Constructing Auxin-Inducible Degron Mutants Using an All-in-One Vector


Abstract
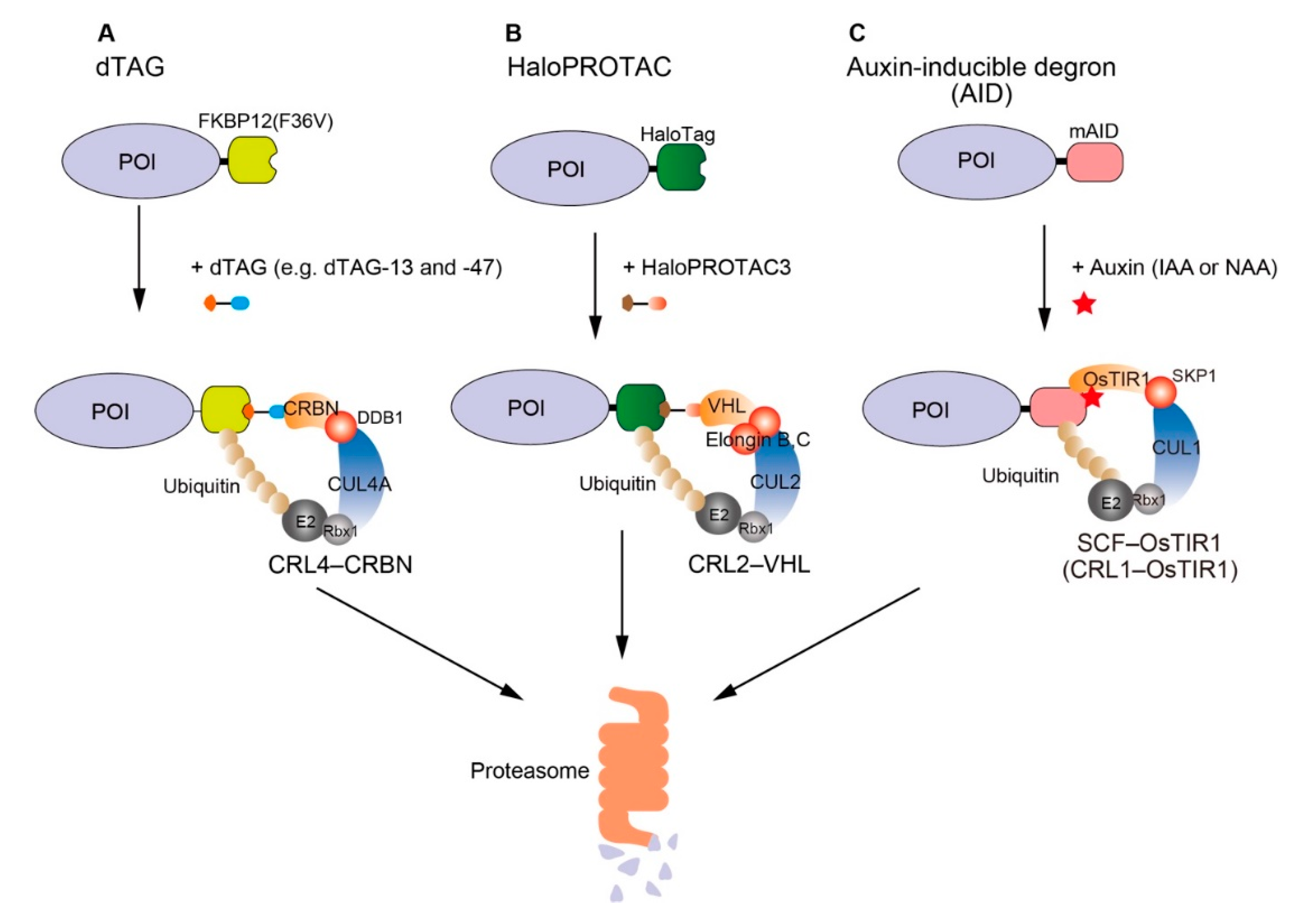
:1. Introduction
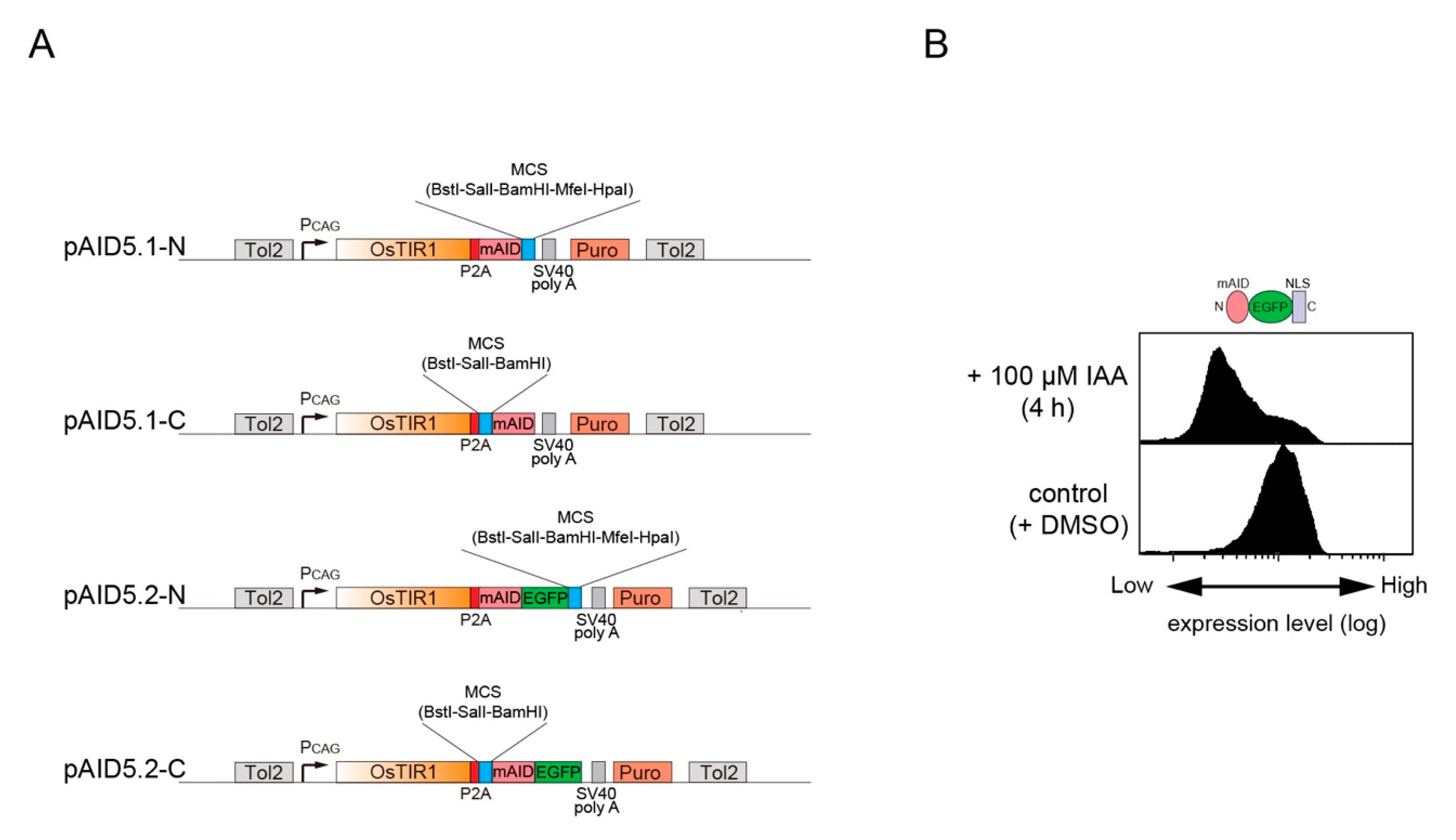
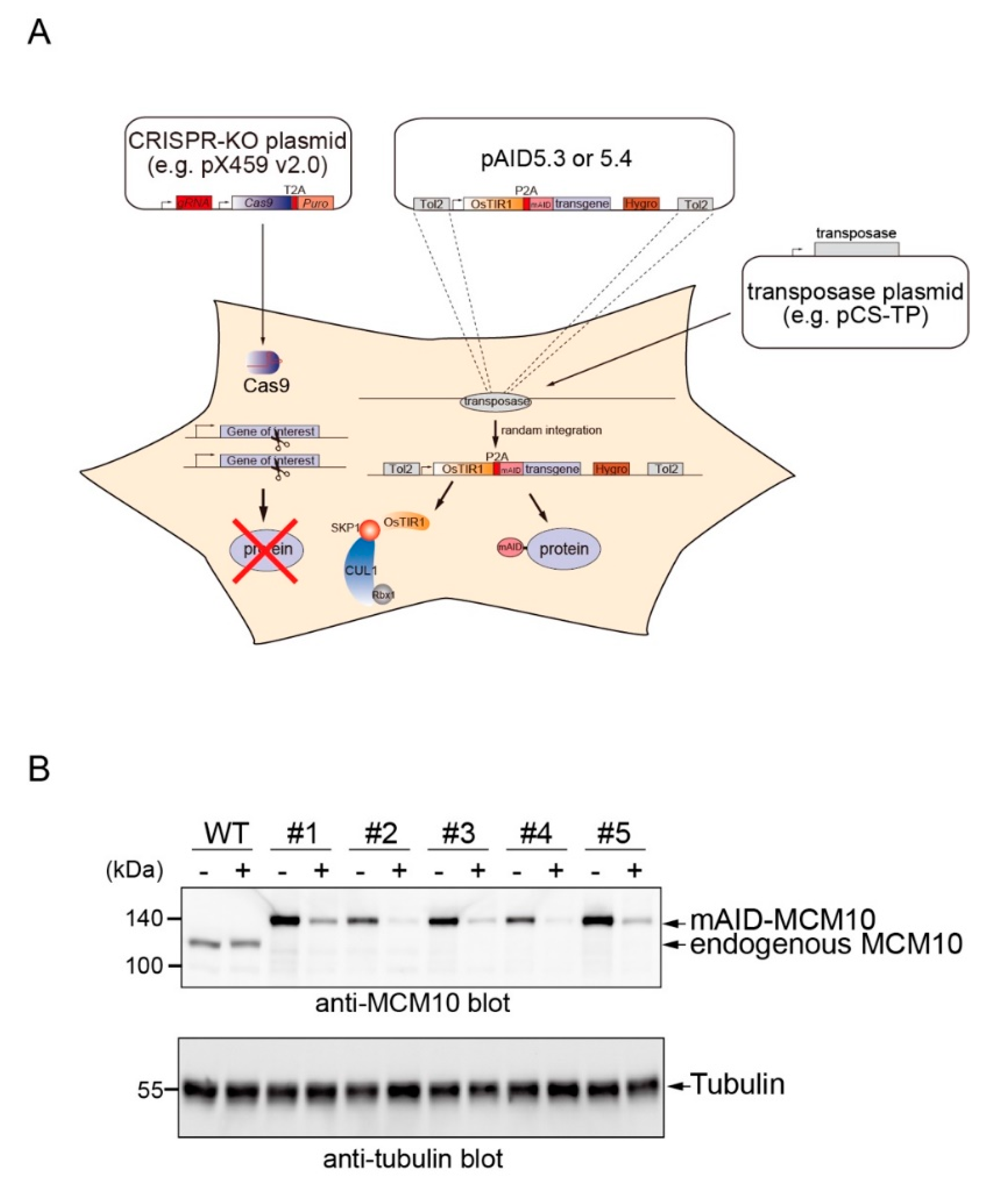
2. Results
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture, Transfection, and Isolation of Clones
4.3. Flow cytometry
4.4. Protein Detection by Western Blotting
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Nat. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Okuhira, K.; Ohoka, N.; Sai, K.; Nishimaki-Mogami, T.; Itoh, Y.; Ishikawa, M.; Hashimoto, Y.; Naito, M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 2011, 585, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, M.; Ohoka, N.; Shibata, N.; Tsukumo, Y. Targeted Protein Degradation by Chimeric Small Molecules, PROTACs and SNIPERs. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-induced genetic degradation as a tool for target validation. Drug Discov. Today Technol. 2019, 31, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Huang, H.T.; Seo, H.S.; Zhang, T.; Wang, Y.; Jiang, B.; Li, Q.; Buckley, D.L.; Nabet, B.; Roberts, J.M.; Paulk, J.; et al. MELK is not necessary for the proliferation of basal-like breast cancer cells. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588 e1528. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512 e499. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Method 2009, 6, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesbolatova, A.; Natsume, T.; Hayashi, K.I.; Kanemaki, M.T. Generation of conditional auxin-inducible degron (AID) cells and tight control of degron-fused proteins using the degradation inhibitor auxinole. Methods 2019, 164–165, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320 e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944 e922. [Google Scholar] [CrossRef] [Green Version]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A pathway for mitotic chromosome formation. Science 2018, 359. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Takeda, H.; Kawakami, N.; Kobayashi, M.; Matsuda, N.; Mishina, M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 2004, 7, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, F.; Sengupta, S.; De Piccoli, G.; Sanchez-Diaz, A.; Labib, K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012, 31, 2195–2206. [Google Scholar] [CrossRef] [Green Version]
- Kanke, M.; Kodama, Y.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012, 31, 2182–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watase, G.; Takisawa, H.; Kanemaki, M.T. Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr. Biol. 2012, 22, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, M.; Mizuno, T.; Yanagi, K.I.; Sugimura, K.; Okumura, K.; Imamoto, N.; Abe, T.; Hanaoka, F. The Mcm2-7-interacting domain of human mini-chromosome maintenance 10 (Mcm10) protein is important for stable chromatin association and origin firing. J. Biol. Chem. 2017, 292, 13008–13021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Fukagawa, T. An efficient method to generate conditional knockout cell lines for essential genes by combination of auxin-inducible degron tag and CRISPR/Cas9. Chromosome Res. 2017, 25, 253–260. [Google Scholar] [CrossRef]
- Zhou, Y.; Aran, J.; Gottesman, M.M.; Pastan, I. Co-expression of human adenosine deaminase and multidrug resistance using a bicistronic retroviral vector. Hum. Gene Ther. 1998, 9, 287–293. [Google Scholar] [CrossRef]
- Wong, E.T.; Ngoi, S.M.; Lee, C.G. Improved co-expression of multiple genes in vectors containing internal ribosome entry sites (IRESes) from human genes. Gene Ther. 2002, 9, 337–344. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Plasmid | Tag | Marker | Transposon |
|---|---|---|---|
| pAID5.1-N | mAID | Puromycin | TOL2 |
| pAID5.1-C | mAID | Puromycin | TOL2 |
| pAID5.2-N | mAID–EGFP | Puromycin | TOL2 |
| pAID5.2-C | mAID–EGFP | Puromycin | TOL2 |
| pAID5.3-N | mAID | Hygromycin | TOL2 |
| pAID5.3-C | mAID | Hygromycin | TOL2 |
| pAID5.4-N | mAID–EGFP | Hygromycin | TOL2 |
| pAID5.4-C | mAID–EGFP | Hygromycin | TOL2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yesbolatova, A.; Saito, Y.; Kanemaki, M.T. Constructing Auxin-Inducible Degron Mutants Using an All-in-One Vector. Pharmaceuticals 2020, 13, 103. https://doi.org/10.3390/ph13050103
Yesbolatova A, Saito Y, Kanemaki MT. Constructing Auxin-Inducible Degron Mutants Using an All-in-One Vector. Pharmaceuticals. 2020; 13(5):103. https://doi.org/10.3390/ph13050103
Chicago/Turabian StyleYesbolatova, Aisha, Yuichiro Saito, and Masato T. Kanemaki. 2020. "Constructing Auxin-Inducible Degron Mutants Using an All-in-One Vector" Pharmaceuticals 13, no. 5: 103. https://doi.org/10.3390/ph13050103
APA StyleYesbolatova, A., Saito, Y., & Kanemaki, M. T. (2020). Constructing Auxin-Inducible Degron Mutants Using an All-in-One Vector. Pharmaceuticals, 13(5), 103. https://doi.org/10.3390/ph13050103