciences, Moulin de la housse and UFR Pharmacie, 51 rue cognacq-jay, 51096 Reims, France
2
Université de Reims-Champagne-Ardenne, EA 4691 Biomatériaux & Inflammation en site OSseux (BIOS), UFR Pharmacie, 51 rue cognacq-jay, 51096 Reims, France
3
Centre International de Recherche en Infectiologie (CIRI), EA7426, Faculté de Médecine Lyon-Sud, 165 chemin du Grand Revoyet, 69921 Oullins, France
In the recent years, cyclic nucleotide phosphodiesterase type 4 (PDE4), which controls intracellular level of cyclic nucleotide cAMP, has emerged as a suitable target for anti-inflammatory therapy (Mulhall, A.M., et al., Expert Opin. Invest. Drugs 2015, 24, 1597–1611; Michalski, J.M., et al., Clin. Pharmacol. Ther. 2012, 91, 134–142).
In continuation of our efforts to develop PDE4 inhibitors for the treatment of respiratory diseases (Gérard, S. et al. Pat. Appl. WO 2016066973 A1 20160506), we describe the design and the synthesis of a new family of compounds possessing pyridazinone scaffold, corresponding molecular modeling study and their evaluation as anti-inflammatory agents. Among these compounds, two 4,5-dihydropyridazinone derivatives possess promising activity, selectivity towards PDE4 isoenzyme and are able to reduce IL-8 production (Barberot, C., et al., Eur. J. Med. Chem. 2018, 146, 139–146).
![Pharmaceuticals 12 00073 i038]()
Acknowledgments: Financial support from the French National Centre for Scientific Research (CNRS), the Regional Council of Champagne–Ardenne (France), the French Ministry of Higher Education and Research (MESR) and the European Regional Development Fund (FEDER) to the PlAneT CPER project is acknowledged. C.B. thanks the Regional Council of Champagne–Ardenne (France) for a Ph.D. fellowship. A.M. Ph.D. fellowship is co-funded by Reims-Metropole and the European Union (Europe invests in Champagne-Ardenne with the European Regional Development Fund). Thanks are given to the Maison de la simulation de Champagne-Ardenne (Reims, France) for the provision of computational facilities.
6.19. Structural and Inhibition Study of Mycobacterium Tuberculosis Phosphoserine Phosphatase (SerB) (P25)
Marie Haufroid *,
Elise Pierson,
Nephtali Callaerts,
Manon Mirgaux
and
Johan Wouters
Laboratoire de Chimie Biologique Structurale (CBS), Université de Namur (UNamur) and Namur Medicine & Drug Innovation Center (NAMEDIC-NARILIS)
Tuberculosis is still the leading cause of death by a single treatable infectious disease, since it kills over 1.5 million people every year. Drug resistant and multi-drug resistant strains are emerging and treatment options are limited. The search for new targets and drugs is needed (Global Tuberculosis Report 2017.
World Health Organization Online,
www.who.int).
In the search for new targets essential for bacterial survival, it was shown that SerB2 (a phosphoserine phosphatase) is secreted within cytoplasm of THP-1 macrophages infected with tuberculosis. The enzyme can dephosphorylate proteins and transcription factors in order to cause cytoskeleton rearrangement and deactivate the host immune response (Sharma, A.K., et al., Indian J. Microbiol. 2016, 1–10). The inhibition of MtSerB2 may help to improve treatment options against the disease.
SerB2 is part of the serine pathway, which produces the amino-acid L-serine from D-3- phosphoglycerate, an intermediate of glycolysis. It catalyses the third and last step of this pathway which is the transformation of phospho-L-serine into L-serine. There are only a few described inhibitors of MtSerB2 and the crystallographic structure of the protein is still unknown (Arora, G., et al., J. Biol. Chem. 2014, 289, 25149–25165).
The purpose of this work is to study the structure of the protein and to conceive new inhibitors. In order to do that, an enzyme inhibition assay was optimized and performed to allow a screening of NAMEDIC’s chemical library. On 125 tested compounds, three showed good inhibitions profile and were found to be competitive inhibitors by kinetic studies. Crystallization assays and docking experiments are still ongoing in order to elucidate the pose of those inhibitors within the active site. Furthermore, in order to assess and design selective inhibitors, human phosphoserine phosphatase (hPSP) was also produced and inhibition and crystallization assays were also performed.
Acknowledgments: The authors thanks members of NAMEDIC who generated NAMEDIC library, in particular Prof B. Masereel and L. Pochet.
6.20. Bioproduction of Polymyxin Derivatives (P26)
Caroline Salsano 1,
Fatoumata Tambadou 1,
J.E. Pereanez Sacarias 2,
Cyrille Barthélémy 1,
Sandrine Didelot 1,
Valérie Sopéna 1,
Valérie Thiéry 1,*
and
Romain Chevrot 1
1
University of La Rochelle, UMR CNRS 7266, La Rochelle, France
2
Autonomous University of Campeche, Department of Environmental Microbiology and Biotechnology, Mexico
With the rise in bacterial resistance and overuse of antibiotics, it is necessary to find efficient treatments against human infections. One strategy is to enhance existing molecules by genetic engineering. As part of our ongoing research (Chevrot, R., et al., Probiotics Antimicrob. Proteins 2013, 5, 18–25; Tambadou, F., et al., Arch. Microbiol, 2015, 197, 521–532), we respectively isolated from the environment and a sputum of a cystic fibrosis patient two novel antibiotic producing strains of Paenibacillus, B-LR and P-32. Paenibacilli are ubiquitous bacteria that produce antibiotics such as polymyxins and fusaricidins. These molecules are obtained from specific enzymes called Nonribosomal Peptide Synthetases (NRPS). The strains B-LR and P-32 inhibit the growth of Pseudomonas aeruginosa and Staphylococcus aureus clinical strains (including methicillin resistant strains). These peptides were purified and characterized by LC-MS. B-LR produces polymyxin E (colistin) and ten different molecules while P-32 synthesizes a new depsipeptide. Two genomic libraries were constructed and screened in order to find the gene clusters responsible for the biosynthesis of these molecules. This screening lead to the discovery of the colistin gene cluster in B-LR. This new gene cluster covers 41 kb and includes 5 ORF. Three of them encode for NRPS involved in the colistin synthesis, whereas two others are ABC transporter-like genes that may release the antibiotic. This study constitutes the first description of the biosynthesis pathway of this commercial antibiotic that could contribute to a better understanding of the biosynthesis. The biotechnological development appears to be a good way the pharmaceutical production of optimized antibacterial treatments.
6.21. Discovery of a Novel Series of Inhibitors of Anti-Apoptotic Proteins for Treatment of Chemorestistant Ovarian Cancers (P29)
Camille Denis 1,*,
Martina De Pascale 1,
Bogdan Marekha 1,
Marie Jouanne 1,
Charline Kieffer 1,
Emilie Brotin 2,
Louis-Bastien Weiswald 2,
Christophe Denoyelle 2,
Laurent Poulain 2,
Asma Bourafai Aziez 3,
Muriel Sebban 3,
Hassan Oulyadi 3,
Ronan Bureau 1
and
Anne Sophie Voisin Chiret 1
1
Centre d’Etudes et de Recherche sur le Médicament de Normandie, Normandie Univ, UNICAEN, CERMN, 14000 Caen, France
2
Normandie Univ, UNICAEN, INSERMN U10186 « ANTICIPE » Axe 2 Bioticla, Centre François Baclesse, Caen, France
3
Normandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA UMR 6014, F-76000 Rouen, France
Protein-protein interactions (PPIs) control many important physiological processes within human cells. A hallmark of cancers is the evasion of apoptosis, which is often associated with the upregulation of the anti-apoptotic members of the Bcl-2 family of proteins. These proteins comprise pro-survival (Bcl-2, Bcl-xL, Mcl-1) and pro-apoptotic members. In many cancers, the balance between the pro- and anti-apoptotic Bcl-2 family members is tipped towards survival. Drugs inhibiting the pro-survival activity of Bcl-2 proteins to restore cell death may therefore be valuable as cancer therapeutics.
Even though potent and selective inhibitors of Bcl-2 and Bcl-xL have been developed, it is not enough to re-establish the apoptosis. Mcl-1 protein has been shown to cause resistance to chemotherapeutics. Consequently, the inhibition of this protein is required to maintain activity. Our laboratory has synthesized a selective inhibitor of Mcl-1 protein, pyridoclax (Gloaguen, C., et al., J. Med. Chem. 2015, 58, 1644–1668). However, the discovery of dual Mcl-1 and Bcl-xL inhibitors would be an important advance in cancer treatment.
Starting from the structure of our lead compound, pyridoclax, fragments have been designed with the objective to retain an activity against Mcl-1 and concomitantly target Bcl-xL. The design, synthesis and the first biological results of these compounds will be presented in this poster.
6.22. β-Lactam Analogues of Combretastatin A-4 Prevent Metabolic Inactivation by Glucuronidation in Chemoresistant HT-29 Colon Cancer Cells (P30)
Azizah Malebari 1,2,*,
Seema M. Nathwani 3,
Darren Fayne 4,
Niamh M. O’Boyle 2
and
Mary J. Meegan 2
1
Department of Pharmaceutical Chemistry, College of Pharmacy, King Abdulaziz University, Jeddah, KSA
2
School of Pharmacy and Pharmaceutical Sciences, Trinity Biomedical Sciences Institute, Trinity College Dublin, Ireland
3
School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Ireland
4
Molecular Design Group, School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Ireland
Glucuronidation by uridine 5-diphosphoglucuronosyl transferase enzymes (UGTs) is a cause of intrinsic drug resistance in cancer cells (Aprile, S., et al., Drug Meta. Dispos. 2007, 35, 2252–2261). Glucuronidation of combretastatin A-4 (CA-4) was previously identified as a mechanism of resistance in hepatocellular cancer cells (Quan, H., et al., J. Pharmacol. Exp. Ther. 2009, 330, 326–333). Herein, we propose chemical manipulation of þ-lactam bridged analogues of Combretastatin A-4 as a novel means of overcoming drug resistance associated with glucuronidation due to the expression of UGTs in the CA-4 resistant human colon cancer HT-29 cells. The alkene bridge of CA-4 is replaced with a þ-lactam ring to circumvent potential isomerisation while the potential sites of glucuronate conjugation are deleted in the novel 3- substituted- 1,4-diaryl-2-azetidinone analogues of CA-4. We hypothesise that glucuronidation of CA-4 is the mechanism of drug resistance in HT-29 cells. Ring B thioether containing 2-azetidinone analogues of CA-4 such as 4-(4-(methylthio)-phenyl)-3-phenyl-1-(3,4,5-trimethoxyphenyl)azetidin-2-one was identified as the most potent inhibitors of tumour cell growth, independent of UGT status, displaying antiproliferative activity in the low nanomolar range. These compounds also disrupted the microtubular structure in MCF-7 and HT-29 cells, and caused G2ƒM arrest and apoptosis. Taken together, these findings highlight the potential of chemical manipulation as a means of overcoming glucuronidation attributed drug resistance in CA-4 resistant human colon cancer HT-29 cells, allowing the development of therapeutically superior analogues.
![Pharmaceuticals 12 00073 i039]()
6.23. The Phosphorylation of the Residues Ser-202, Thr-205 and Ser-208 of the Protein Tau Is the Driving Force for Its Aggregation, an Observation of Clinical Interest in the Context of Alzheimer Disease (P33)
Clément Despres 1,
Cillian Byrne 2,3,
Haoling Qi 1,
François-Xavier Cantrelle 1,
Isabelle Huvent 1,
Béatrice Chambraud 2,4,
Etienne-Emile Baulieu 2,4,*,
Yves Jacquot 3,*,
Isabelle Landrieu 1,
Guy Lippens 5,*
and
Caroline Smet-Nocca 1,*
1
Unité de Glycobiologie Structurale et Fonctionnelle, Centre National de la Recherche Scientifique (CNRS), UMR8576, Université de Lille, Institut National de la Recherche Agronomique (INRA), USC1409, Lille, France
2
Institut Baulieu, Université Paris-Saclay, Institut National de la Santé et de la Recherche Médicale (INSERM), UMR1195, Kremlin-Bicêtre, France
3
Laboratoire des biomolécules, Université Pierre et Marie Curie—Paris 6, École normale supérieure (ENS), Centre National de la Recherche Scientifique (CNRS), UMR7203 Paris, France
4
Petites molécules de neuroprotection, neurodégénération, neurogénération et remyélinisation, Institut National de la Santé et de la Recherche Médicale (INSERM), UMR1195, Le Kremlin-Bicêtre, France
5
Laboratoire d’ingénierie des Systèmes Biologiques et des Procédés - Institut National de la Recherche Agronomique (INRA), UMR0792, Institut National des Sciences Appliquées—Centre National de la Recherche Scientifique (CNRS), UMR5504, Toulouse, France
Alzheimer’s disease is characterized by two important cytological hallmarks: (i) extraneuronal Aβ amyloid fibers and (ii) intraneuronal aggregates (tangles) that are principally composed of the hyperphosphorylated (pathological) form of Tau. In a physiological context, the protein Tau is in charge of the stabilization of microtubules and is thus considered as a microtubule-associated protein (MAP). Briefly, Tau is composed of 441 amino acids amongst which are 85 real or potential phosphorylation sites (85 serines / threonines). Phosphorylation sites seem to be of prime importance pathologically. For example, the doubly phosphorylations at the Ser-202 and Thr-205, which induce a turn-like structure, are components of the epitope that is recognized by the AT8 antibody, which is used to diagnose Alzheimer disease post-mortem (Gandhi, N.S., et al. Angew. Chem. Int. Ed. 2015, 54, 6819–6823).
We show here not only that hyperphosphorylation is a switch for Tau aggregation, but also that the phosphorylation at the three positions Ser-202, Thr-205 and Ser-208, with absence of phosphorylation at the Ser-262, is sufficient to induce Tau aggregation, in vitro.
Whereas the Thr-205 is phosphorylated by the serine/threonine kinase ERK2, the Ser-208 is phosphorylated by others kinases. Thus, targeting specifically the intraneuronal kinases that are in charge of the phosphorylation of these three residues could be of therapeutic interest (Despres, C., et al. Proc. Natl. Acad. Sci. USA 2017, 114, 9080–9085).
Acknowledgments: This work was supported by grants from LabEx DISTALZ, the Région Hauts-de-France, the Centre National de la Recherche Scientifique, the TGE RMN THC, the Fédération de Recherche FRABio, MetaToul (MetabolHUB-AR-11-INBS-0010) and the Institut Baulieu.
6.24. Modulation of Solubility of Trisubstituted Harmine Derivatives Using Crystallization Engineering (P33)
Sébastien Marx *,
Laurie Bodart,
Céline Meinguet,
Nikolay Tumanov
and
Johan Wouters
Namur Medicine & Drug Innovation Center (NAMEDIC-NARILIS) Département de Chimie, Laboratoire de Chimie Biologique Structurale (CBS), Université de Namur, 5000 Namur, Belgium
Harmine is a natural -carboline compound which can be isolated from Peganum harmala, presenting antiproliferative activity. Trisubstituted harmine derivatives were shown to be more potent than mono- and di-substituted ones (Meinguet, C., et al., Eur. J. Med. Chem., 2015, 94, 45–55; Meinguet, C., et al., Eur. J. Pharm. Sci., 2015, 77, 135–140). Here, new trisubstituted harmine derivatives were designed and synthetized in our group with the aim to overcome the intrinsic resistance of cancer cells to apoptotic stimuli.
However, trisubstituted harmine derivatives often present a moderate solubility at pH 7.4. In this context cyclodextrin complexes with CM16 lead compounds were already prepared in our group to improve compounds solubility (Meinguet, C., et al. Eur. J. Pharm. Sci. 2015, 77, 135–140). Here we propose a new approach to reach this objective. Indeed, the third substituent chosen to be placed on the harmine core contains a pyridine moiety in order to improve compound solubility in comparison with benzyl moiety. Moreover, pyridine increases the tendency towards cocrystal/salt formation which is also considered as a potential method to modulate the solubility of the new analogs of CM16 (Pharmaceutical Salts and Co-crystals, J. Wouters and L. Quéré, eds. RSC Publishing: city, country, 2012, pp. 116–119). The aim of this work is to obtain novel druggable harmine-based molecules combining antiproliferative activity in micromolar to submicromolar range and a high solubility at physiological pH in order to enable intravenous injection of these compounds.
Acknowledgments: SM and LB thank the Fonds National pour la Recherche Scientifique-FNRS for financial support and the PC2 technological platform of UNamur.
Funding: The authors would like to acknowledge the Labex SynOrg, Sponsors & Exhibitors for financial support. For more details, please visit:
http://gp2a.org/.