Drug Repurposing: Dipeptidyl Peptidase IV (DPP4) Inhibitors as Potential Agents to Treat SARS-CoV-2 (2019-nCoV) Infection

 ,
, 

Abstract
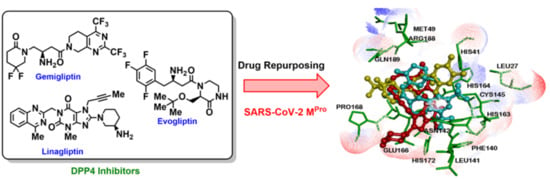
1. Introduction
2. Results
2.1. Interaction of DPP4 Inhibitors in the SARS-CoV-2 Mpro Protomer
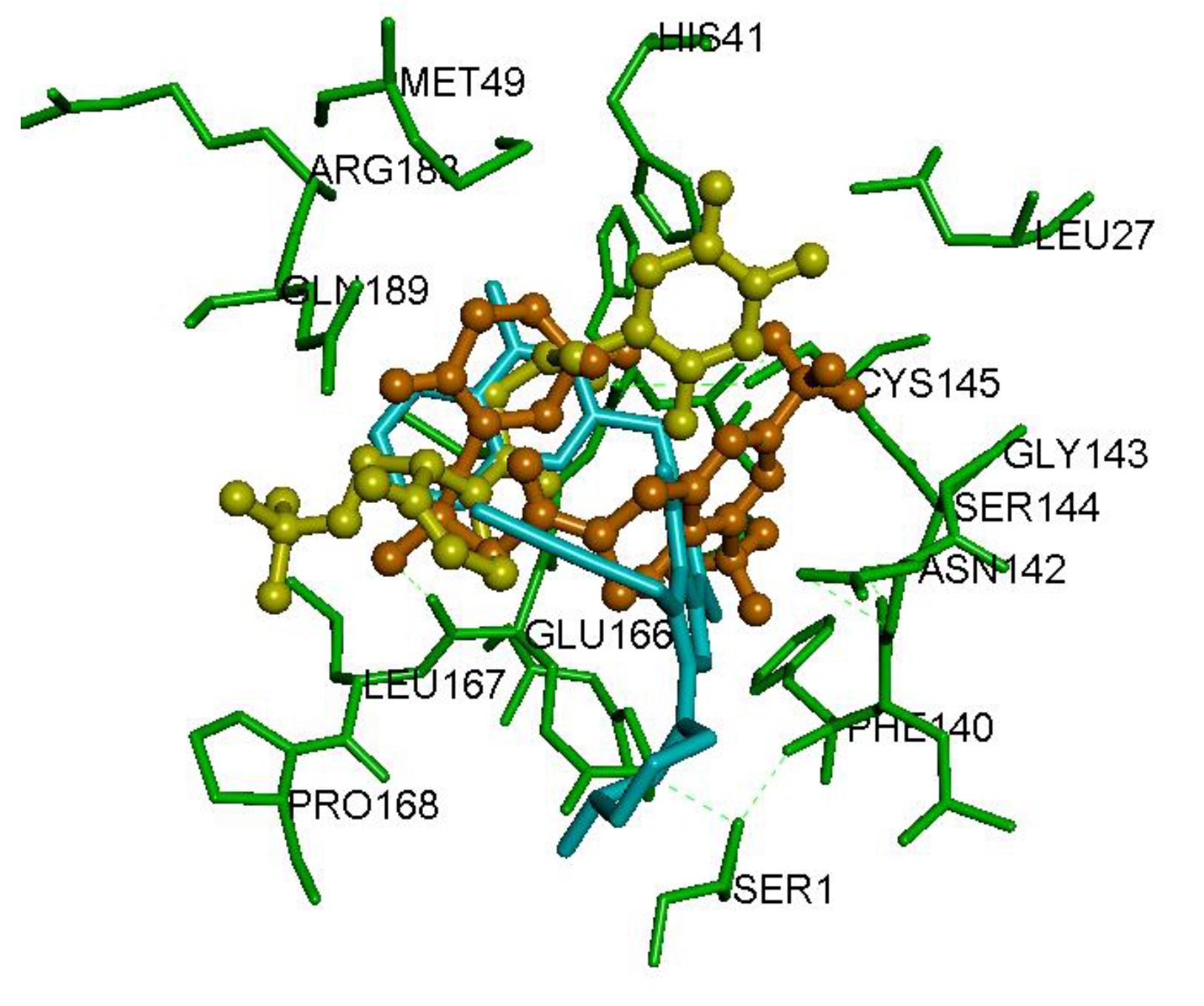
2.2. Interaction of DPP4 Inhibitors Gemigliptin, Linagliptin and Evogliptin in the SARS-CoV-2 Mpro Dimer
2.3. Interaction of DPP4 Inhibitors Gemigliptin, Linagliptin and Evogliptin in the SARS-CoV Mpro Dimer
2.4. Interaction of DPP4 Inhibitors Gemigliptin, Linagliptin and Evogliptin in the MERS-CoV CLpro Dimer
2.5. Pharmacophore Model of DPP4 Inhibitors toward SARS-CoV-2 Mpro Dimer
2.6. Physicochemical Properties of DPP4 Inhibitors
3. Discussion
4. Materials and Methods
4.1. Preparation of Ligands
4.2. Preparation of Target Proteases, the SARS-CoV-2 Mpro Protomer, SARS-CoV-2 Mpro Dimer, SARS-CoV Mpro Dimer, MERS-CoV CLpro Dimer and DPP4 Dimer
4.3. Molecular Docking Studies of DPP4 Inhibitors with Viral Proteases
4.4. Pharmacophore Modeling
4.5. Molecular Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). COVID-19 Weekly Epidemiological Update. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update—15-December-2020 (accessed on 17 December 2020).
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in hospitalized patients with Covid-19-preliminary report. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.O.; et al. Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19: The CoDEX randomized clinical trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef]
- Pushpakom, S.; Lorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palu, G.; Loregian, A. Drug repurposing for viral infectious diseases: How far are we? Trends Microbiol. 2018, 10, 865–876. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Wang, Y.H.; Zhang, F.; Diao, H.; Wu, R. Covalent inhibition mechanism of antidiabetic drugs—Vildagliptin and saxagliptin. ACS Catal. 2019, 9, 2292–2302. [Google Scholar] [CrossRef]
- Ahren, B.; Schweizer, A.; Dejager, S.; Villhauer, E.B.; Dunning, B.E.; Foley, J.E. Mechanisms of action of the dipeptidyl peptidase-4 inhibitor vildagliptin in humans. Diabetes Obes. Metab. 2011, 13, 775–783. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanism of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Rohrborn, D.; Wronkowitz, N.; Eckel, J. DPP4 in diabetes. Front. Immunol. 2015, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.; Klemann, C.; Stephan, M.; Horsten, S.V. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin. Exp. Immunol. 2016, 184, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Thiel, V.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Song, W.; Wang, Y.; Wang, N.; Wang, D.; Guao, J.; Fu, L.; Shi, X. Identification of residues on human receptor DPP4 critical for MERS-CoV binding and entry. Virology 2014, 471–473, 49–53. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Lyroni, K.; Aznaourova, M.; Tseliou, M.; Al-Anazi, M.R.; Al-Ahdal, M.N.; Alkahtani, S.; Sourvinos, G.; Tsatsanis, C. Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ. Oncotarget 2017, 8, 9053–9066. [Google Scholar] [CrossRef]
- Reinhold, D.; Brocke, S. DPP4-Directed therapeutic strategies for MERS-CoV. Lancet Infect. Dis. 2014, 14, 100–101. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the cornonavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72,314 cases from the Chinese center for disease control and prevention. JAMA 2020, 324, 1239–1242. [Google Scholar] [CrossRef]
- Sinclair, A.; Saeedi, P.; Kaundal, A.; Karuranga, S.; Malanda, B.; Williams, R. Diabetes and global ageing among the 65–99-year-old adults: Findings from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108078. [Google Scholar] [CrossRef]
- Iacobellis, G. COVID-19 and diabetes: Can DPP4 inhibition play a role? Diabetes Res. Clin. Pract. 2020, 162, 108125. [Google Scholar] [CrossRef]
- Apicella, M.; Campopiano, M.C.; Mantuano, M.; Mazoni, L.; Coppelli, A.; Prato, S.D. COVID-19 in people with diabetes: Understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol. 2020, 8, P782–P792. [Google Scholar] [CrossRef]
- Solerte, S.B.; D’Addio, F.; Trevisa, R.; Lovati, E.; Rossi, A.; Pastore, I.; Dell’Acqua, M.; Ippolito, E.; Scaranna, C.; Bellante, R.; et al. Sitagliptin treatment at the time of hospitalization was associated with reduced mortality in patients with type 2 diabetes and COVID-19: A multicenter, case-control, retrospective, observational study. Diabetes Care 2020, 43, 2999–3006. [Google Scholar] [CrossRef]
- Groves, M.; Domling, A.; Moreno, A.J.R.; Romero, A.R.; Neochoritis, C.; Velazquez, V.M. Gliptin repurposing for COVID-19. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Demmler, G.; Ligon, B.L. Severe acute respiratory syndrome (SARS): A review of the history, epidemiology, prevention, and concerns for the future. Semin. Pediatr. Infect. Dis. 2003, 14, 240–244. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.J.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome (SARS-CoV) 3CL protease inhibitors: Peptidomimetic and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Tan, J.; Verschueren, K.H.G.; Anand, K.; She, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. pH dependent conformational flexibility of the SARS-CoV main proteinase (MPro) dimer: Molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Kilianski, A.; Mielech, A.M.; Deng, X.; Baker, S.C. Assessing activity and inhibition of middle east respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013, 87, 11955–11962. [Google Scholar] [CrossRef]
- Ho, B.L.; Cheng, S.C.; Shi, L.; Wang, T.Y.; Ho, K.I.; Chou, C.Y. Critical assessment of the important residues involved in the dimerization and catalysis of MERS coronavirus main protease. PLoS ONE 2015, 10, e0144865. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Wang, L.; Zhang, N.; Deng, X.; Su, M.; Su, Y.; Hu, L.; He, C.; Ying, T.; Jiang, S.; et al. Development of small-molecule MERS-CoV inhibitors. Viruses 2018, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Brunn, A.V.; Leyssen, P.; Lanko, K.; Neyts, L.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Tomar, S.; Johnston, M.L.; John, S.E.T.; Osswald, H.L.; Nyalapatla, P.R.; Paul, L.N.; Ghosh, A.K.; Denison, M.R.; Mesecar, A.D. Ligand-induced dimerization of middle east respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro). J. Biol. Chem. 2015, 290, 19403–19422. [Google Scholar] [CrossRef]
- Kankanamalage, A.C.G.; Kim, Y.; Damalanka, V.C.; Rathnayake, A.D.; Fehr, A.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Lushington, G.H.; Perlman, S.; et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL-protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018, 150, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, M.; Langkopf, E.; Mark, M.; Tadayyon, M.; Thomas, L.; Nar, H.; Pfrengle, W.; Guth, B.; Lotz, R.; Sieger, P.; et al. 8-(3-(R)-Aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitors for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. [Google Scholar] [CrossRef]
- Arulmozhiraja, S.; Matsuo, N.; Ishitsubo, E.; Okazaki, S.; Shimano, H.; Tokiwa, H. Comparative binding analysis of dipeptidyl peptidase IV (DPP-4) with antidiabetic drugs—An ab initio molecular orbital study. PLoS ONE 2016, 11, e0166275. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Weber, H.K.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Ryskjaer, J.; Deacon, C.F.; Carr, R.D.; Krarup, T.; Madsbad, S.; Holst, J.; Vilsboll, T. Plasma dipeptidyl peptidase-IV activity in patients with type-2 diabetes mellitus correlates positively with HbAlc levels, but is not acutely affected by food intake. Eur. J. Endocrinol. 2006, 155, 485–493. [Google Scholar] [CrossRef]
- Bosch, B.J.; Smits, S.L.; Haagmans, B.L. Membrane ectopeptidases targeted by human coronaviruses. Curr. Opin. Virol. 2014, 6, 5560. [Google Scholar] [CrossRef]
- Rao, P.N.P.; Mohamed, T.; Teckwani, K.; Tin, G. Curcumin binding to beta amyloid: A computational study. Chem. Biol. Drug Des. 2015, 86, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | CDOCKER Energy in kcal/mol 1 | CDOCKER Interaction Energy in kcal/mol 1 |
|---|---|---|
| Anagliptin | −27.50 | −45.29 |
| Trelagliptin | −22.32 | −46.04 |
| Sitagliptin | −7.41 | −40.13 |
| Linagliptin | −34.15 | −50.46 |
| Gemigliptin | −39.55 | −48.54 |
| Tenegliptin | −16.74 | −41.14 |
| Evogliptin | −33.95 | −39.96 |
| Gosogliptin | −8.16 | −37.98 |
| 1 | −56.14 | −70.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, P.P.N.; Pham, A.T.; Shakeri, A.; El Shatshat, A.; Zhao, Y.; Karuturi, R.C.; Hefny, A.A. Drug Repurposing: Dipeptidyl Peptidase IV (DPP4) Inhibitors as Potential Agents to Treat SARS-CoV-2 (2019-nCoV) Infection. Pharmaceuticals 2021, 14, 44. https://doi.org/10.3390/ph14010044
Rao PPN, Pham AT, Shakeri A, El Shatshat A, Zhao Y, Karuturi RC, Hefny AA. Drug Repurposing: Dipeptidyl Peptidase IV (DPP4) Inhibitors as Potential Agents to Treat SARS-CoV-2 (2019-nCoV) Infection. Pharmaceuticals. 2021; 14(1):44. https://doi.org/10.3390/ph14010044
Chicago/Turabian StyleRao, Praveen P. N., Amy Trinh Pham, Arash Shakeri, Amna El Shatshat, Yusheng Zhao, Rahul C. Karuturi, and Ahmed A. Hefny. 2021. "Drug Repurposing: Dipeptidyl Peptidase IV (DPP4) Inhibitors as Potential Agents to Treat SARS-CoV-2 (2019-nCoV) Infection" Pharmaceuticals 14, no. 1: 44. https://doi.org/10.3390/ph14010044
APA StyleRao, P. P. N., Pham, A. T., Shakeri, A., El Shatshat, A., Zhao, Y., Karuturi, R. C., & Hefny, A. A. (2021). Drug Repurposing: Dipeptidyl Peptidase IV (DPP4) Inhibitors as Potential Agents to Treat SARS-CoV-2 (2019-nCoV) Infection. Pharmaceuticals, 14(1), 44. https://doi.org/10.3390/ph14010044