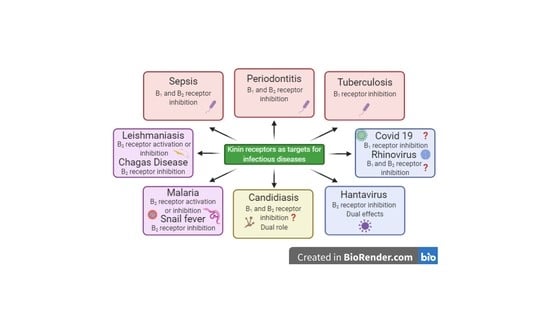
Kinins and Their Receptors in Infectious Diseases


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bacterial Infections and Kinins
2.1. Bacterial Endotoxins and Kinin Responses
2.2. The Kinin System in Systemic Inflammatory Response Syndrome (SIRS)
2.3. Periodontitis and Its Relationship with Kinins
2.4. Kinins and Tuberculosis
3. Viral Infections and Kinins
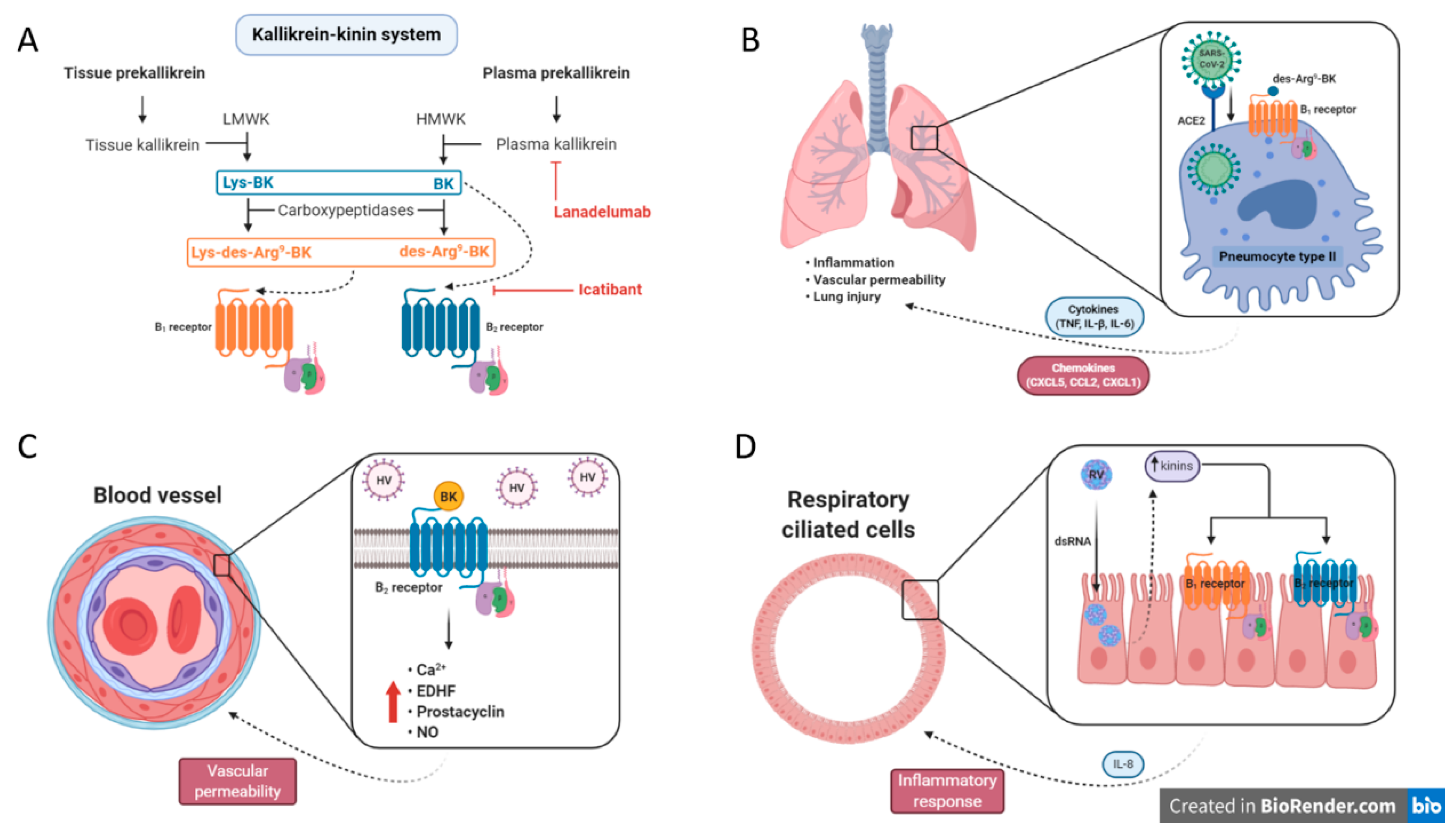
3.1. Crosstalk between SARS-CoV-2 and the Kinin System
3.2. Hantavirus Infection and BK Signaling
3.3. Interplay between Rhinovirus and Kinin Pathways
3.4. Viral Infections and Coagulation–KKS Axis
4. Kinins and Their Implication in Protozoan and Parasite Infections
4.1. Kinins in Leishmaniasis Pathophysiology
4.2. Kinins Set the Tone in Trypanosoma Infections
4.3. The Kinin System in Malaria
4.4. Kinins and Snail Fever
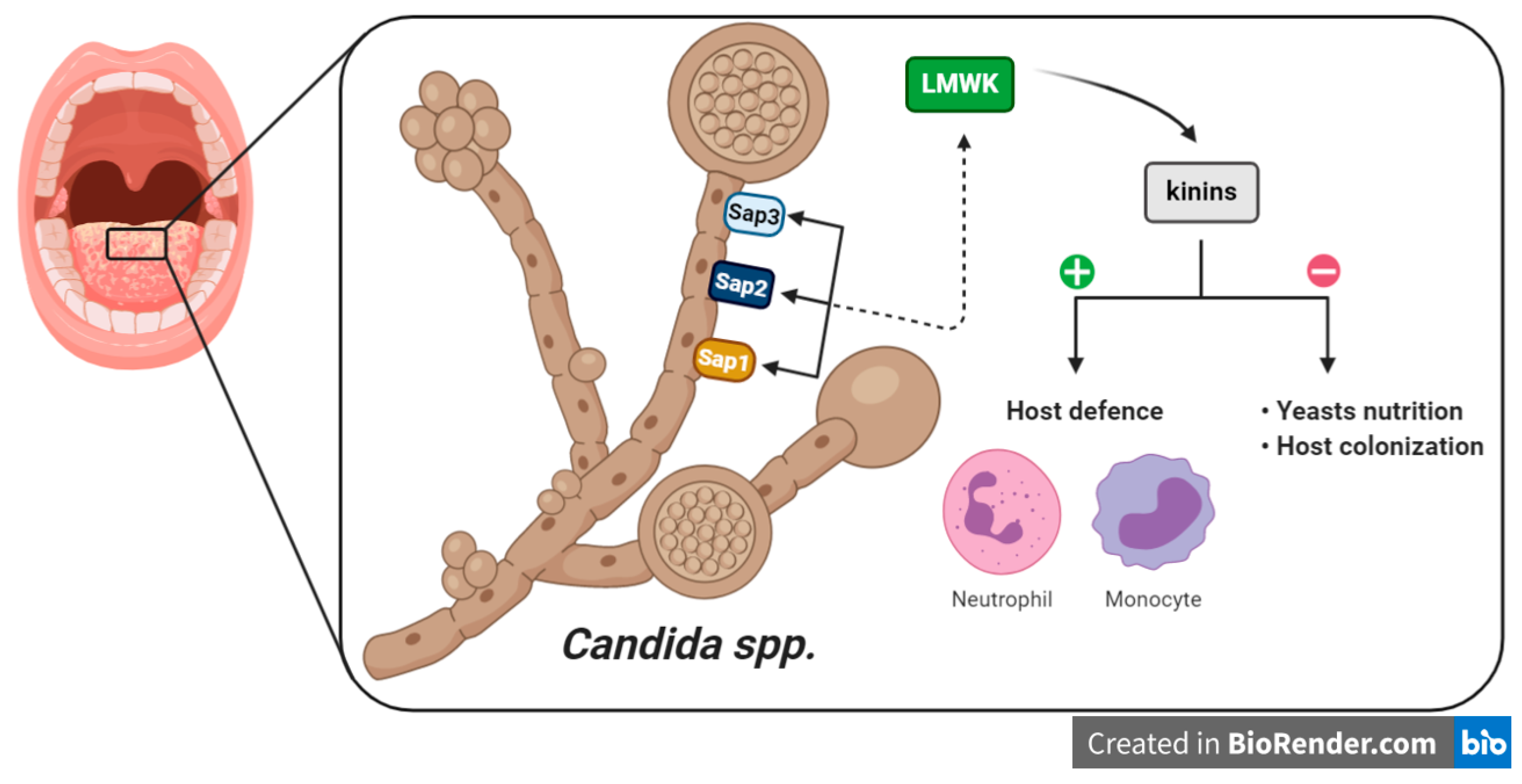
5. Kinins and Fungal Infections
6. Conclusions
Funding
Conflicts of Interest
References
- Kaplan, A.P.; Joseph, K.; Silverberg, M. Pathways for bradykinin formation and inflammatory disease. J. Allergy Clin. Immunol. 2002, 109, 195–209. [Google Scholar] [CrossRef]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Couture, R.; Harrisson, M.; Vianna, R.M.J.; Cloutier, F. Kinin receptors in pain and inflammation. Eur. J. Pharmacol. 2001, 429, 161–176. [Google Scholar] [CrossRef]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852. [Google Scholar] [CrossRef]
- Leeb-Lundberg, L.M.F.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L.; Leeb-Lundberg, L.M.F. International Union of Pharmacology. XLV. Classification of the Kinin Receptor Family: From Molecular Mechanisms to Pathophysiological Consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [Green Version]
- Costa-Neto, C.M.; Dillenburg-Pilla, P.; Heinrich, T.A.; Parreiras-E-Silva, L.T.; Pereira, M.G.; Reis, R.I.; Souza, P.P.C. Participation of kallikrein–kinin system in different pathologies. Int. Immunopharmacol. 2008, 8, 135–142. [Google Scholar] [CrossRef]
- Turner, A.J.; Hooper, N.M. The angiotensin–converting enzyme gene family: Genomics and pharmacology. Trends Pharmacol. Sci. 2002, 23, 177–183. [Google Scholar] [CrossRef]
- Calixto, J.B.; Cabrini, D.A.; Ferreira, J.; Campos, M.M. Kinins in pain and inflammation. Pain 2000, 87, 1–5. [Google Scholar] [CrossRef]
- Campos, M.M.; Leal, P.C.; Yunes, R.A.; Calixto, J.B. Non-peptide antagonists for kinin B1 receptors: New insights into their therapeutic potential for the management of inflammation and pain. Trends Pharmacol. Sci. 2006, 27, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Asraf, K.; Torika, N.; Danon, A.; Fleisher-Berkovich, S. Involvement of the Bradykinin B1 Receptor in Microglial Activation: In vitro and in vivo Studies. Front. Endocrinol. 2017, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The Kallikrein-Kinin System: Current and Future Pharmacological Targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Takasawa, R.; Kubo, D.; Iwanaga, N.; Fujita, S.; Suzuki, K.; Suzuki, H.; Kamiya, H.; Chiba, K. Improved Tag-Assisted Liquid-Phase Peptide Synthesis: Application to the Synthesis of the Bradykinin Receptor Antagonist Icatibant Acetate. Org. Process. Res. Dev. 2019, 23, 2576–2581. [Google Scholar] [CrossRef]
- Lesage, A.; Gibson, C.; Marceau, F.; Ambrosi, H.-D.; Saupe, J.; Katzer, W.; Loenders, B.; Charest-Morin, X.; Knolle, J. In vitro Pharmacological Profile of a New Small Molecule Bradykinin B2 Receptor Antagonist. Front. Pharmacol. 2020, 11, 916. [Google Scholar] [CrossRef] [PubMed]
- Bhatwadekar, A.D.; Kansara, V.S.; Ciulla, T.A. Investigational plasma kallikrein inhibitors for the treatment of diabetic macular edema: An expert assessment. Expert Opin. Investig. Drugs 2020, 29, 237–244. [Google Scholar] [CrossRef]
- Levi, M.; Cohn, D.M.; Zeerleder, S. Hereditary angioedema: Linking complement regulation to the coagulation system. Res. Pract. Thromb. Haemost. 2019, 3, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.N. Activation of the Bradykinin System by Angiotensin-Converting Enzyme Inhibitors. Eur. J. Inflamm. 2010, 8, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Ramos, P.I.P.; Barral-Netto, M. G Protein-Coupled Kinin Receptors and Immunity against Pathogens. In Advances in Immunology; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 29–84. [Google Scholar]
- Fein, A.M. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA J. Am. Med. Assoc. 1997, 277, 482–487. [Google Scholar] [CrossRef]
- Bagnaresi, P.; de Barros, N.M.; Assis, D.M.; Melo, P.M.S.; Fonseca, R.G.; Juliano, M.A.; Pesquero, J.B.; Juliano, L.; Rosenthal, P.J.; Carmona, A.K.; et al. Intracellular proteolysis of kininogen by malaria parasites promotes release of active kinins. Malar. J. 2012, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Nguyen, D.T.M.; Li, Y.; Lyu, J.; Graviss, E.A.; Hu, T.Y. Predictive value of serum bradykinin and desArg9-bradykinin levels for chemotherapeutic responses in active tuberculosis patients: A retrospective case series. Tuberculosis 2016, 101, S109–S118. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Insel, P.A. A hypothesis for pathobiology and treatment of COVID-19: The centrality of ACE1/ACE2 imbalance. Br. J. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Frick, I.-M.; Björck, L.; Herwald, H. The dual role of the contact system in bacterial infectious disease. Thromb. Haemost. 2007, 98, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Available online: https://apps.who.int/neglected_diseases/ntddata/sch/sch.html (accessed on 21 July 2020).
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regoli, D.C.; Marceau, F.; Lavigne, J. Induction of B1-receptors for kinins in the rabbit by a bacterial lipopolysaccharide. Eur. J. Pharmacol. 1981, 71, 105–115. [Google Scholar] [CrossRef]
- Marceau, F.; Lussier, A.; St-Pierre, S. Selective Induction of Cardiovascular Responses to des-Arg9-Bradykinin by Bacterial Endotoxin. Pharmacology 1984, 29, 70–74. [Google Scholar] [CrossRef]
- Bouthillier, J.; Deblois, D.; Marceau, F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vitro. Br. J. Pharmacol. 1987, 92, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Saban, R. Lipopolysaccharide upregulates bradykinin 1 receptors in the isolated mouse bladder. J. Urol. 1998, 160, 2267–2273. [Google Scholar]
- Campos, M.M.; Souza, G.E.P.; Calixto, J.B. Upregulation of B1 receptor mediating des-Arg9-BK-induced rat paw oedema by systemic treatment with bacterial endotoxin. Br. J. Pharmacol. 1996, 117, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.M.; Oliveira, C.R.; Pajolla, G.P.; Calixto, J.B.; Pelá, I.R. Central involvement of kinin B1 and B2 receptors in the febrile response induced by endotoxin in rats. Br. J. Pharmacol. 1997, 121, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Passos, G.F.; Fernandes, E.S.; Campos, M.M.; Araújo, J.G.V.C.; Pesquero, J.L.; Souza, G.E.P.; Avellar, M.C.W.; Teixeira, M.M.; Calixto, J.B. Kinin B1 Receptor Up-Regulation after Lipopolysaccharide Administration: Role of Proinflammatory Cytokines and Neutrophil Influx. J. Immunol. 2004, 172, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paya, D.; Stoclet, J.C. Involvement of bradykinin and nitric oxide in the early hemodynamic effects of lipopolysaccharide in rats. Shock 1995, 3, 376–379. [Google Scholar] [PubMed]
- Luo, S.-F.; Wang, C.-C.; Chiu, C.-T.; Chien, C.-S.; Hsiao, L.-D.; Lin, C.-H.; Yang, C.-M. Lipopolysaccharide enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Br. J. Pharmacol. 2000, 130, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, P.G.; Young, S.K.; Poch, K.R.; Nick, J.A.; Falk, S.; Schrier, R.W.; Worthen, G.S. Systemic Inhibition of the Angiotensin-Converting Enzyme Limits Lipopolysaccharide-Induced Lung Neutrophil Recruitment through Both Bradykinin and Angiotensin II-Regulated Pathways. J. Immunol. 2006, 177, 7233–7241. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg 9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Anton, E.L.; Fernandes, D.; Assreuy, J.; Silva-Santos, J.E. Bradykinin increases BP in endotoxemic rat: Functional and biochemical evidence of angiotensin II AT1/bradykinin B2 receptor heterodimerization. Br. J. Pharmacol. 2019, 176, 2608–2626. [Google Scholar] [CrossRef]
- Oehmcke-Hecht, S.; Köhler, J. Interaction of the Human Contact System with Pathogens—An Update. Front. Immunol. 2018, 9, 312. [Google Scholar] [CrossRef]
- Bengtson, S.H. Kinin receptor expression during Staphylococcus aureus infection. Blood 2006, 108, 2055–2063. [Google Scholar] [CrossRef]
- Geng, L.; Wang, S.; Zhao, Y.; Hu, H. Gene expression profile in mouse bacterial chronic rhinosinusitis. Exp. Ther. Med. 2019, 17, 3451–3458. [Google Scholar] [CrossRef] [Green Version]
- Waack, U.; Warnock, M.; Yee, A.; Huttinger, Z.; Smith, S.; Kumar, A.; Deroux, A.; Ginsburg, D.; Mobley, H.L.T.; Lawrence, D.A.; et al. CpaA Is a Glycan-Specific Adamalysin-like Protease Secreted by Acinetobacter baumannii That Inactivates Coagulation Factor XII. mBio 2018, 9, e01606-18. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Scicluna, B.P.; Stroo, I.; Yang, J.; Roelofs, J.J.T.H.; Boer, O.J.; Vos, A.F.; Nürnberg, P.; Revenko, A.S.; Crosby, J.; et al. Prekallikrein inhibits innate immune signaling in the lung and impairs host defense during pneumosepsis in mice. J. Pathol. 2020, 250, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Stroo, I.; Zeerleder, S.; Ding, C.; Luken, B.; Roelofs, J.; de Boer, O.; Meijers, J.; Castellino, F.; van’t Veer, C.; van der Poll, T. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb. Haemost. 2017, 117, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; van’t Veer, C.; Roelofs, J.J.T.H.; Shukla, M.; McCrae, K.R.; Revenko, A.S.; Crosby, J.; van der Poll, T. Limited role of kininogen in the host response during gram-negative pneumonia-derived sepsis. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L397–L405. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Yang, J.; van’t Veer, C.; van der Poll, T. Bradykinin receptor deficiency or antagonism do not impact the host response during gram-negative pneumonia-derived sepsis. Intensive Care Med. Exp. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Köhler, J.; Maletzki, C.; Koczan, D.; Frank, M.; Trepesch, C.; Revenko, A.S.; Crosby, J.R.; MacLeod, A.R.; Mikkat, S.; Oehmcke-Hecht, S. The contact system proteases play disparate roles in streptococcal sepsis. Haematologica 2020, 105, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, R.; Rosenheinrich, M.; Kreikemeyer, B.; Oehmcke-Hecht, S. Streptococcus pyogenes Triggers Activation of the Human Contact System by Streptokinase. Infect. Immun. 2015, 83, 3035–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Xie, Z.; Wang, B.; Colman, R.W.; Dai, J.; Wu, Y. An essential role of high-molecular-weight kininogen in endotoxemia. J. Exp. Med. 2017, 214, 2649–2670. [Google Scholar] [CrossRef] [PubMed]
- Pixley, R.A.; De La Cadena, R.; Page, J.D.; Kaufman, N.; Wyshock, E.G.; Chang, A.; Taylor, F.B.; Colman, R.W. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vitro use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J. Clin. Investig. 1993, 91, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunathan, V.; Zilberman-Rudenko, J.; Olson, S.R.; Lupu, F.; McCarty, O.J.T.; Shatzel, J.J. The contact pathway and sepsis. Res. Pract. Thromb. Haemost. 2019, 3, 331–339. [Google Scholar] [CrossRef]
- Niederwanger, C.; Bachler, M.; Hell, T.; Linhart, C.; Entenmann, A.; Balog, A.; Auer, K.; Innerhofer, P. Inflammatory and coagulatory parameters linked to survival in critically ill children with sepsis. Ann. Intensive Care 2018, 8, 111. [Google Scholar] [CrossRef]
- Ridings, P.C.; Sugerman, H.J.; Blocher, C.R.; Fisher, B.J.; Fowler, A.A. Hemodynamic Effects of Bradykinin Antagonism in Porcine Gram-Negative Sepsis. J. Investig. Surg. 1995, 8, 115–122. [Google Scholar] [CrossRef]
- Tidjane, N.; Hachem, A.; Zaid, Y.; Merhi, Y.; Gaboury, L.; Girolami, J.-P.; Couture, R. A primary role for kinin B1 receptor in inflammation, organ damage, and lethal thrombosis in a rat model of septic shock in diabetes. Eur. J. Inflamm. 2015, 13, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugesan, P.; Jung, B.; Lee, D.; Khang, G.; Doods, H.; Wu, D. Kinin B1 Receptor Inhibition with BI113823 Reduces Inflammatory Response, Mitigates Organ Injury, and Improves Survival among Rats with Severe Sepsis. J. Infect. Dis. 2016, 213, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, V.F.; Todiras, M.; Campos, L.A.; Saul, V.; Popova, E.; Baltatu, O.C.; Pesquero, J.B.; Bader, M. Increased susceptibility to endotoxic shock in transgenic rats with endothelial overexpression of kinin B1 receptors. J. Mol. Med. 2008, 86, 791–798. [Google Scholar] [CrossRef]
- Ni, A.; Yin, H.; Agata, J.; Yang, Z.; Chao, L.; Chao, J. Overexpression of kinin B1 receptors induces hypertensive response to Des-Arg9-bradykinin and susceptibility to inflammation. J. Biol. Chem. 2003, 278, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Tapping, R.I.; Akashi, S.; Miyake, K.; Godowski, P.J.; Tobias, P.S. Toll-Like Receptor 4, But Not Toll-Like Receptor 2, Is a Signaling Receptor for Escherichia and Salmonella Lipopolysaccharides. J. Immunol. 2000, 165, 5780–5787. [Google Scholar] [CrossRef] [Green Version]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting Edge: TLR2 Is Required for the Innate Response to Porphyromonas gingivalis: Activation Leads to Bacterial Persistence and TLR2 Deficiency Attenuates Induced Alveolar Bone Resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Collyer, C.A. Gingipains from Porphyromonas gingivalis—Complex domain structures confer diverse functions. Eur. J. Microbiol. Immunol. 2011, 1, 41–58. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Scovino, A.; Raposo, S.; Gaze, V.M.; Cruz, C.; Svensjö, E.; Narciso, M.S.; Colombo, A.P.; Pesquero, J.B.; Feres-Filho, E.; et al. Kinin Danger Signals Proteolytically Released by Gingipain Induce Fimbriae-Specific IFN-γ-and IL-17-Producing T Cells in Mice Infected Intramucosally with Porphyromonas gingivalis. J. Immunol. 2009, 183, 3700–3711. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, I.; Potempa, J.; Travis, J.; Gao, X.-P. Mechanisms Mediating Porphyromonas gingivalis Gingipain RgpA-Induced Oral Mucosa Inflammation in vitro. Infect. Immun. 2001, 69, 1199–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.-W.; Huang, C.-H.; Huang, H.-C.; Lai, Y.-Y.; Lin, Y.-Y. Transvascular dissemination of Porphyromonas gingivalis from a sequestered site is dependent upon activation of the kallikrein/kinin pathway. J. Periodontal Res. 2006, 41, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Dornelles, F.N.; Santos, D.S.; Van Dyke, T.E.; Calixto, J.B.; Batista, E.L.; Campos, M.M. In vitro Up-Regulation of Kinin B1 Receptors after Treatment with Porphyromonas gingivalis Lipopolysaccharide in Rat Paw. J. Pharmacol. Exp. Ther. 2009, 330, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.P.C.; Lundberg, P.; Lundgren, I.; Magalhães, F.A.C.; Costa-Neto, C.M.; Lerner, U.H. Activation of Toll-like receptor 2 induces B1 and B2 kinin receptors in human gingival fibroblasts and in mouse gingiva. Sci. Rep. 2019, 9, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza, K.; Kalinska, M.; Bochenska, O.; Meyer-Hoffert, U.; Wu, Z.; Fischer, J.; Falkowski, K.; Sasiadek, L.; Bielecka, E.; Potempa, B.; et al. Gingipains of Porphyromonas gingivalis Affect the Stability and Function of Serine Protease Inhibitor of Kazal-type 6 (SPINK6), a Tissue Inhibitor of Human Kallikreins. J. Biol. Chem. 2016, 291, 18753–18764. [Google Scholar] [CrossRef] [Green Version]
- Karkowska-Kuleta, J.; Surowiec, M.; Gogol, M.; Koziel, J.; Potempa, B.; Potempa, J.; Kozik, A.; Rapala-Kozik, M. Peptidylarginine Deiminase of Porphyromonas gingivalis Modulates the Interactions between Candida albicans Biofilm and Human Plasminogen and High-Molecular-Mass Kininogen. Int. J. Mol. Sci. 2020, 21, 2495. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Tuberculosis Report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 25 July 2020).
- Campos, M.M.; Henriques, M.G.M.O.; Calixto, J.B. The role of B1 and B2 kinin receptors in oedema formation after long-term treatment with Mycobacterium bovis bacillus Calmette-Guérin (BCG). Br. J. Pharmacol. 1997, 120, 502–508. [Google Scholar] [CrossRef] [PubMed]
- de Campos, R.O.; Henriques, M.G.M.; Calixto, J. Systemic treatment with Mycobacterium bovis bacillus calmette-guerin (BCG) potentiates kinin B1 receptor agonist-induced nociception and oedema formation in the formalin test in mice. Neuropeptides 1998, 32, 393–403. [Google Scholar] [CrossRef]
- Sabir, N.; Hussain, T.; Liao, Y.; Wang, J.; Song, Y.; Shahid, M.; Cheng, G.; Mangi, M.H.; Yao, J.; Yang, L.; et al. Kallikrein 12 Regulates Innate Resistance of Murine Macrophages against Mycobacterium bovis Infection by Modulating Autophagy and Apoptosis. Cells 2019, 8, 415. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Junior, V.S.; Pail, P.B.; Villela, A.D.; Falcão, V.C.A.; Dadda, A.S.; Abbadi, B.L.; Pesquero, J.B.; Santos, D.S.; Basso, L.A.; Campos, M.M. Effect of the bradykinin 1 receptor antagonist SSR240612 after oral administration in Mycobacterium tuberculosis-infected mice. Tuberculosis 2018, 109, 1–7. [Google Scholar] [CrossRef]
- McArthur, D.B. Emerging Infectious Diseases. Nurs. Clin. North. Am. 2019, 54, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.M.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Khaiboullina, S.; Morzunov, S.; St. Jeor, S. Hantaviruses: Molecular Biology, Evolution and Pathogenesis. Curr. Mol. Med. 2005, 5, 773–790. [Google Scholar] [CrossRef]
- Simpson, S.Q.; Spikes, L.; Patel, S.; Faruqi, I. Hantavirus Pulmonary Syndrome. Infect. Dis. Clin. N. Am. 2010, 24, 159–173. [Google Scholar] [CrossRef]
- Zaki, S.R.; Greer, P.W.; Coffield, L.M.; Goldsmith, C.S.; Nolte, K.B.; Foucar, K.; Feddersen, R.M.; Zumwalt, R.E.; Miller, G.L.; Khan, A.S.; et al. Hantavirus pulmonary syndrome: Pathogenesis of an emerging infectious disease. Am. J. Pathol. 1995, 146, 552–579. [Google Scholar]
- Krüger, D.H.; Schönrich, G.; Klempa, B. Human pathogenic hantaviruses and prevention of infection. Hum. Vaccines 2011, 7, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, T.D.; Sander, G.E.; Nossaman, B.D.; Kadowitz, P.J. Impaired Vasodilation in the Pathogenesis of Hypertension: Focus on Nitric Oxide, Endothelial-Derived Hyperpolarizing Factors, and Prostaglandins. J. Clin. Hypertens. 2012, 14, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Wahl-Jensen, V.; Copeland, A.M.; Jahrling, P.B.; Schmaljohn, C.S. Endothelial Cell Permeability during Hantavirus Infection Involves Factor XII-Dependent Increased Activation of the Kallikrein-Kinin System. PLoS Pathog. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Laine, O.; Leppänen, I.; Koskela, S.; Antonen, J.; Mäkelä, S.; Sinisalo, M.; Vaheri, A.; Mustonen, J. Severe Puumala virus infection in a patient with a lymphoproliferative disease treated with icatibant. Infect. Dis. 2015, 47, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Antonen, J.; Leppänen, I.; Tenhunen, J.; Arvola, P.; Mäkelä, S.; Vaheri, A.; Mustonen, J. A severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Scand. J. Infect. Dis. 2013, 45, 494–496. [Google Scholar] [CrossRef]
- To, K.K.W.; Yip, C.C.Y.; Yuen, K.-Y. Rhinovirus—From bench to bedside. J. Formos. Med. Assoc. 2017, 116, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.R.; Hewson, C.A.; Laza-Stanca, V.; Lau, H.-T.H.; Mukaida, N.; Hershenson, M.B.; Johnston, S.L. Protein kinase R, IκB kinase-β and NF-κB are required for human rhinovirus induced pro-inflammatory cytokine production in bronchial epithelial cells. Mol. Immunol. 2007, 44, 1587–1597. [Google Scholar] [CrossRef]
- Custovic, A.; Belgrave, D.; Lin, L.; Bakhsoliani, E.; Telcian, A.G.; Solari, R.; Murray, C.S.; Walton, R.P.; Curtin, J.; Edwards, M.R.; et al. Cytokine Responses to Rhinovirus and Development of Asthma, Allergic Sensitization, and Respiratory Infections during Childhood. Am. J. Respir. Crit. Care Med. 2018, 197, 1265–1274. [Google Scholar] [CrossRef]
- Shelfoon, C.; Shariff, S.; Traves, S.L.; Kooi, C.; Leigh, R.; Proud, D. Chemokine release from human rhinovirus–infected airway epithelial cells promotes fibroblast migration. J. Allergy Clin. Immunol. 2016, 138, 114–122.e4. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, P.; McConn-Walsh, R.; Walsh, M.; Costello, R.W. Subjects with non-allergic non-infectious perennial rhinitis do not show nasal hyper-responsiveness to bradykinin. Eur. Arch. Oto-Rhino-Laryngol. 2007, 264, 33–37. [Google Scholar] [CrossRef]
- Naclerio, R.M.; Proud, D.; Lichtenstein, L.M.; Kagey-Sobotka, A.; Hendley, J.O.; Sorrentino, J.; Gwaltney, J.M. Kinins are Generated During Experimental Rhinovirus Colds. J. Infect. Dis. 1988, 157, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, S.C.; Proud, D.; Sarnoff, R.B.; Juergens, U.; Cochrane, C.G.; Zuraw, B.L. Elevation of Tissue Kallikrein and Kinin in the Airways of Asthmatic Subjects after Endobronchial Allergen Challenge. Am. Rev. Respir. Dis. 1992, 145, 900–905. [Google Scholar] [CrossRef]
- Christiansen, S.C.; Eddleston, J.; Bengtson, S.H.; Jenkins, G.R.; Sarnoff, R.B.; Turner, R.B.; Gwaltney, J.M.; Zuraw, B.L. Experimental rhinovirus infection increases human tissue kallikrein activation in allergic subjects. Int. Arch. Allergy Immunol. 2008, 147, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, S.H.; Eddleston, J.; Christiansen, S.C.; Zuraw, B.L. Double-stranded RNA increases kinin B1 receptor expression and function in human airway epithelial cells. Int. Immunopharmacol. 2007, 7, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Massberg, S. Blood coagulation in immunothrombosis—At the frontline of intravascular immunity. Semin. Immunol. 2016, 28, 561–569. [Google Scholar] [CrossRef]
- Agraz-Cibrian, J.M.; Giraldo, D.M.; Mary, F.-M.; Urcuqui-Inchima, S. Understanding the molecular mechanisms of NETs and their role in antiviral innate immunity. Virus Res. 2017, 228, 124–133. [Google Scholar] [CrossRef]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.E.A.; Meijers, J.C.M.; Brandjes, D.P.M.; van Gorp, E.C.M. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef]
- Gershom, E.S.; Sutherland, M.R.; Lollar, P.; Pryzdial, E.L.G. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J. Thromb. Haemost. 2010, 8, 1037–1043. [Google Scholar] [CrossRef]
- Antoniak, S.; Tatsumi, K.; Hisada, Y.; Milner, J.J.; Neidich, S.D.; Shaver, C.M.; Pawlinski, R.; Beck, M.A.; Bastarache, J.A.; Mackman, N. Tissue factor deficiency increases alveolar hemorrhage and death in influenza A virus-infected mice. J. Thromb. Haemost. 2016, 14, 1238–1248. [Google Scholar] [CrossRef]
- Claborn, D. The biology and control of leishmaniasis vectors. J. Glob. Infect. Dis. 2010, 2, 127. [Google Scholar] [CrossRef]
- Steverding, D. The history of leishmaniasis. Parasit. Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- WHO. Available online: https://apps.who.int/neglected_diseases/ntddata/leishmaniasis/leishmaniasis.html (accessed on 14 July 2020).
- Thakur, S.; Joshi, J.; Kaur, S. Leishmaniasis diagnosis: An update on the use of parasitological, immunological and molecular methods. J. Parasit. Dis. 2020, 44, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, P.H.; Xu, X.; Oliveira, F.; Chagas, A.C.; Nascimento, C.R.; Francischetti, I.M.B.; Juliano, M.A.; Juliano, L.; Scharfstein, J.; Valenzuela, J.G.; et al. Novel Family of Insect Salivary Inhibitors Blocks Contact Pathway Activation by Binding to Polyphosphate, Heparin, and Dextran Sulfate. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2759–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Mishra, M.; Rajput, S.K.; Bajpai, S.; Singh, R.K. Detection and diagnostic applicability of human urinary kininogen in kala-azar patients. Parasitol. Res. 2012, 111, 1851–1855. [Google Scholar] [CrossRef] [PubMed]
- Svensjö, E.; Batista, P.R.; Brodskyn, C.I.; Silva, R.; Lima, A.P.C.A.; Schmitz, V.; Saraiva, E.; Pesquero, J.B.; Mori, M.A.S.; Müller-Esterl, W.; et al. Interplay between parasite cysteine proteases and the host kinin system modulates microvascular leakage and macrophage infection by promastigotes of the Leishmania donovani complex. Microbes Infect. 2006, 8, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Nico, D.; Feijó, D.; Maran, N.; Morrot, A.; Scharfstein, J.; Palatnik, M.; Palatnik-de-Sousa, C. Resistance to visceral leishmaniasis is severely compromised in mice deficient of bradykinin B2-receptors. Parasit. Vectors 2012, 5, 261. [Google Scholar] [CrossRef] [Green Version]
- Eschenlauer, S.C.P.; Faria, M.S.; Morrison, L.S.; Bland, N.; Ribeiro-Gomes, F.L.; DosReis, G.A.; Coombs, G.H.; Lima, A.P.C.A.; Mottram, J.C. Influence of parasite encoded inhibitors of serine peptidases in early infection of macrophages with Leishmania major. Cell. Microbiol. 2009, 11, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.S.; Reis, F.C.G.; Azevedo-Pereira, R.L.; Morrison, L.S.; Mottram, J.C.; Lima, A.P.C.A. Leishmania Inhibitor of Serine Peptidase 2 Prevents TLR4 Activation by Neutrophil Elastase Promoting Parasite Survival in Murine Macrophages. J. Immunol. 2011, 186, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.S.; Calegari-Silva, T.C.; Vivarini, A.D.C.; Mottram, J.C.; Lopes, U.G.; Lima, A.P.C.A. Role of protein kinase R in the killing of Leishmania major by macrophages in response to neutrophil elastase and TLR4 via TNFα and IFNβ. FASEB J. 2014, 28, 3050–3063. [Google Scholar] [CrossRef]
- Svensjö, E.; Nogueira de Almeida, L.; Vellasco, L.; Juliano, L.; Scharfstein, J. Ecotin-Like ISP of L. major Promastigotes Fine-Tunes Macrophage Phagocytosis by Limiting the Pericellular Release of Bradykinin from Surface-Bound Kininogens: A Survival Strategy Based on the Silencing of Proinflammatory G-Protein Coupled Kinin B2 and B1. Mediat. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Maxfield, L.; Bermudez, R. Trypanosomiasis (Trypansomiasis); StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Waseem, M. Chagas Disease (American Trypanosomiasis); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Dunn, N.; Wang, S.; Adigun, R. African Trypanosomiasis (Sleeping Sickness); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kato, H.; Jochim, R.C.; Gomez, E.A.; Sakoda, R.; Iwata, H.; Valenzuela, J.G.; Hashiguchi, Y. A repertoire of the dominant transcripts from the salivary glands of the blood-sucking bug, Triatoma dimidiata, a vector of Chagas disease. Infect. Genet. Evol. 2010, 10, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isawa, H.; Orito, Y.; Jingushi, N.; Iwanaga, S.; Morita, A.; Chinzei, Y.; Yuda, M. Identification and characterization of plasma kallikrein-kinin system inhibitors from salivary glands of the blood-sucking insect Triatoma infestans. FEBS J. 2007, 274, 4271–4286. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, D.; Tabbabi, A.; Yamamoto, D.S.; Kien, L.T.; Kato, H. Salivary gland transcriptome of the Asiatic Triatoma rubrofasciata. Acta Trop. 2020, 210, 105473. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Jochim, R.C.; Gomez, E.A.; Tsunekawa, S.; Valenzuela, J.G.; Hashiguchi, Y. Salivary gland transcripts of the kissing bug, Panstrongylus chinai, a vector of Chagas disease. Acta Trop. 2017, 174, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Boreham, P.F.L. Kinin release and the immune reaction in human trypanosomiasis caused by Trypanosoma rhodesiense. Trans. R. Soc. Trop. Med. Hyg. 1970, 64, 394–400. [Google Scholar] [CrossRef]
- Richards, W.H.G. Pharmacologically active substances in the blood, tissues and urine of mice infected with Trypanosoma brucei. Br. J. Pharmacol. Chemother. 1965, 24, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Boreham, P.F.L. Pharmacologically Active Peptides produced in the Tissues of the Host during Chronic Trypanosome Infections. Nature 1966, 212, 190–191. [Google Scholar] [CrossRef]
- Morty, R.E.; Vadász, I.; Bulau, P.; Dive, V.; Oliveira, V.; Seeger, W.; Juliano, L. Tropolysin, a New Oligopeptidase from African Trypanosomes. Biochemistry 2005, 44, 14658–14669. [Google Scholar] [CrossRef]
- Scharfstein, J. Subverting bradykinin-evoked inflammation by co-opting the contact system. Curr. Opin. Hematol. 2018, 25, 347–357. [Google Scholar] [CrossRef]
- Scharfstein, J.; Gomes, J.D.A.S.; Correa-Oliveira, R. Back to the future in Chagas disease: From animal models to patient cohort studies, progress in immunopathogenesis research. Mem. Inst. Oswaldo Cruz 2009, 104, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfstein, J. Parasite cysteine proteinase interactions with α2-macroglobulin or kininogens: Differential pathways modulating inflammation and innate immunity in infection by pathogenic trypanosomatids. Immunobiology 2006, 211, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Scharfstein, J.; Andrade, D. Infection-Associated Vasculopathy in Experimental Chagas Disease. In Advances in Parasitology Volume 57; Elsevier BV: Amsterdam, The Netherlands, 2011; pp. 101–127. [Google Scholar]
- Scharfstein, J.; Monteiro, A.C.; Schmitz, V.; Svensjö, E. Angiotensin-converting enzyme limits inflammation elicited by Trypanosoma cruzi cysteine proteases: A peripheral mechanism regulating adaptive immunity via the innate kinin pathway. Biol. Chem. 2008, 389, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Serveau, C.; Lalmanach, G.; Juliano, M.A.; Scharfstein, J.; Juliano, L.; Gauthier, F. Investigation of the substrate specificity of cruzipain, the major cysteine proteinase of Trypanosoma cruzi, through the use of cystatin-derived substrates and inhibitors. Biochem. J. 1996, 313, 951–956. [Google Scholar] [CrossRef]
- Del Nery, E.; Juliano, M.A.; Lima, A.P.C.A.; Scharfstein, J.; Juliano, L. Kininogenase Activity by the Major Cysteinyl Proteinase (Cruzipain) from Trypanosoma cruzi. J. Biol. Chem. 1997, 272, 25713–25718. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Morrot, A. A role for extracellular amastigotes in the immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 1999, 94, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.P.C.A.; Almeida, P.C.; Tersariol, I.L.S.; Schmitz, V.; Schmaier, A.H.; Juliano, L.; Hirata, I.Y.; Müller-Esterl, W.; Chagas, J.R.; Scharfstein, J. Heparan Sulfate Modulates Kinin Release by Trypanosoma cruzi through the Activity of Cruzipain. J. Biol. Chem. 2002, 277, 5875–5881. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Schmitz, V.; Morandi, V.; Capella, M.M.A.; Lima, A.P.C.A.; Morrot, A.; Juliano, L.; Müller-Esterl, W. Host Cell Invasion by Trypanosoma cruzi is Potentiated by Activation of Bradykinin B2 Receptors. J. Exp. Med. 2000, 192, 1289–1300. [Google Scholar] [CrossRef]
- Morris, S.; Tanowitz, H.; Hatcher, V.; Bilezikian, J.; Wittner, M. Alterations in intracellular calcium following infection of human endothelial cells with Trypanosoma cruzi. Mol. Biochem. Parasitol. 1988, 29, 213–221. [Google Scholar] [CrossRef]
- Mijares, A.; Espinosa, R.; Adams, J.; Lopez, J.R. Increases in [IP3]i aggravates diastolic [Ca2+] and contractile dysfunction in Chagas’ human cardiomyocytes. PLoS Negl. Trop. Dis. 2020, 14, e0008162. [Google Scholar] [CrossRef]
- Todorov, A.G.; Andrade, D.; Pesquero, J.B.; Carvalho Araujo, R.; Bader, M.; Stewart, J.; Gera, L.; Müller-Esterl, W.; Morandi, V.; Goldenberg, R.C.S.; et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor subtypes (B1/B2). FASEB J. 2003, 17, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.C.; Schmitz, V.; Morrot, A.; de Arruda, L.B.; Nagajyothi, F.; Granato, A.; Pesquero, J.B.; Müller-Esterl, W.; Tanowitz, H.B.; Scharfstein, J. Bradykinin B2 Receptors of Dendritic Cells, Acting as Sensors of Kinins Proteolytically Released by Trypanosoma cruzi, Are Critical for the Development of Protective Type-1 Responses. PLoS Pathog. 2007, 3, e185. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Schmitz, V.; Svensjo, E.; Gazzinelli, R.T.; Almeida, I.C.; Todorov, A.; de Arruda, L.B.; Torrecilhas, A.C.T.; Pesquero, J.B.; Morrot, A.; et al. Cooperative Activation of TLR2 and Bradykinin B2 Receptor Is Required for Induction of Type 1 Immunity in a Mouse Model of Subcutaneous Infection by Trypanosoma cruzi. J. Immunol. 2006, 177, 6325–6335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfstein, J.; Schmitz, V.; Svensjö, E.; Granato, A.; Monteiro, A.C. Kininogens Coordinate Adaptive Immunity through the Proteolytic Release of Bradykinin, an Endogenous Danger Signal Driving Dendritic Cell Maturation. Scand. J. Immunol. 2007, 66, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Coelho dos Santos, J.S.; Menezes, C.A.S.; Villani, F.N.A.; Magalhães, L.M.D.; Scharfstein, J.; Gollob, K.J.; Dutra, W.O. Captopril increases the intensity of monocyte infection by Trypanosoma cruzi and induces human T helper type 17 cells. Clin. Exp. Immunol. 2010, 162, 528–536. [Google Scholar] [CrossRef]
- Schmitz, V.; Svensjö, E.; Serra, R.R.; Teixeira, M.M.; Scharfstein, J. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J. Leukoc. Biol. 2009, 85, 1005–1014. [Google Scholar] [CrossRef]
- Coates, B.M.; Sullivan, D.P.; Makanji, M.Y.; Du, N.Y.; Olson, C.L.; Muller, W.A.; Engman, D.M.; Epting, C.L. Endothelial Transmigration by Trypanosoma cruzi. PLoS ONE 2013, 8, e81187. [Google Scholar] [CrossRef] [Green Version]
- Tanowitz, H.B. Role of vasoactive mediators in the pathogenesis of Chagas disease. Front. Biosci. 2003, 8, 1103. [Google Scholar] [CrossRef] [Green Version]
- Camargos, E.R.S.; Machado, C.R.S.; Teixeira, A.L.; Rocha, L.L.V.; Ferreira, A.J.; Almeida, A.P.; Barton, M.; Teixeira, M.M. Role of endothelin during experimental Trypanosoma cruzi infection in rats. Clin. Sci. 2002, 103, 64S–67S. [Google Scholar] [CrossRef]
- Andrade, D.; Serra, R.; Svensjö, E.; Lima, A.P.C.; Ramos Junior, E.S.; Fortes, F.S.; Morandini, A.C.F.; Morandi, V.; Soeiro, M.D.N.; Tanowitz, H.B.; et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: A converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 2012, 165, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- D’Orléans-Juste, P.; Bkaily, G.; Rae, G.A. Endothelin and bradykinin: ‘brothers-in-arms’ in Chagas vasculopathies? Br. J. Pharmacol. 2012, 165, 1330–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, V.; Almeida, L.N.; Svensjö, E.; Monteiro, A.C.; Köhl, J.; Scharfstein, J. C5a and Bradykinin Receptor Cross-Talk Regulates Innate and Adaptive Immunity in Trypanosoma cruzi Infection. J. Immunol. 2014, 193, 3613–3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, C.R.; Andrade, D.; Carvalho-Pinto, C.E.; Serra, R.R.; Vellasco, L.; Brasil, G.; Ramos-Junior, E.S.; da Mota, J.B.; Almeida, L.N.; Andrade, M.V.; et al. Mast Cell Coupling to the Kallikrein–Kinin System Fuels Intracardiac Parasitism and Worsens Heart Pathology in Experimental Chagas Disease. Front. Immunol. 2017, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Dellalibera-Joviliano, R.; Bestetti, R.B.; Lopes, G.S.; Furlan-Daniel, R.; Lopes, K.C.; Faria-Junior, M.; Junior, N.I. Kinins and nitric oxide in patients with chronic chagas disease and systemic arterial hypertension. Cardiovasc. Pathol. 2020, 49, 107257. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2019. Available online: https://www.who.int/publications/i/item/world-malaria-report-2019 (accessed on 20 July 2020).
- Isawa, H.; Yuda, M.; Orito, Y.; Chinzei, Y. A Mosquito Salivary Protein Inhibits Activation of the Plasma Contact System by Binding to Factor XII and High Molecular Weight Kininogen. J. Biol. Chem. 2002, 277, 27651–27658. [Google Scholar] [CrossRef] [Green Version]
- Isawa, H.; Orito, Y.; Iwanaga, S.; Jingushi, N.; Morita, A.; Chinzei, Y.; Yuda, M. Identification and characterization of a new kallikrein-kinin system inhibitor from the salivary glands of the malaria vector mosquito Anopheles stephensi. Insect Biochem. Mol. Biol. 2007, 37, 466–477. [Google Scholar] [CrossRef]
- Maegraith, B.; Fletcher, A. The Pathogenesis of Mammalian Malaria. Adv. Parasitol. 1972, 10, 49–75. [Google Scholar]
- Maegraith, B. Other pathological processes in malaria. Bull. World Health Organ. 1974, 50, 187–193. [Google Scholar]
- Onabanjo, A.O.; Maegraith, B.G. Inflammatory changes in small blood vessels induced by kallikrein (kininogenase) in the blood of Macaca mulatta, infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 1970, 64, 227–236. [Google Scholar] [CrossRef]
- Onabanjo, A.O.; Maegraith, B.G. Pathological lesions produced in the brain by kallikrein (kininogenase) in Macaca mulatta, infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 1970, 64, 237–242. [Google Scholar] [CrossRef]
- Onabanjo, A.O.; Bhabani, A.R.; Maegraith, B.G. The significance of kinin-destroying enzymes activity in Plasmodium knowlesi malarial infection. Br. J. Exp. Pathol. 1970, 51, 534–540. [Google Scholar] [PubMed]
- Ohtomo, H.; Katori, M. Realible evidence of involvement of the kinin system in mouse malaria. Jpn. J. Pharmacol. 1972, 22, 493–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, I.G. The kallikrein-kinin system and its role in the hypotensive shock syndrome of animals infected with the haemoprotozoan parasites Babesia, Plasmodium and Trypanosoma. Gen. Pharmacol. Vasc. Syst. 1979, 10, 319–325. [Google Scholar] [CrossRef]
- Cotrin, S.S.; Gouvêa, I.E.; Melo, P.M.S.; Bagnaresi, P.; Assis, D.M.; Araújo, M.S.; Juliano, M.A.; Gazarini, M.L.; Rosenthal, P.J.; Juliano, L.; et al. Substrate specificity studies of the cysteine peptidases falcipain-2 and falcipain-3 from Plasmodium falciparum and demonstration of their kininogenase activity. Mol. Biochem. Parasitol. 2013, 187, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.F.; Alves, F.L.; Pedron, C.N.; Torres, M.D.T.; Silva, L.S.; Pinheiro, A.A.S.; Miranda, A.; Oliveira, V.X. Anti-plasmodial activity of bradykinin and analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3311–3313. [Google Scholar] [CrossRef]
- Silva, L.D.S.; Peruchetti, D.D.B.; Silva, C.T.F.-D.; Ferreira-DaSilva, A.T.; Perales, J.; Caruso-Neves, C.; Pinheiro, A.A.S. Interaction between bradykinin B2 and Ang-(1–7) Mas receptors regulates erythrocyte invasion by Plasmodium falciparum. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2438–2444. [Google Scholar] [CrossRef]
- de Moraes, L.V.; Barateiro, A.; Sousa, P.M.; Penha-Gonçalves, C. Bradykinin Sequestration by Plasmodium berghei Infected Erythrocytes Conditions B2R Signaling and Parasite Uptake by Fetal Trophoblasts. Front. Microbiol. 2018, 9, 3106. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.S.; Pinheiro, A.S.; Teixeira, D.E.; Silva-Aguiar, R.P.; Peruchetti, D.B.; Scharfstein, J.; Caruso-Neves, C.; Pinheiro, A.A.S. Kinins Released by Erythrocytic Stages of Plasmodium falciparum Enhance Adhesion of Infected Erythrocytes to Endothelial Cells and Increase Blood Brain Barrier Permeability via Activation of Bradykinin Receptors. Front. Med. 2019, 6, 1–9. [Google Scholar] [CrossRef]
- Ventura, P.D.S.; Carvalho, C.P.F.; Barros, N.M.T.; Martins-Silva, L.; Dantas, E.O.; Martinez, C.; Melo, P.M.S.; Pesquero, J.B.; Carmona, A.K.; Nagaoka, M.R.; et al. Malaria infection promotes a selective expression of kinin receptors in murine liver. Malar. J. 2019, 18, 213. [Google Scholar] [CrossRef]
- Clemens, R.; Pramoolsinsap, C.; Lorenz, R.; Pukrittayakamee, S.; Bock, H.L.; White, N.J. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br. J. Haematol. 1994, 87, 100–105. [Google Scholar] [CrossRef]
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 28 July 2020).
- Nelwan, M.L. Schistosomiasis: Life Cycle, Diagnosis, and Control. Curr. Ther. Res. 2019, 91, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Verjee, M.A. Schistosomiasis: Still a Cause of Significant Morbidity and Mortality. Res. Rep. Trop. Med. 2020, 10, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoukian, N.; Borges, D.R. Prealbumina, precalicreína e protrombina na forma hepatesplênica da esquistossomose: Catabolismo aumentado de proteínas da coagulação? Rev. Inst. Med. Trop. Sao Paulo 1984, 26, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omran, S.A.; Amer, A.M.; El-Kaliouby, A.H.; Eldin, A.A.S. Study of contact activation in endemic hepatosplenomegaly. Blood Coagul. Fibrinolysis 1991, 2, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Bentley, A.A.; Merkulov, S.M.; Peng, Y.; Rozmarynowycz, R.; Qi, X.; Pusztai-Carey, M.; Merrick, W.C.; Yee, V.C.; McCrae, K.R.; Komar, A.A. Chimeric Glutathione S-Transferases Containing Inserts of Kininogen Peptides. J. Biol. Chem. 2012, 287, 22142–22150. [Google Scholar] [CrossRef] [Green Version]
- Cocude, C.; Pierrot, C.; Cetre, C.; Godin, C.; Capron, A.; Khalife, J. Molecular characterization of a partial sequence encoding a novel Schistosoma mansoni serine protease. Parasitology 1997, 115, 395–402. [Google Scholar] [CrossRef]
- Cocude, C.; Pierrot, C.; Cetre, C.; Fontaine, J.; Godin, C.; Capron, A.; Khalife, J. Identification of a developmentally regulated Schistosoma mansoni serine protease homologous to mouse plasma kallikrein and human factor I. Parasitology 1999, 118, 389–396. [Google Scholar] [CrossRef]
- Da’dara, A.; Skelly, P.J. Manipulation of vascular function by blood flukes? Blood Rev. 2011, 25, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Grabe, K.; Haas, W. Navigation within host tissues: Schistosoma mansoni and Trichobilharzia ocellata schistosomula respond to chemical gradients. Int. J. Parasitol. 2004, 34, 927–934. [Google Scholar] [CrossRef]
- Ranasinghe, S.L.; Fischer, K.; Gobert, G.N.; McManus, D.P. Functional expression of a novel Kunitz type protease inhibitor from the human blood fluke Schistosoma mansoni. Parasit. Vectors 2015, 8, 408. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Da’dara, A.A.; Skelly, P.J. The blood fluke Schistosoma mansoni cleaves the coagulation protein high molecular weight kininogen (HK) but does not generate the vasodilator bradykinin. Parasit. Vectors 2018, 11, 182. [Google Scholar] [CrossRef] [Green Version]
- Leontovyč, A.; Ulrychová, L.; O’Donoghue, A.J.; Vondrášek, J.; Marešová, L.; Hubálek, M.; Fajtová, P.; Chanová, M.; Jiang, Z.; Craik, C.S.; et al. SmSP2: A serine protease secreted by the blood fluke pathogen Schistosoma mansoni with anti-hemostatic properties. PLoS Negl. Trop. Dis. 2018, 12, e0006446. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.M.; Doenhoff, M.J.; McNeice, C.; Williams, T.J.; Hellewell, P.G. Mechanisms of the inflammatory response induced by extracts of Schistosoma mansoni larvae in guinea pig skin. J. Immunol. 1993, 151, 5525–5534. [Google Scholar] [PubMed]
- Fallon, P.G.; Teixeira, M.M.; Neice, C.M.; Williams, T.J.; Hellewell, P.G.; Doenhoff, M.J. Enhancement of Schistosoma mansoni Infectivity by Intradermal Injections of Larval Extracts: A Putative Role for Larval Proteases. J. Infect. Dis. 1996, 173, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Parreira, N.A.; Ramalho, F.S.; Augusto, M.J.; Silva, D.M.; Prado, C.M.; Elias Júnior, J.; Rodrigues, V.; Ramalho, L.N.Z. The comparative efficacy of renin-angiotensin system blockers in schistosomal hepatic fibrosis. Exp. Parasitol. 2018, 191, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, Candida parapsilosis and Candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [Green Version]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Gogol, M.; Bochenska, O.; Karkowska-Kuleta, J.; Wolak, N.; Kamysz, W.; Aoki, W.; Ueda, M.; Faussner, A.; Rapala-Kozik, M. Kinin release from human kininogen by 10 aspartic proteases produced by pathogenic yeast Candida albicans. BMC Microbiol. 2015, 15, 60. [Google Scholar] [CrossRef] [Green Version]
- Bras, G.; Bochenska, O.; Rapala-Kozik, M.; Guevara-Lora, I.; Faussner, A.; Kamysz, W.; Kozik, A. Release of biologically active kinin peptides, Met-Lys-bradykinin and Leu-Met-Lys-bradykinin from human kininogens by two major secreted aspartic proteases of Candida parapsilosis. Peptides 2013, 48, 114–123. [Google Scholar] [CrossRef]
- Bras, G.; Bochenska, O.; Rapala-Kozik, M.; Guevara-Lora, I.; Faussner, A.; Kozik, A. Extracellular aspartic protease SAP2 of Candida albicans yeast cleaves human kininogens and releases proinflammatory peptides, Met-Lys-bradykinin and des-Arg9-Met-Lys-bradykinin. Biol. Chem. 2012, 393, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Karkowska-Kuleta, J.; Kedracka-Krok, S.; Rapala-Kozik, M.; Kamysz, W.; Bielinska, S.; Karafova, A.; Kozik, A. Molecular determinants of the interaction between human high molecular weight kininogen and Candida albicans cell wall: Identification of kininogen-binding proteins on fungal cell wall and mapping the cell wall-binding regions on kininogen molecule. Peptides 2011, 32, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Seweryn, K.; Karkowska-Kuleta, J.; Wolak, N.; Bochenska, O.; Kedracka-Krok, S.; Kozik, A.; Rapala-Kozik, M. Kinetic and thermodynamic characterization of the interactions between the components of human plasma kinin-forming system and isolated and purified cell wall proteins of Candida albicans. Acta Biochim. Pol. 2015, 62, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Garg, A.V.; Jawale, C.V.; Conti, H.R.; Whibley, N.; Jackson, E.K.; Shiva, S.S.; Horne, W.; Kolls, J.K.; Gaffen, S.L.; et al. The Kallikrein-Kinin System: A Novel Mediator of IL-17-Driven Anti-Candida Immunity in the Kidney. PLoS Pathog. 2016, 12, e1005952. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagnino, A.P.A.; Campos, M.M.; Silva, R.B.M. Kinins and Their Receptors in Infectious Diseases. Pharmaceuticals 2020, 13, 215. https://doi.org/10.3390/ph13090215
Dagnino APA, Campos MM, Silva RBM. Kinins and Their Receptors in Infectious Diseases. Pharmaceuticals. 2020; 13(9):215. https://doi.org/10.3390/ph13090215
Chicago/Turabian StyleDagnino, Ana Paula A., Maria M. Campos, and Rodrigo B. M. Silva. 2020. "Kinins and Their Receptors in Infectious Diseases" Pharmaceuticals 13, no. 9: 215. https://doi.org/10.3390/ph13090215
APA StyleDagnino, A. P. A., Campos, M. M., & Silva, R. B. M. (2020). Kinins and Their Receptors in Infectious Diseases. Pharmaceuticals, 13(9), 215. https://doi.org/10.3390/ph13090215