4.1. Chemistry
4.1.1. General Methods
The progress of the reactions was checked by thin layer chromatography (TLC) using silica gel aluminum plates (Merck 60 F254) with visualization under UV light (254 nm) and/or by immersion in a 10% H2SO4 solution in ethanol or in a solution of cerium (IV) sulfate (0.2% w/v) and ammonium molybdate (5% w/v) in H2SO4 (6% aq.) followed by heating (200 °C). Microwave experiments were carried out in a CEM Discover SP Microwave Synthesizer. Flash column chromatography was performed on silica gel 60 G (0.040–0.063 mm, E. Merck). Optical rotations (589 nm, 20 °C) were measured on a Perkin–Elmer 343 polarimeter. NMR spectra were acquired using a BRUKER Avance 400 spectrometer operating at 400.13 MHz for 1H or 100.62 MHz for 13C. The chemical shifts are given in parts per million (ppm). The spectra were calibrated with internal TMS (in the case of 1H NMR spectra in CDCl3) or with the respective solvent residual peak. Coupling constants (J) are given in hertz (Hz). Assignments were made with the help of 2D experiments (COSY, HSQC, HMBC). HRMS spectra were acquired with a QqTOF Impact II mass spectrometer (Bruker Daltonics) equipped with an electrospray ion source (ESI) and were recorded in positive mode.
Synthesis of compound
2 was previously described [
19].
4.1.2. General Procedure for the Mitsunobu Coupling of 3-O-Octyl/Dodecyl-1,2-O-isopropylidene-α-D-xylo/glucofuranose (2, 17) with Purine Derivatives/Uracil
To a solution of partially protected 1,2-O-isopropylidene-α-D-xylo/glucofuranose (1 mmol) in THF (28 mL) or DMF (10 mL) under nitrogen atmosphere, PPh3 (2 equiv.), diethyl azodicarboxylate (DEAD, 2 equiv.) and purine derivative/uracil (2 equiv.) were added. The mixture was stirred under the conditions indicated further. The mixture was concentrated under vacuum and the crude residue was subjected to column chromatography.
4.1.3. General Microwave-Assisted Procedure for the Mitsunobu Coupling of 3-O-Octyl/Dodecyl-1,2-O-isopropylidene-α-D-xylofuranose (2, 17) with Theobromine/Adenine
To a solution of partially protected 1,2-O-isopropylidene-α-D-xylofuranose (0.2 mmol) in THF/DMF (1:1, 2 mL) or DMF (2 mL), PPh3 (2 equiv.), diethyl azodicarboxylate (DEAD, 2 equiv.) and theobromine (2 equiv.) were sequentially added. The mixture was stirred under microwave irradiation (150 W max., P max = 250 Psi) at 65 °C for 30–50 min. The solvent was evaporated, and the crude residue was subjected to column chromatography on silica-gel.
4.1.4. 1-(5-Deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (3)
Obtained according to the general procedure for Mitsunobu coupling, starting from compound 2 (328 mg, 0.916 mmol), theobromine (340 mg, 1.89 mmol) and using PPh3 (502 mg, 1.91 mmol) and DEAD (40% wt. soln. in toluene, 0.89 mL, 1.95 mmol) in THF (25 mL). The reaction mixture was stirred at 50 °C for 48 h. Purification by column chromatography (from AcOEt/hexane 1:1 to 4:1) afforded 5 (76 mg, 16%) as a yellow oil.
Alternatively, the title compound could be obtained using the MW-assisted procedure, starting from compound 2 (79 mg, 0.219 mmol), theobromine (80 mg, 0.44 mmol) and using PPh3 (115 mg, 0.43 mmol) and DEAD (40% wt. soln. in toluene, 0.2 mL, 0.44 mmol) in THF/DMF (1:1, 2 mL), within a reaction mixture of 30 min and in 44% yield (50 mg). = −5 (c = 1, in CH2Cl2). 1H NMR (CDCl3, 400 MHz): δ 7.49 (s, 1H, H-8), 5.97 (d, 1 H, H-1′, J1′,2′ = 3.9), 4.74 (dd, 1 H, H-5′a, J4′,5′a = 8.8, J5′a,5′b = 14.1), 4.59–4.52 (m, 2 H, H-2’, H-4′), 3.99–3.91 (m, 4 H, CH3, N7, H-5′b, J4′,5′b = 1.8), 3.90 (d, 1 H, H-3′, J3′,4′ = 3.4), 3.69–3.59 (m, 1 H, H-1″a), 3.56 (s, 3 H, CH3-N3), 3.48–3.38 (ddd, H-1″b), 1.62–1.51 (m 2 H, CH2-2″), 1.43 (s, 3 H, CH3, i-Pr), 1.38–1.16 (m, 21 H, CH2-3″ to CH2-11″, CH3, i-Pr), 0.86 (t, 3 H, CH3-12″, J = 6.7). 13C NMR (CDCl3, 100 MHz): δ 155.4 (C-6), 151.7 (C-2), 148.9 (C-4), 141.5 (C-8), 111.5 (Cq, i-Pr), 107.9 (C-5), 105.3 (C-1′), 83.5 (C-3′), 82.5 (C-2′), 78.6 (C-4′), 70.5 (CH2-1″), 40.0 (C-5′), 33.7 (CH3, N7), 32.1 (CH2-2″), 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 26.8 (CH3, N3, CH2-3″ to CH2-10″), 26.3, 26.3 (2 × CH3, i-Pr), 22.8 (CH2-11″), 14.3 (CH3-12″). HRMS: calcd for C27H44N4O6 [M + H]+ 521.3334, found 521.3333; calcd for C27H44N4O6 [M + Na]+ 543.3153, found 543.3149.
4.1.5. 9-(5-Deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)adenine (4)
Obtained according to the general MW-assisted procedure for Mitsunobu coupling, starting from compound 2 (80 mg, 0.22 mmol), adenine (60 mg, 0.44 mmol) and using PPh3 (117 mg, 0.44 mmol) and DEAD (40% wt. soln. in toluene, 0.2 mL, 0.44 mmol), in THF/DMF (1:1, 2 mL). The reaction mixture was stirred for 50 min. Purification by column chromatography (from AcOEt/cyclohexane 2:1 to AcOEt) afforded 4 (33 mg, 31%) as a colorless oil. 1H NMR (CDCl3, 400 MHz): δ 8.36 (s, 1 H, H-2), 7.93 (s, 1 H, H-8), 5.96 (d, 1 H, H-1′, J1’,2’ = 3.6), 5.69 (br.s, 2 H, NH2), 4.52 (d, 1 H, H-1′, J1’,2’ = 3.2), 4.64–4.50 (m, 3 H, H-2′, H-5′a, H-4′), 4.35 (dd, 1 H, H-5′b, J5’a,5’b = 15.9, J4’,5’b = 10.0), 3.87 (d, 1 H, H-3′, J2′,3′ = 2.8 ), 3.69–3.60 (m, 1 H, H-1″a), 3.47–3.38 (m, 1 H, H-1″b), 1.62–1.53 (m 2 H, CH2-2″), 1.42–1.20 (m, 26 H, CH2-3″ to CH2-11″, 2 × CH3, i-Pr), 0.87 (t, 4.2 H, CH3-12″, J = 6.7). 13C NMR (CDCl3, 100MHz): δ 155.4 (C-6), 153.0 (C-2), 150.3 (C-4), 141.7 (C-8), 119.6 (C-5), 112.0 (Cq, i-Pr), 105.3 (C-1′), 82.7 (C-3′), 82.2 (C-2′), 78.4 (C-4′), 70.6 (CH2-1″), 42.9 (C-5′), 32.1 (CH2-2″), 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 26.9, 26.3, 26.3, 22.8 (CH2-3″ to CH2-11″, 2 × CH3, i-Pr), 14.3 (CH2-12″). HRMS: calcd for C25H41N5O4 [M + H]+ 476.3231, found 476.3226. calcd for C25H41N5O4 [M + Na]+ 498.3051, found 498.3043.
4.1.6. 2-Acetamido-6-chloro-9-(5-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)purine (5)
Obtained according to the general procedure for Mitsunobu coupling, starting from compound 2 (337 mg, 0.94 mmol), 2-acetamido-6-chloropurine (399 mg, 1.88 mmol) and using PPh3 (504 mg, 1.92 mmol) and DEAD (40% wt. soln. in toluene, 0.82 mL, 1.8 mmol) in THF (25 mL). The reaction mixture was stirred at 50 °C for 16 h. Purification by column chromatography (from AcOEt/petroleum ether 1:2) afforded 5 (265 mg, 51%) as a yellow oil.
= −14 (c = 1, in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1 H, H-8), 5.95 (d, 1 H, H-1’, J1′,2′ = 3.7), 4.60 (d, 1 H, H-2′), 4.56–4.46 (m, 2 H, H-4′, H-5′a), 4.39 (dd, 1 H, H-5′b, J4′,5′b = 9.0, J5′a,5′b = 15.0), 3.85 (d, 1 H, H-3′, J3′,4′ = 3.0), 3.68–3.58 (m, 1 H, H-1″a), 3.45–3.35 (ddd, H-1″b), 2.54 (s, 3 H, CH3, NHAc), 1.60–1.49 (m 2 H, CH2-2″), 1.40 (s, 3 H, CH3, i-Pr), 1.36–1.16 (m, 21 H, CH2-3″ to CH2-11″, CH3, i-Pr), 0.87 (t, 3 H, CH3-12″, J = 6.7). 13C NMR (100 MHz, CDCl3) δ: 153.0 (C-4), 152.0, 151.3 (C-2, C-6), 145.7 (C-8), 128.0 (C-5), 112.2 (Cq, i-Pr), 105.3 (C-1′), 82.6 (C-3′), 82.0 (C-2′), 77.8 (C-4′), 70.6 (CH2-1″), 43.2 (C-5′), 32.0 (CH2-2″), 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 26.9 (CH2-3″ to CH2-10″), 26.3, 26.2 (2 × CH3, i-Pr), 25.3 (CH3, NHAc), 22.8 (CH2-11″), 14.3 (CH3-12″). HRMS: calcd for C27H42ClN5O5 [M + H]+ 552.2947, found 552.2957; calcd for C27H42ClN5O5 [M + Na]+ 574.2767, found 574.2778.
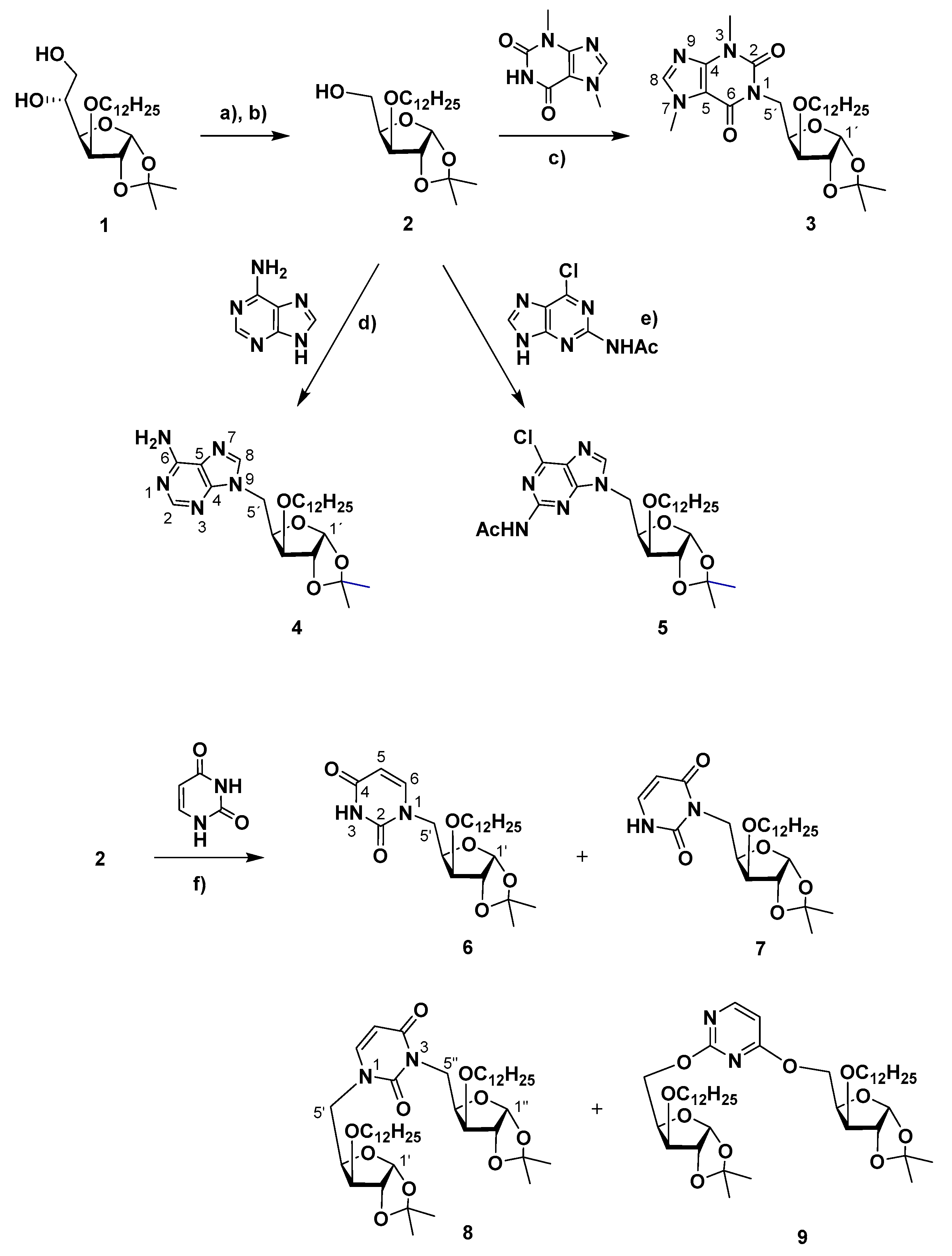
4.1.7. 1-(5-Deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)uracil (6), 3-(5-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)uracil (7), 1,3-bis-(5-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)uracil (8) and 2,4-bis-O-(5-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)uracil (9)
Obtained according to the general procedure for Mitsunobu coupling, starting from compound 2 (286 mg, 0.798 mmol), uracil (182 mg, 1.62 mmol) and using PPh3 (425 mg, 1.62 mmol) and DEAD (40% wt. soln. in toluene, 0.7 mL, 1.54 mmol) in THF (25 mL). The reaction mixture was stirred at 50 °C for 18 h. Purification by column chromatography (from AcOEt/petroleum ether 1:5 to AcOEt) afforded 9 (17 mg, 5%), 8 (91 mg, 29%), 6 (42 mg, 12%) and 7 (46 mg, 13%) as colorless oils.
Compound 6: = −2 (c = 1, in CH2Cl2). 1H NMR (CDCl3, 400 MHz): δ 8.99 (s, 1 H, NH), 7.30 (d, 1 H, H-6, J5,6 = 7.9), 5.91 (d, 1 H, H-1’, J1’,2’ = 3.7), 5.66 (d, 1 H, H-5), 4.56 (d, 1 H, H-2’), 4.41 (dt, 1 H, H-4′, J4′,5′b = 8.8), 4.33 (dd, 1 H, H-5′a, J4′,5′a = 2.6, J5′a,5′b = 14.6), 3.85 (d, 1 H, H-3′, J3′,′ = 3.2), 3.69–3.57 (m, 2 H, H-1″a, H-5′b), 3.44–3.34 (ddd, H-1″b), 1.60–1.50 (m 2 H, CH2-2″), 1.46 (s, 3 H, CH3, i-Pr), 1.34–1.18 (m, 21 H, CH2-3″ to CH2-11″, CH3, i-Pr), 0.87 (t, 3 H, CH3-12″, J = 6.7). 13C NMR (CDCl3, 100MHz): δ 163.8 (C-4), 151.1 (C-2), 145.7 (C-6), 112.1 (Cq, i-Pr), 105.2 (C-1′), 102.1 (C-5), 82.9 (C-3′), 82.2 (C-2′), 78.1 (C-4′), 70.6 (CH2-1″), 48.1 (C-5′), 32.0 (CH2-2″), 29.8, 29.8, 29.8, 29.7, 29.7, 29.5, 29.5, 26.8 (CH2-3″ to CH2-10″) 26.3, 26.2 (2 × CH3, i-Pr), 22.8 (CH2-11″), 14.3 (CH3-12″). HRMS: calcd for C24H40N2O6 [M + Na]+ 475.2779, found 475.2782; calcd for C24H40N2O6 [M + H]+ 453.2959, found 453.2962.
Compound 7: = +6 (c = 1, in CH2Cl2).1H NMR (CDCl3, 400 MHz): δ 9.98 (br.d, 1 H, NH), 7.20 (dd, 1 H, H-6, JNH,6 = 5.8, J5,6 = 7.6), 5.94 (d, 1 H, H-1′, J1′,2′ = 3.8), 5.72 (dd, 1 H, H-5, JNH,5 = 1.2, J5,6 = 7.6 ), 4.59–4.46 (m, 3 H, H-2′, H-4′, H-5′a), 4.00–3.90 (m, 1 H, H-5′b) 3.88 (d, 1 H, H-3′, J3′,4′ = 2.4), 3.68–3.59 (m, 2 H, H-1″a), 3.47–3.37 (ddd, H-1″b), 1.64–1.51 (m 2 H, CH2-2″), 1.45 (s, 3 H, CH3, i-Pr), 1.37–1.16 (m, 21 H, CH2-3″ to CH2-11″, CH3, i-Pr), 0.86 (t, 3 H, CH3-12″, J = 6.7). 13C NMR (CDCl3, 400 MHz): δ 163.6 (C-4), 153.0 (C-2), 139.1 (C-6), 111.7 (Cq, i-Pr), 105.2 (C-1′), 102.0 (C-5), 83.3 (C-3′), 82.4 (C-2′), 78.2 (C-4′), 70.6 (CH2-1″), 40.1 (C-5′), 32.0 (CH2-2″), 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 26.8 (CH2-3″ to CH2-10″), 26.3, 26.2 (2 × CH3, i-Pr), 22.8 (CH2-11″), 14.3 (CH3-12″). HRMS: calcd for C24H40N2O6 [M + Na]+ 475.2779, found 475.2788; calcd for C24H40N2O6 [M + H]+ 453.2959, found 453.2969.
Compound 8: = −4 (c = 1, in CH2Cl2). 1H NMR (CDCl3, 400 MHz): δ 7.24 (d, 1 H, H-6, J5,6 = 7.9), 5.93, 5.90 (2 d, 2 H, H-1′, H-1″, J1′,2′ = 3.8, J1″,2″ = 3.8 ), 5.69 (d, 1 H, H-5), 4.63–4.46 (m, 4 H, H-5″a, H-2′ H-2″, H-4″), 4.44 (ddd, 1 H, H-4′), 4.31 (dd, 1 H, H-5′a, J5′a,5′b = 14.7, J4′,5′a = 2.6), 3.92 (dd, 1 H, H-5″b, J5″a,5″b = 13.8, J4″,5″a = 1.6), 3.86 (d, 1 H, H-3″, J3″,4″ = 3.3), 3.82 (d, 1 H, H-3′, J3′,4′ = 3.1), 3.68–3.55 (m, 3 H, H-5’b, H-1′′′a, H-1″″a), 3.46–3.32 (m, 2 H, H-1′′′b, H-1″″b), 1.60–1.49 (m 4 H, CH2-2′′′, CH2-2″″) 1.45, 1.43 (2 s, 2 × 3 H, 2 × CH3, i-Pr), 1.35–1.16 (m, 42 H, CH2-3′′′ to CH2-11′′′, CH2-3″″ to CH2-11″″, 2 × CH3, i-Pr), 0.86 (t, 3 H, CH3-12′′′, CH3-12″″, J = 6.7). 13C NMR (CDCl3, 100MHz): δ 163.2 (C-4), 151.9 (C-2), 143.4 (C-6), 112.0, 111.5 (Cq, i-Pr), 105.3, 105.3, (C-1′, C-1″), 101.8 (C-5), 83.5 (C-3″), 82.9 (C-3′), 82.4, 82.1 (C-2′, C-2″), 78.3 (C-4′), 78.1 (C-4″), 70.5 (CH2-1′′′, CH2-1″″), 49.0 (C-5′), 40.8 (C-5″), 32.0 (CH2-2′′′, CH2-2″″), 29.8, 29.8, 29.8, 29.7, 29.7, 29.7, 29.7, 29.6, 29.5, 29.5, 29.5, 26.8 (CH2-3′′′ to CH2-10′′′, CH2-3″″ to CH2-10″″), 26.3, 26.3, 26.2, 26.2 (4 × CH3, i-Pr), 22.8 (CH2-11′′′, CH2-11″″), 14.3 (CH3-12′′′, CH3-12″″). HRMS: calcd for C44H76N2O10 [M + Na]+ 815.5392, found 815.5420; calcd for C44H76N2O10 [M + H]+ 793.5573, found 793.5584.
Compound 9: = −31 (c = 1, in CH2Cl2). 1H NMR (CDCl3, 400 MHz): δ = 8.18 (d, 1H, H-6, J = 5.7), 6.41 (d, 1 H, H-5), 5.97–5.92 (m, 2 H, H-1′, H-1″, J = 3.9, J = 4.2), 4.69 (dd, 1H, H-5′a, J5a,5b = 10.5, J4,5a = 3.9), 4.63–4.42 (m, 7 H, H-2′, H-2″, H-4′, H-4″, H-5′b, H-5″a, H-5″b), 3.95, 3.89 (2 d, 2 × 1 H, H-3′, H-3″, J = 2.2 Hz, J = 2.8), 3.66–3.52 (m, 2 H, H-1′′′a, H-1″″a), 3.48–3.36 (m, 2 H, H-1′′′b, H-1″″b), 1.59–1.45 (m, 10 H, 2 × CH3, i-Pr, CH2-2′′′, CH2-2″″); 1.36–1.14 (m, 42 H, 2 × CH3, i-Pr, CH2-3′′′ to CH2-11′′′, CH2-3′′′ to CH2-11″″); 0,87 (t, 6 H, CH3-12′′′, CH3-12″″, J = 6.5) ppm. 13C NMR (CDCl3, 400 MHz): δ 170.9 (C-4), 164.8 (C-2), 158.7 (C-6), 111.9, 111.8 (2 × Cq, i-Pr), 105.4, 105.3 (C-1′, C-1″), 102.54 (C5), 82.7, 82.6, 82.4, 82.4 (C-2′, C-2″, C-3′, C3″), 78.4, 78.3 (C-4′, C4″), 70.8, 70.7 (CH2-1′′′/ CH2-1″″), 64.7, 64.4 (C-5′, C-5″), 32.1, 29.8, 29.8, 29.7, 29.7, 29.5, 27.0, 26.5, 26.4, 26.2, 26.1 (4 × CH3, i-Pr, CH2-2′′′ to CH2-10′′′, CH2-2″″ to CH2-10″″), 22.8 (CH2-11′′′, CH2-11″″), 14.4 (CH3-12′′′, CH3-12″″) ppm. HRMS: calcd for C44H76N2O10 [M + H]+ 793.5573, found 793.5592; calcd for C44H76N2O10 [M + Na]+ 815.5392, found 815.5414.
4.1.8. 1-(5-Deoxy-3-O-dodecyl-α,β-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (10)
A solution of 1-(5-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (3, 39 mg, 0.075 mmol) in aq. trifluoroacetic acid (TFA, 60%, 3 mL) was stirred at room temp. for 2 h. The solvents were co-evaporated with toluene and the residue was subjected to column chromatography (from AcOEt/hexane, 10:1 to AcOEt/MeOH, 12:1) to afford 10 (30 mg, 83%, anomeric mixture, α/β ratio, 1:0.9) as colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.54 (s, 1.9 H, H-8 α, β), 5.56 (br.s, 1 H, H-1′ α), 5.11 (1, 0.9 H, H-1′ β), 4.67–4.38 (m, 3.8 H, H-4′ α, H-4′ β, H-5′a α, H-5′a β), 4.34–4.19 (m, 1.9 H, H-2′ α, H-2′ β), 4.08–3.90 (m, 9.5 H, H-3′ α, H-3′ β, H-5′b α, H-5’b β, CH3-N7 α, β), 3.71–3.42 (m, 9.5 H, CH2-1″ α, CH2-1″ β, CH3-N3 α, β) 1.70–1.60 (m 3.8 H, CH2-2″ α, CH2-2″ β), 1.37–1.20 (m, 34.2 H, CH2-3″ to CH2-11″), 0.87 (t, 5.7 H, CH3-12″, J = 6.7). 13C NMR (100 MHz, CDCl3) δ: 155.9 (C-6 α, β), 152.0, 151.9 (C-2 α, β), 149.1, 148.9 (C-4 α, β), 142.1 (C-8 α, β), 107.9, 107.8 (C-5 α, β), 103.5 (C-1′ β), 96.4 (C-1′ α), 84.6 (C-3’ β), 84.1 (C-3’ α), 80.1 (C-2′ α), 76.8 (C-4′ α, β), 75.2 (C-2′ β), 71.5 (CH2-1″ α), 70.9 (CH2-1″ β), 43.0 (C-5′, α, β), 33.8, 33.7 (CH3, N7, α, β), 32.1 (CH2-2″, α, β), 30.1, 29.9, 29.9, 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.6, 29.5, 26.2, 22.8, (CH3, N3, CH2-3″ to CH2-11″, α, β), 14.3 (CH2-12″, α, β). HRMS: calcd for C24H40N4O6 [M + Na]+ 503.2840, found 503.2836; calcd for C24H40N4O6 [M + H]+ 481.3021, found 481.3018.
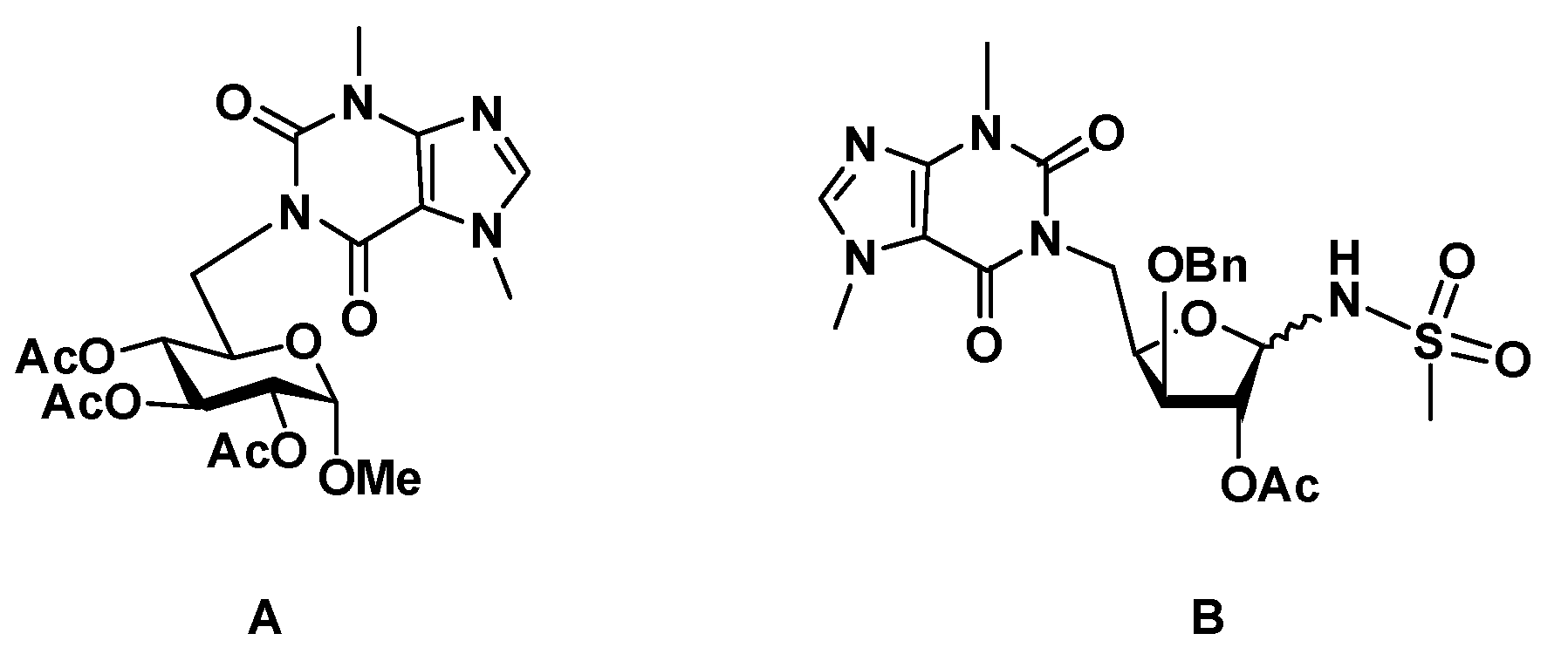
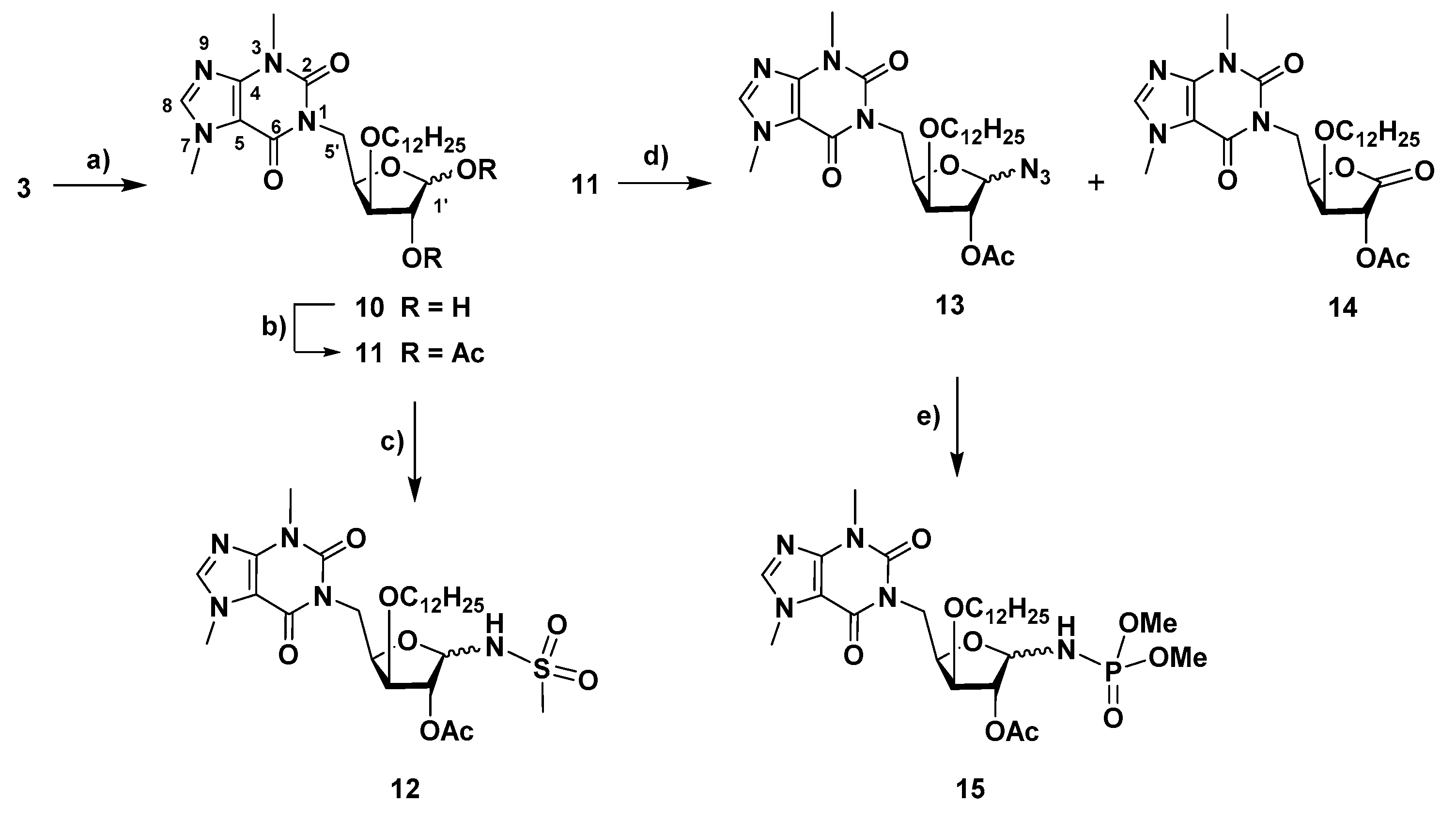
4.1.9. 1-(1,2-Di-O-acetyl-5-deoxy-3-O-dodecyl-α,β-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (11)
A solution of 1-(5-deoxy-3-O-dodecyl-α,β-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (10, 41 mg, 0.078 mmol) in pyridine (3 mL) and acetic anhydride (1.5 mL) was stirred at room temp. for 1.5 h. After co-evaporation of the solvents with toluene, the residue was subjected to column chromatography (AcOEt/hexane, 5:1) to give 11 (33 mg, 69%, anomeric mixture, α/β ratio, 1:0.5) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.50, 7.48 (2 s, 1.5 H, H-8 α, β), 6.38 (dd, 1 H, H-1′ α, J1’,2’ α = 4.5), 5.99 (1, 0.5 H, H-1′ β), 5.27 (t, 1 H, H-2′ α), 5.23 (s, 0.5 H, H-2′ β), 4.84–4.58 (m, 3 H, H-4′ α, H-4′ β, H-5′a α, H-5′a β), 4.19 (t, 1 H, H-3, J2’,3’ α ~ J2’,4’ α = 5.5), 4.01 (br.d, 0.5 H, H-3′ β, J3’,4’ β = 5.0), 3.99–3.84 (m, 6 H, CH3-N7 α, β, H-5′b α, H-5′b β), 3.74–3.65 (m, 0.5 H, H-1′′a β), 3.65–3.44 (m, 7 H, CH2-1″ α, H-1″b β, CH3-N3 α, β), 2.18 (s, 1.5 H, CH3, Ac, β), 2.05 (s, 4.5 H, CH3, Ac, α, β), 1.99 (s, 3 H, CH3, Ac, α), 1.63–1.50 (m, 3 H, CH2-2″, α, β), 1.41–1.16 (m, 27 H, CH2-3″ to CH2-11″), 0.85 (t, 4.5 H, CH3-12″, J = 6.8). 13C NMR (100 MHz, CDCl3) δ: 170.5, 169.8 (CO, Ac, β), 169.7, 169.4 (CO, Ac, α), 155.3, 155.3 (C-6 α, β), 151.7 (C-2 α, β), 148.9, 148.8 (C-4 α, β), 141.6, 141.4 (C-8 α, β), 107.8 (C-5 α, β), 99.0 (C-1′ β), 84.2 (C-1′ α), 82.0 (C-3’ β), 80.7 (C-3’ α), 80.2 (C-4′ β), 79.8 (C-2′ β), 76.5 (C-2’ α), 76.2 (C-4′ α), 71.2, 71.0 (CH2-1″, α, β), 41.9 (C-5′ β), 41.3 (C-5′ α), 33.7, 33.7 (CH3, N7, α, β), 32.0 (CH2-2″, α, β), 29.9, 29.8, 29.8, 29.8, 29.7, 29.7, 29.6, 29.5, 26.3, 26.2, 22.8, (CH3, N3, CH2-3″ to CH2-11″, α, β), 21.5, 21.0, 20.9, 20.7 (CH3, Ac, α, β), 14.2 (CH2-12″, α, β).
4.1.10. N-[2-O-Acetyl-1,5-dideoxy-5-(3,7-dimethyl-3,7-dihydro-2,6-dioxo-1H-purin-1-yl)-3-O-dodecyl-α,β-D-xylofuranos-1-yl]methanesulfonamide (12)
To a solution of 1-(1,2-di-O-acetyl-5-deoxy-3-O-dodecyl-α,β-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (11, 22 mg, 0.039 mmol) in CH2Cl2/acetonitrile (1.2 mL, 4:1) under nitrogen and at 0 °C, BF3·Et2O (0.03 mL, 0.24 mmol) and methanesulfonamide (21 mg, 0.22 mmol) were added. The mixture was stirred at room temp. for 2 h. Then, it was diluted with CH2Cl2 and washed with a satd. aq. NaHCO3 soln. The aqueous phase was extracted with CH2Cl2 (2×) and the combined organic layers were dried with anhydrous MgSO4. After filtration and evaporation of the solvent, the residue was subjected to column chromatography (AcOEt/hexane, 15:1) to afford 12 (19 mg, 81%, anomeric mixture, β/α ratio, 1:0.4) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.52, 7.51 (2 s, 1.4 H, H-8 α, β), 5.73 (d, 1 H, NH, β, J1’,NH β = 11.1), 5.61 (dd, 0.4 H, H-1′ α, J1’,2’ α = 3.5, J1’,NH α = 11.3), 5.23 (d, 1 H, H-1′ β), 5.20 (s, 1 H, H-2′ β), 5.14 (dd, 0.4 H, H-2′ α, J1’,2’ α = 3.5, J2’,3’ α = 0.9), 5.07 (d, 0.4 H, NH α), 4.72 (dd, 1 H, H-5′a β, J5’a,5’b β = 13.9, J4’,5’a β = 9.5), 4.63–4.45 (m, 2.2 H, H-5′a α, H-5′b α, H-4′ α, H-4′ β, J5’a,5’b α = 13.8, J4’,5’a α = 8.7), 4.02–3.91 (m, 6.6 H, H-3′ α, H-3′ β, H-5’b β, CH3-N7 α, β), 3.81–3.72 (m, 1.4 H, H-1′′a α, H-1″a β), 3.61–3.49 (m, 5.6 H, H-1″b α, H-1″b β, CH3-N3 α, β), 3.11 (s, 3 H, S-CH3, β), 3.03 (s, 1.2 H, S-CH3, α), 2.11 (s, 1.2 H, CH3, Ac, α), 2.07 (s, 3 H, CH3, Ac, β), 1.70–1.60 (m 2.8 H, CH2-2″ α, CH2-2″ β), 1.37–1.20 (m, 25.2 H, CH2-3″ to CH2-11″, α, β), 0.87 (t, 4.2 H, CH3-12″, α, β, J = 6.7). 13C NMR (100 MHz, CDCl3) δ: 169.6, 168.9 (CO, Ac, α, β) 155.3 (C-6 α, β), 151.7 (C-2 α, β), 149.1, 149.0 (C-4 α, β), 141.8, 141.7 (C-8 α, β), 107.7 (C-5 α, β), 88.0 (C-1′ β), 82.9 (C-1′ α), 81.5 (C-3’ α), 81.0 (C-3’ β), 79.4 (C-4′ β), 78.9 (C-2′ β), 71.8 (CH2-1″, α, β), 43.0 (SCH3, β), 42.3, 41.7 (SCH3, α, C-5′ β), 33.8 (CH3-N7, α, β), 32.0 (CH2-2″, α, β), 29.9, 29.9, 29.8, 29.8, 29.7, 29.7, 29.6, 29.6, 29.5, 26.1, 22.8 (CH3-N3, CH2-3″ to CH2-11″, α, β) 21.0, 21.0 (CH3, Ac, α, β), 14.3 (CH2-12″, α, β). HRMS: calcd for C27H45N5O8S [M + H]+ 600.3062, found 600.3058; calcd for C27H45N5O8S [M + Na]+ 622.2881, found 622.2878.
4.1.11. 1-(2-O-Acetyl-1-azido-1,5-dideoxy-3-O-dodecyl-α-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (13) and 1-(2-O-acetyl-5-deoxy-3-O-dodecyl-D-xylono-1,4-lacton-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (14)
To a solution of 1-(1,2-di-O-acetyl-5-deoxy-3-O-dodecyl-α,β-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (11, 30 mg, 0.053 mmol) in acetonitrile (5 mL), trimethylsilyl azide (TMSN3, 0.06 mL, 0.43 mmol) and trimethylsilyl triflate (TMSOTf, 0.08 mL, 0.044 mmol) were sequentially added. The mixture was stirred at 65 °C under microwave irradion (150 W, Pmax = 250 Psi) for 1 h 40 min. The solution was then diluted with CH2Cl2 and washed with a sat. aq. NaHCO3 soln. The aqueous phase was extracted with CH2Cl2 (3×) and the combined organic phases were dried with anhydrous MgSO4. After filtration and evaporation, the residue was subjected to column chromatography (from AcOEt/cyclohexane, 1:1 to AcOEt) to afford 13-α (11 mg, 38%) and a mixture (18 mg) containing 13-β and the xylonolactone derivative 14 in a 3.3:1 ratio (corresponding to 14 mg of 13-β (48%) and 4 mg of 14 (14%)).
Data for 13-α: = +74 (c = 1, in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 7.52 (s, 1 H, H-8), 5.58 (d, 1 H, H′-1, J1′,2′ = 4.7), 5.12 (t, 1 H, H′-2, J1′,2′ ~ J2′,3′), 4.71–4.56 (m, 2 H, H′-4, H′-5a), 4.12 (t, 1 H, H′-3, J2′,3′ ~ J3′,4′ ~ 4.5), 3.99 (s, 3 H, CH3-N7), 3.91 (d, 1 H, H′-5b, J = 12.2), 3.66–3.53 (m, 4 H, H-1″a, CH3-N3), 3.54–3.45 (m, 1 H, H-1″b), 2.13 (s, 3 H, CH3, OAc), 1.62–1.52 (m, 2 H, CH2-2″), 1.38–1.18 (m, 18 H, CH2-3″ to CH2-11″), 0.87 (t, 3 H, CH3-12″, J = 6.7). 13C NMR (100 MHz, CDCl3) δ: 170.1 (CO, OAc), 155.4 (C-6), 151.7 (C-2), 149.0 (C-4), 141.7 (C-8), 107.8 (C-5), 89.6 (C-1′), 81.4 (C-3′), 77.7 (C′-2), 76.5 (C′-4); 71.2 (C-1″), 41.2 (C-5ʹ), 33.8 (CH3-N7), 32.1, 29.9, 29.9, 29.8, 29.8, 29.7, 29.6, 29.5, 26.2, 22.8 (CH3-N3, CH2-2″ to CH2-11″), 20.8 (CH3, OAc), 14.3 (C-12″). HRMS: calcd for C26H41N7O6 [M + H]+ 548.3191, found 548.3187; calcd for C26H41N7O6 [M + Na]+ 570.3011, found 570.3004.
Data for 13-β: 1H NMR (400 MHz, CDCl3) δ: 7.50 (s, 1 H, H-8), 5.11 (s, 1 H, Hʹ-2), 5.02 (s, 1 H, H′-1) 4.83 (dd, 1 H, H-5′a, J5′a,5′b = 14.3, J5′a,4′ = 8.3), 4.59 (ddd, 1 H, H-4′), 4.07 (dd, 1 H, H-5′b, J5′b,4′ = 1.9), 3.99 (s, 3 H, CH3-N7), 3.91 (d, 1 H, H′-3, J3′,4′ = 4.3), 3.79–3.70 (m, 1 H, H-1″a), 3.58 (s, 3 H, CH3-N3), 3.54–3.45 (m, 1 H, H-1″b), 2.07 (s, 3 H, CH3, OAc), 1.65–1.56 (m, 2 H, CH2-2″), 1.44–1.17 (m, 18 H, CH2-3″ to CH2-11″), 0.86 (t, 3 H, CH3-12″, J = 6.7)*. 13C NMR (100 MHz, CDCl3) δ: 169,7 (CO, OAc), 155.3 (C-6), 151.8 (C-2), 149.0 (C-4), 141.6 (C-8), 107.8 (C-5), 93.3 (C-1′), 81.9 (C-4′), 81.4 (C′-3), 79.8 (C′-2); 70.9 (C-1″), 41.2 (C-5′), 33.8 (CH3-N7), 32.1, 29.9, 29.8, 29.8, 29.7, 29.6, 29.5, 26.2, 22.8 (CH3-N3, CH2-2″ to CH2-11″), 20.8 (CH3, OAc), 14.3 (C-12″)*. HRMS: calcd for C26H41N7O6 [M + H]+ 548.3191, found 548.3186.
Data for 14: 1H NMR (400 MHz, CDCl3) δ: 7.51 (s, 1 H, H-8), 5.59 (d, 1 H, H′-2, J2′,3′ = 6.7), 5.07 (ddd, 1 H, H′-4), 4.83 (dd, 1 H, H-5′a, J5′a,5′b = 14.3, J5′a,4′ = 10.9), 4.37 (t, 1 H, H′-3, J2′,3′ ~ J3′,4′), 4.07 (dd, 1 H, H-5′b, J5′b,4′ = 3.0), 3.97 (s, 3 H, CH3-N7), 3.65–3.53 (m, 5 H, CH3-N3, CH2-1″), 2.17 (s, 3 H, CH3, OAc), 1.66–1.54 (m, 2 H, CH2-2″), 1.47–1.15 (m, 18 H, CH2-3″ to CH2-11″), 0.87 (t, 3 H, CH3-12″, J = 6.7)*. 13C NMR (100 MHz, CDCl3) δ: 170.0 (C-1′), 169.4 (CO, Ac), 155.1 (C-6), 151.6 (C-2), 149.1 (C-4), 141.8 (C-8), 107.7 (C-5), 78.7 (C-3′), 75.9 (C-4′), 72.2 (C′-2), 71.3 (C-1″), 40.5 (C-5′), 33.8 (CH3-N7), 32.1, 29.9, 29.8, 29.8, 29.7, 29.5, 29.5, 26.1, 22.8 (CH3-N3, CH2-2″ to CH2-11″), 20.7 (CH3, OAc), 14.3 (C-12″)*. HRMS: calcd for C26H40N4O7 [M + H]+ 521.2970, found 521.2966.
* Data extracted from the spectrum of a mixture containing 13-β/14.
4.1.12. Dimethyl N-[2-O-acetyl-1,5-dideoxy-3-O-dodecyl -5-(3,7-dimethyl-3,7-dihydro-2,6-dioxo-1H-purin-1-yl)-α,β-D-xylofuranos-1-y]phosphoramidate (15)
To a solution of 1-(2-O-acetyl-1-azido-1,5-dideoxy-3-O-dodecyl-α-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (13-α, 11 mg, 0.02 mmol) in CH2Cl2 (2 mL), trimethyl phosphite (0.03 mL, 0.25 mmol) was added. The solution was stirred at room temperature for 23 h. The solution was then concentrated under vacuum and the residue was subjected to column chromatography (from AcOEt/hexane, 15:1 to AcOEt/MeOH, 15:1) to afford 15 (7 mg, 56%, anomeric mixture, α/β ratio, 1:0.3) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.50 (s, 1.3 H, H-8 α, β), 5.33 (ddd, 1 H, H-1′ α, J1′,2′ α = 3.9 Hz, J1′,P α = 7.5 Hz, J1′,NH α = 11.6), 5.14 (s, 0.3 H, H-2′ β), 5.06 (d, 1 H, H-2′ α, J1′,2′ α = 3.4 Hz), 4.96 (dd, 0.3 H, H-1′ β, J1′,P β = 7,8 Hz, J1′,NH β = 11.2), 4.73–4.57 (m, 1.3 H, H-5′a, α, β, J5′a,5′b α = 13.9, J4′,5′a α = 9.1, J5′a,5′b β = 13.8, J4′,5′a β = 8.9), 4.54–4.42 (m, 1.3 H, H-4′, α, β), 4.15 (t, 0.3 H, NH, β, J1′,NH β ~ JNH,P β = 11.2 Hz), 4.05–3.85 (m, 6.5 H, H-5′b α, β, H-3′ α, β, CH3-N7 α, β), 3.77–3.46 (m, 15.6 H, 2 × OCH3 α, 2 × OCH3 β, CH2-1″ α, β, H-3′ α, β, CH3-N7 α, β), 2.11 (s, 3 H, CH3, Ac, α), 2.06 (s, 0.9 H, Ac, β), 1.66–1.54 (m, 2.6 H, CH2-2″ α, β), 1.38–1.17 (m, 23.4 H, CH2-3″ to CH2-11″ α, β), 0,87 (t, 3.9 H, CH2-12″ α, β, J = 6.7). 13C NMR (100 MHz, CDCl3) δ: 169.9, 169.7 (CO, Ac α, β), 155.4 (C-6 α, β), 151.7 (C-2 α, β); 149.0, 148.9 (C-4 α, β), 141.6, 141.5 (C-8 α, β), 107.8 (C-5 α, β), 87.4 (C-1′ β), 82.4 (C-1ʹ α), 82.2, 81.5 (C-3′ α, β), 79.9 (C-2′ β); 78.4 (C-4′ α or β), 76.2, 76.0, 76.0 (C-2′ α, C-4′ α or β), 71.4, 71.3 (CH2-1″ α, β); 53.4, 53.4, 53.3, 53.2 (4 d, 2 × OCH3 α, β, JC,P = 4.8), 41.7, 40-8 (C-5′ α, β), 33.7 (CH3-N7 α, β), 32.1, 29.8, 29.8, 29.8, 29.6, 29.6, 29.5, 26.2, 26.2, 22.8 (CH3-N3 α, β, CH2-2″ to CH2-11″), 21.1, 21.0 (CH3, Ac, β), 14.3 (CH3-12″ α, β). 31P NMR (162 MHz, CDCl3) δ: 8.49. HRMS: calcd for C28H48N5O9P [M + H]+ 630.3262, found 630.3270; calcd for C28H48N5O9P [M + Na]+ 652.3082, found 652.3090.
4.1.13. 3-O-Octyl-1,2-O-isopropylidene-α-D-glucofuranose (16)
To a solution of 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (15, 3.0 g, 11.53 mmol) in anhydrous DMF (30 mL) under nitrogen atmosphere and at 0 °C and, NaH (60%, 0.69 g, 17.25 mmol) was added. The suspension was stirred at 0 °C for 10 min. Then, octyl bromide (2.38 mL, 13.79 mmol) was added and the mixture was stirred at room temperature for 22 h. It was then diluted with CH2Cl2 and washed with water and brine solution. The aqueous phase was extracted with CH2Cl2 (3×) and the combined organic layers were dried with anhydrous MgSO4, filtered and concentrated. To the resulting residue, aq. acetic acid (70% soln., 38 mL) was added and the resulting solution was stirred at room temperature for 26 h. After co-evaporation with toluene, the residue was subjected to column chromatography (EtOAc/hexane, 1:3) to afford 16 (3.37 g, 88%, 2 steps) as a colorless oil. 1H NMR (CDCl3, 400 MHz): 5.82 (d, 1 H, H-1, J1,2 = 3.7), 4.47 (d, 1 H, H-2), 4.03 (dd, 1 H, H-4, J3,4 = 3.2, J4,5 = 7.8), 3.95–3.85 (m, 2 H, H-3, H-5), 3.73 (dd, part A of AB system, H-6a, J5,6a = 3.0, J6a,6b = 11.6), 3.63 (dd, part B of AB system, H-6b, J5,6b = 5.6), 3.59–3.49 (m, 1 H, H-1′a), 3.49–3.14 (m, 3 H, H-1′b, OH-5, OH-6), 1.57–1.44 (m, 2 H, CH2-2′), 1.41 (s, 3 H, CH3, i-Pr), 1.31–1.12 (m, 13 H, CH2-3′ to CH2-7′, CH3, i-Pr), 0.81 (t, 3 H, CH3-8′, J = 6.6). 13C NMR (CDCl3, 100 MHz): 111.6 (Cq, i-Pr), 105.0 (C-1), 82.6 (C-3), 82.1 (C-2), 79.7 (C-4), 70.6 (C-1′), 69.3 (C-5), 64.2 (C-6), 31.8, 29.7, 29.3, 29.2, 26.7, 26.2, 26.0, 22.6 (C-2′ to C-7′, 2 × CH3, i-Pr), 14.1 (C-8′). HRMS: calcd for C17H32O6 [M + H]+ 333.2272, found 333.2266; calcd for C17H32O6 [M + Na]+ 355.2091, found 355.2081.
4.1.14. 3-O-Octyl-1,2-O-isopropylidene-α-D-xylofuranose (17)
To a solution of 3-O-octyl-1,2-O-isopropylidene-α-D-glucofuranose (16, 3.14 g, 9.45 mmol) in 60% aq. THF (22 mL), at 0 °C, sodium metaperiodate (4.85 g, 22.68 mmol) was added. The mixture was stirred for 4 h at room temperature. Then, it was diluted with EtOAc and washed with water and brine solution. The aqueous phase was extracted with EtOAc (3×) and the combined organic layers were dried with anhydrous MgSO4. After filtration and concentration under vacuum, the residue was dissolved in EtOH/H2O (57 mL, 2:1). To the resulting solution at 0 °C, NaBH4 (0.461 g, 12 mmol) was added and the mixture was stirred at room temperature for 1.5 h. Then, EtOAc was added. The mixture was washed with brine soln. and the aqueous phase was extracted with EtOAc (2×). The combined organic layers were dried with anhydrous MgSO4, filtered and concentrated. The residue was subjected to column chromatography (EtOAc/petroleum ether, 2:1) to afford 17 (1.85 g, 65%, 2 steps) as a colorless oil. 1H NMR (CDCl3, 400 MHz): 5.98 (d, 1 H, H-1, J1,2 = 3.8), 4.57 (d, 1 H, H-2), 4.27 (br. q, 1 H, H-4), 4.00–3.85 (m, 2 H, H-3, H-5a, H-5b, J3,4 = 3.3, J4,5a = 4.6, J4,5b = 4.4, J5a,5b = 12.3), 3.68–3.58 (m, 1 H, H-1′a), 3.47–3.38 (m, 1 H, H-1′b), 1.61–1.51 (m, 2 H, CH2-2′), 1.49 (s, 3 H, CH3, i-Pr), 1.38–1.19 (m, 13 H, CH2-3′ to CH2-7′, CH3, i-Pr), 0.88 (t, 3 H, CH3-8′, J = 6.6). 13C NMR (CDCl3, 100 MHz): 111.8 (Cq, i-Pr), 105.2 (C-1), 84.5 (C-3), 82.5 (C-2), 79.9 (C-4), 70.6 (C-1′), 61.3 (C-5), 31.9, 29.8, 29.5, 29.3, 27.0, 26.5, 26.2, 22.8 (C-2′ to C-7′, 2 × CH3, i-Pr), 14.2 (C-8′). HRMS: calcd for C16H30O5 [M + H]+ 303.2166, found 303.2156; calcd for C16H30O5 [M + Na]+ 325.1985, found 325.1974.
4.1.15. 1-(5-Deoxy-3-O-dodecyl-α-D-xylofuranos-5-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (18)
A solution of 3-O-octyl-1,2-O-isopropylidene-α-D-xylofuranose (17, 80 mg, 0.26 mmol) in DMF (2 mL) was subjected to the MW-assisted protocol for Mitsunobu coupling, according to the general procure, using PPh3 (138 mg, 0.526 mmol), DEAD (40 % wt soln in toluene, 0.2 mL, 0.44 mmol) and theobromine (95 mg, 0.527 mmol). The mixture was stirred for 30 min. Column chromatography was performed using (AcOEt/petroleum ether, 2:1 as eluent. To the obtained residue, aq. TFA (60% soln., 3 mL) was added and the resulting solution was stirred at room temperature for 4 h. After co-evaporation of the solvents with toluene, the residue was subjected to column chromatography (from AcOEt/petroleum ether, 20:1 to AcOEt/MeOH, 12:1) to afford 10 (22 mg, 20%, 2 steps, anomeric mixture, α/β ratio, 1:1) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.54, 7.51 (s, 2 H, H-8 α, β), 5.56 (d, 1 H, H-1′ α, J1′,2′α = 4.0), 5.11 (d, 1 H, H-1′ β, J 1′,2′β = 1.5), 4.68–4.41 (m, 4 H, H-4′ α, H-4′ β, H-5′a α, H-5′a β), 4.30 (dd, 1 H, H-2′ β, J 2′,3′β = 3.6), 4.22 (br.t, 1 H, H-2′ α), 4.08–3.91 (m, 10 H, H-3′ α, H-3′ β, H-5′b α,H-5’b β, CH3-N7 α, β), 3.71–3.61 (m, 2 H, H-1″a α, H-1″a β), 3.60–3.43 (m, 8 H, H-1″b α, H-1″b β, CH3-N3 α, β) 1.66–1.50 (m 4 H, CH2-2″ α, CH2-2″ β), 1.41–1.16 (m, 20 H, CH2-3″ to CH2-7″), 0.87 (t, 6 H, CH3-8″, J = 6.6). 13C NMR (100 MHz, CDCl3) δ: 155.9, 155.5 (C-6 α, β), 152.0, 151.9 (C-2 α, β), 149.2, 149.0 (C-4 α, β), 142.0, 141.8 (C-8 α, β), 107.9, 107.8 (C-5 α, β), 103.4 (C-1′ β), 96.2 (C-1′ α), 84.6 (C-3’ β), 84.2 (C-3’ α), 80.3 (C-2′ β), 77.1 (C-4′ α, β), 75.3 (C-2′ α), 71.5 (CH2-1″ α), 70.9 (CH2-1″ β), 43.1, 41.5 (C-5′, α, β), 33.8, 33.8 (CH3, N7, α, β), 32.0 (CH2-2″, α, β), 30.0, 30.0, 29.9, 29.6, 29.4, 29.4, 26.2, 26.2, 22.8, (CH3, N3, CH2-3″ to CH2-7″, α, β), 14.3 (CH2-8″, α, β). HRMS: calcd for C20H32N4O6 [M + H]+ 425.2395, found 425.2390; calcd for C20H32N4O6 [M + Na]+ 447.2214, found 447.2208.
4.1.16. 1-(6-Deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-glucofuranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (19)
A solution of 3-O-octyl-1,2-O-isopropylidene-α-D-glucofuranose (16, 80 mg, 0.241 mmol) in DMF (2 mL) was subjected to the protocol for Mitsunobu coupling, according to the general procure, using PPh3 (138 mg, 0.526 mmol), DEAD (40 % wt soln in toluene, 0.24 mL, 0.526 mmol) and theobromine (95 mg, 0.527 mmol). The mixture was stirred under reflux for 20 h. Purification by column chromatography (from AcOEt/petroleum ether 3:1 to 4:1) afforded 19 (19 mg, 16%; isolated yield: 11 mg, 9%) as a colorless oil. 1H NMR (CDCl3, 400 MHz): 7.51 (s, 1 H, H-8), 5.97 (d, 1 H, H-1′, J1,2 = 3.7), 4.54 (d, 1 H, H-2′), 4.43 (dd, part A of AB system, H-6′a, J5′,6′a = 7.3, Ja,b = 17.4), 4.29–4.19 (m, 2 H, H-5′, H-6′b), 4.11 (dd, 1 H, H-4′, J3′,4′ = 3.2, J4′,5′ = 6.3), 4.03 (d, 1 H, H-3′), 3.98 (s, 3 H, CH3-N7), 3.67–3.47 (m, 5 H, CH2-1″, CH3-N3), 1.72–1.52 (m, 2 H, CH2-2″), 1.50 (s, 3 H, CH3, i-Pr), 1.36–1.16 (m, 13 H, CH2-3″ to CH2-7″, CH3, i-Pr), 0.86 (t, 3 H, CH3-8″, J = 6.6). 13C NMR (CDCl3, 100 MHz): 153.4 (C-6), 149.9 (C-4), 141.7 (C-8), 111.7 (Cq, i-Pr), 105.5 (C-1′), 83.1 (C-3′), 82.4 (C-2′), 81.3 (C-4′), 70.9 (C-1″), 69.0 (C-5′), 45.2 (C-6′), 33.8 (CH3, N7), 31.9, 30.0, 29.9, 29.8, 29.6, 29.5, 29.3, 27.0, 26.5, 26.2, 22.8 (CH3, N3, C-2″ to C-7″, 2 × CH3, i-Pr), 14.2 (C-8″). HRMS: calcd for C24H38N4O7 [M + H]+ 495.2813, found 495.2808; calcd for C24H38N4O7 [M + Na]+ 517.2633, found 517.2626.
4.1.17. 1-(6-Deoxy-3-O-dodecyl-α-D-glucopyranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (20)
A solution of 1-(6-deoxy-3-O-dodecyl-1,2-O-isopropylidene-α-D-glucofuranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (19, 20 mg, 0.04 mmol) in aq. trifluoroacetic acid (TFA, 60%, 3 mL) was stirred at room temp. for 5 h. The solvents were co-evaporated of with toluene and the residue was subjected to column chromatography (from AcOEt/petroleum ether 15:1 to AcOEt/MeOH, 20:1) to afford 20 (16 mg, 87%, anomeric mixture, α/β ratio, 1:0.9) as colorless oil. 1H NMR (CDCl3, 400 MHz): 7.62, 7.61 (2 s, 1.9 H, H-8, α, β), 5.28 (br.s, 1 H, H-1′α), 4.61 (d, 0.9 H, H-1′β, J1′,2′ β = 7.5), 4.52 (dd, 0.9 H, H-6′a β, J5′,6′a β = 2.1, J6a,6b β = 14.0), 4.45 (dd, 1 H, H-6′a α, J5′,6′a α = 3.5, J6a,6b α = 14.4), 4.38–4.28 (m, 1.9 H, H-26′b α, H-6′b β), 4.13 (dt, 1 H, H-5′α, J4′,5′ α = 9.7, J5′,6′a α = J5′,6′b α = 3.5), 4.01, 4.00 (2 s, 5.7 H, CH3-N7), 3.95–3.73 (m, 3.8 H, CH2-1″, α, β), 3.67–3.57 (s, 7.6 H, H-3′ β, H-4′ β, CH3-N3), 3.54–3.49 (m, 2.9 H, H-2′ α, H-3′ α, H-5 β), 3.46–3.01 (m, 7.6 H, H-′ β, H-4′ α, OH-1, OH-2, OH-4), 1.66–1.55 (m, 3.8 H, CH2-2″, α, β), 1.39–1.20 (m, 19 H, CH2-3″ to CH2-7″, α, β), 0.86 (t, 5.7 H, CH3-8″, α, β, J = 6.5). 13C NMR (CDCl3, 100 MHz): 156.0 (C-6 α, β), 152.4 (C-2 α, β), 149.0 (C-4 α, β), 142.1 (C-8 α, β), 107.7 (C-5 α, β), 107.7 (C-5 α, β), 97.2 (C-1′ β), 92.6 (C-1′ α), 83.5 (C-3′ β), 81.2 (C-3′ α), 75.1 (C-4′ β), 74.8 (C-2′ β), 73.8 (C-1″), 73.7 (C-5′ β), 72.2 (C-2′ α), 72.1 (C-2′ β), 72.1 (C-4′ α), 71.3 (C-5′ α), 41.6 (C-6′ α, β), 33.9 (CH3, N7, α, β), 32.0, 31.0, 30.2, 29.9, 29.6, 29.4, 26.2, 22.8 (CH3, N3, C-2″ to C-7″, α, β), 14.3 (C-8″, α, β). HRMS: calcd for C21H34N4O7 [M + H]+ 455.2500, found 455.2494; calcd for C21H34N4O7 [M + Na]+ 477.2320, found 477.2313.
4.1.18. 1-(1,2,4-Tri-O-acetyl-6-deoxy-3-O-dodecyl-α-d-glucopyranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (21)
A solution of 1-(6-deoxy-3-O-dodecyl-α-D-glucopyranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (20, 27 mg, 0.059 mmol) in pyridine (1.6 mL) and acetic anhydride (1 mL) was stirred at room temp. for 2.5 h. After co-evaporation with toluene, the residue was subjected to column chromatography (AcOEt/hexane, 15:1 to AcOEt) to give 21 (29 mg, 84%, anomeric mixture, β/α ratio, 1:0.7) as a yellow oil. 1H NMR (CDCl3, 400 MHz): 7.53, 7.52 (2 s, 1.7 H, H-8, α, β), 6.24 (br.s, 0.7 H, H-1′α, J1′,2′ α = 3.6), 5.51 (d, 1 H, H-1′β, J1′,2′ β = 8.1), 5.16–4.97 (m, 3.4 H, H-2′α, H-2′β, H-4′α, H-4′β, J2′,3′ α = 9.9, J3′,4′ α = 9.8, J2′,3′ β ~ J3′,4′ β ~ J4′,5′ β ~ 9.5), 4.63–4.49 (m, 1.7 H, H-6′a α, H-6′a β, J6a,6b β = 13.8, J6a,6b α = 14.0, J5,6a α = 9.8, J5,6a β = 9.3), 4.33 (td, 0.7 H, H-5′ α, J4′,5′ α = J5′,6′a α = 9.8, J5′,6′b α = 2.1), 4.04–3.94 (m, 6.1 H, CH3-N7, H-5′ β), 3.90–3.75 (m, 2.4 H, H-6′b α, H-6′b β, H-3 α), 3.66–3.50 (m, 9.5 H, CH2-1″, α, β, H-3′ β, CH3-N3), 2.18, 2.16, 2.08, 2.07, 2.04, 2.04 (6 s, 15.3 H, 3 x CH3, 3 × Ac, α, β), 1.55–1.42 (m, 3.4 H, CH2-2″, α, β), 1.37–1.19 (m, 17 H, CH2-3″ to CH2-7″, α, β), 0.86 (t, 5.1 H, CH3-8″, α, β, J = 6.7). 13C NMR (CDCl3, 100 MHz): 169.9, 169.6, 169.4. 169.1, 169.0 (CO, Ac, α, β), 155.2, 155.1 (C-6 α, β), 151.6, 151.6 (C-2 α, β), 149.0, 148.9 (C-4 α, β), 141.7, 141.7 (C-8 α, β), 107.7, 107.7 (C-5 α, β), 92.3 (C-1′ β), 89.6 (C-1′ α), 80.4 (C-3′ β), 77.0 (C-3′ α), 73.1, 72.9 (C-1″ α, β), 72.6 (C-5′ β), 72.0, 72.0, 71.6, 71.5 (C-2′, C-4′, α, β), 69.6 (C-5′ α), 42.0, 41.9 (C-6′ α, β), 33.8, 33.7 (CH3, N7, α, β), 32.0, 31.1, 30.4, 30.3, 29.9, 29.8, 29.6, 29.5, 29.4, 29.4, 26.1, 26.1, 22.8 (CH3, N3, C-2″ to C-7″, α, β), 21.1, 21.1, 20.9, 20.8 (CH3, Ac, α, β), 14.2 (C-8″, α, β). HRMS: calcd for C27H40N4O10 [M + H]+ 581.2817, found 581.2819; calcd for C27H40N4O10 [M + Na]+ 603.2637, found 603.2633.
4.1.19. N-[2,4-Di-O-acetyl-1,6-dideoxy-6-(3,7-dimethyl-3,7-dihydro-2,6-dioxo-1H-purin-1-yl)-3-O-dodecyl-α,β-D-glucopyranos-1-yl]methanesulfonamide (22)
To a solution of 1-(1,2,4-tri-O-acetyl-6-deoxy-3-O-dodecyl-α-d-glucopyranos-6-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (21, 20 mg, 0.034 mmol) in CH2Cl2/acetonitrile (1.2 mL, 4:1) under nitrogen and at 0 °C, BF3·Et2O (0.02 mL, 0.16 mmol) and methanesulfonamide (19 mg, 0.19 mmol) were added. The mixture was stirred at room temp. for 3.5 h. Then, it was diluted with CH2Cl2 and washed with a satd. aq. NaHCO3 soln. The aqueous phase was extracted with CH2Cl2 (2×) and the combined organic layers were dried with anhydrous MgSO4. After filtration and evaporation of the solvent, the residue was subjected to column chromatography (AcOEt/petroleum ether, 10:1) to afford 22 (8 mg, 38%, anomeric mixture, β/α ratio, 1:0.4) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.53 (s, 1.4 H, H-8 α, β), 6.21 (br.s, 0.4 H, NH α), 5.52 (d, 1 H, NH, β, J1’,NH β = 9.6), 5.06–4.89 (m, 1.8 H, H-4′α, H-4′β, H-1 α), 4.82 (t,1 H, H-2 ′ β, J1’,2’ β ~J2’,3’ α ~ 9.3), 4.61–4.51 (m, 1.4 H, H-1′ β, H-2 α), 4.46 (dd, 1 H, H-6′a β, J6a,6b β = 13.8, J5,6a β = 9.8), 4.37–4.25 (m, 0.8 H, H-5′ α, H-6′a α), 3.89 (d, 1 H, H-6′b β), 3.98, 3.95 (2 s, 4.2 H, CH3-N7 α, β), 3.82–3.73 (m, 1.8 H, H-5′ β, H-6′b α, H-3′ α), 3.66–3.47 (m, 8 H, H-3′ β, CH3-N3 α, β, CH2-1″, α, β), 2.85 (s, 4.2 H, S-CH3, α, β), 2.18, 2.11, 2.06, 2.01 (s, 8.4 H, CH3, Ac, α), 1.52–1.42 (m 2.8 H, CH2-2″ α, CH2-2″ β), 1.36–1.14 (m, 14 H, CH2-3″ to CH2-7″, α, β), 0.88 (t, 4.2 H, CH3-8″, α, β, J = 6.6). 13C NMR (100 MHz, CDCl3) δ: 171.0, 170.1, 169.0 (CO, Ac, α, β), 155.1 (C-6 α, β), 151.5 (C-2 α, β), 149.2 (C-4 α, β), 142.1 (C-8 α, β), 107.6 (C-5 α, β), 83.1 (C-1′ β), 80.4 (C-3’ β), 74.3 (C-5′ β), 73.4 (CH2-1′′, α, β), 72.0 (C-2′ β), 71.8 (C-4′ β), 43.2 (SCH3, β), 42.1, 42.1 (C-6′ β, C-6′ α), 42.0 (SCH3, α), 33.7 (CH3, N7, α, β), 32.0, 30.3, 29.9, 29.9, 29.8, 29.6, 29.4, 26.1, 22.8 (CH3, N3, C-2″ to C-7″, α, β), 21.1, 21.0 (CH3, Ac, α), 14.2 (C-8″, α, β). HRMS: calcd for C26H41N5O10S [M + H]+ 616.2647, found 616.2645; calcd for C26H41N5O10S [M + Na]+ 638.2466, found 638.2464.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}