The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
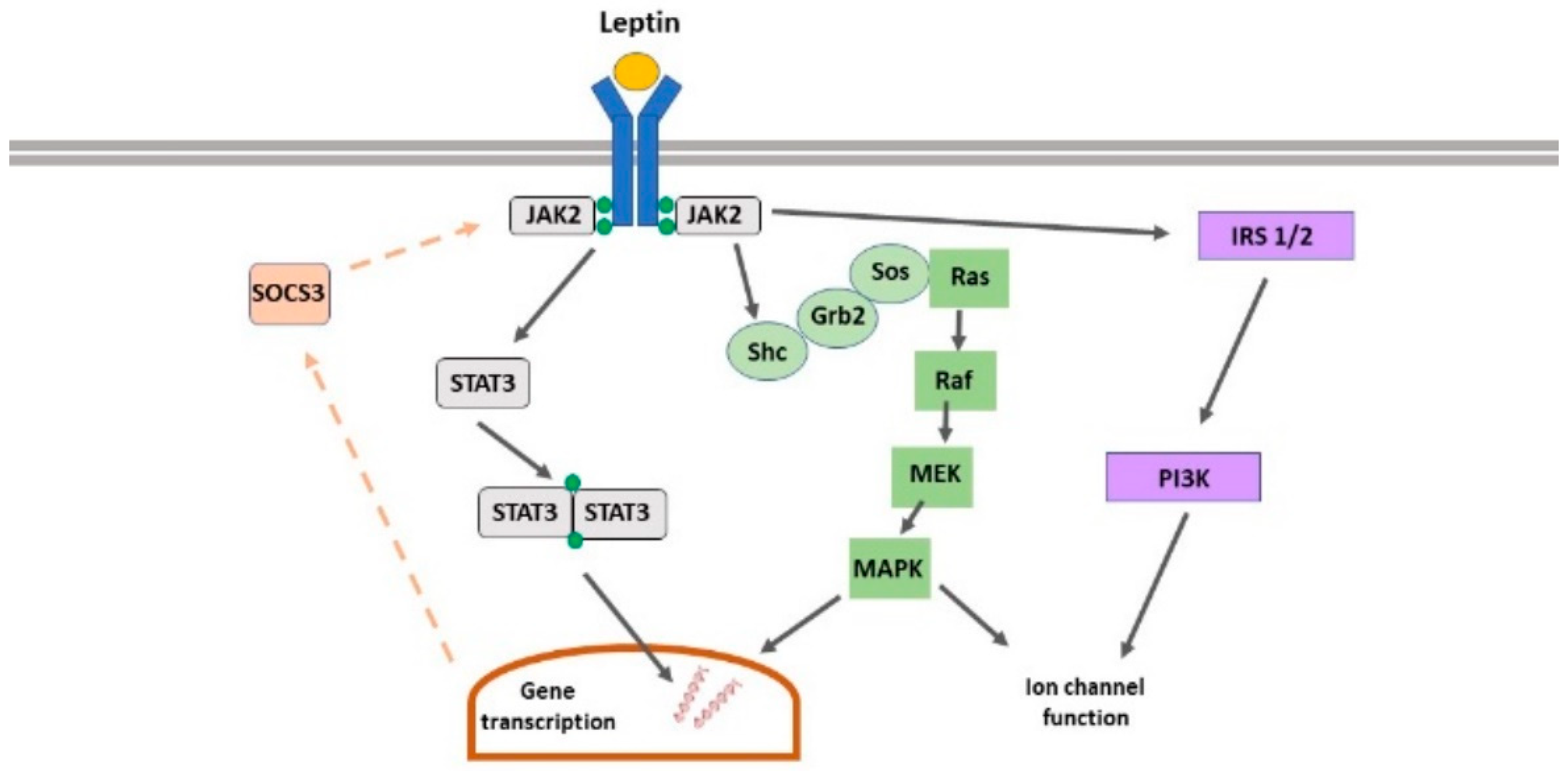
2. Leptin Receptors
2.1. Leptin Receptor Isoforms
2.2. Leptin Receptor-Driven Signalling
2.3. Leptin Receptor Expression in the CNS
3. Leptin Regulation of Hippocampal Excitatory Synaptic Function
3.1. SC-CA1 Synapses
3.2. TA-CA1 Synapses
3.3. Leptin Regulation of AMPA Receptor Trafficking
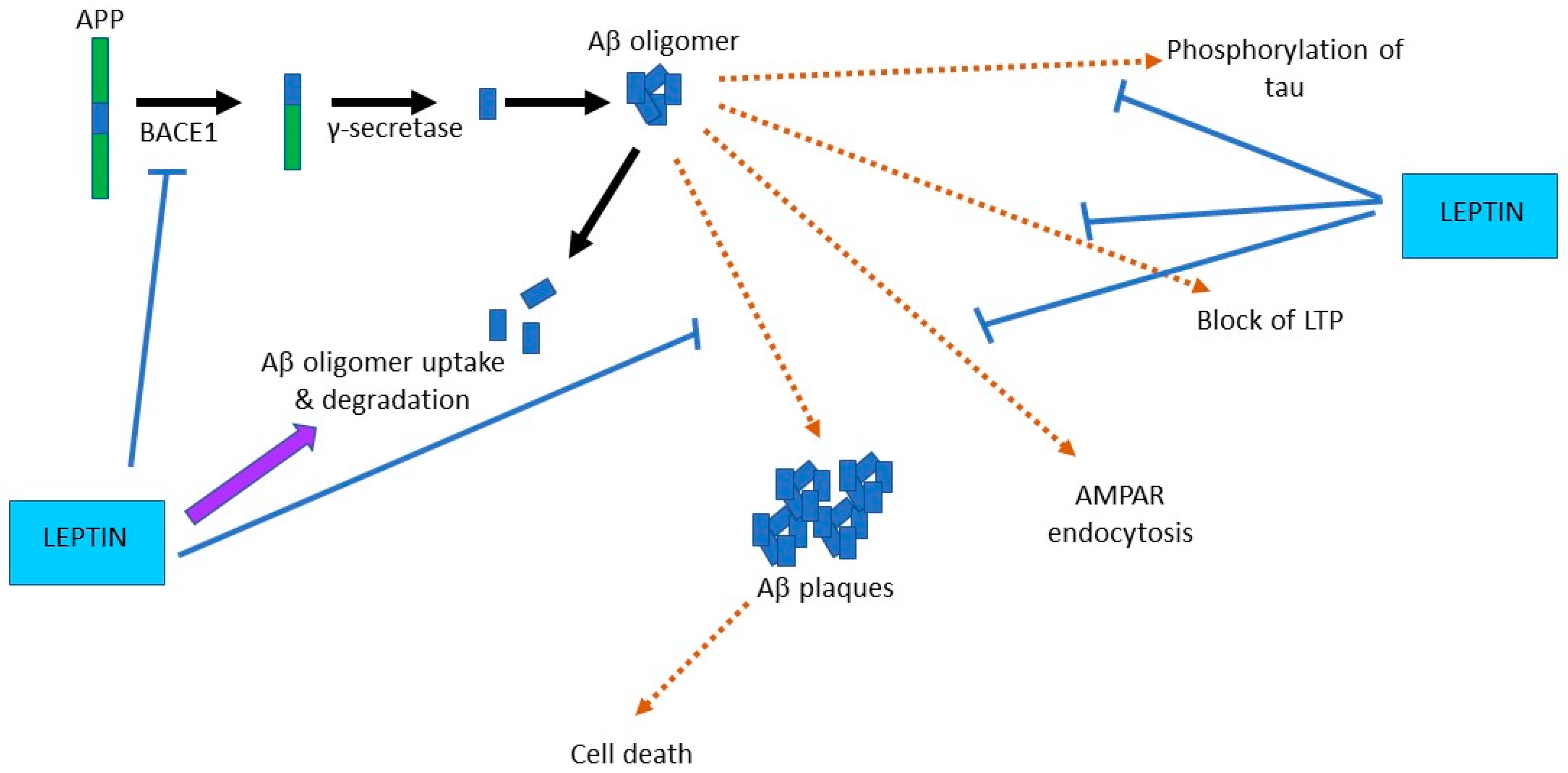
4. Leptin and the Ageing Brain
4.1. Neuroprotective Actions of Leptin
4.2. Leptin Prevents Aβ-Driven Impairments at Hippocampal Synapses
4.3. Leptin Improves Hippocampus-Dependent Learning and Memory
5. Targeting the Leptin System as a Novel Therapeutic in AD?
6. Protective Actions of Leptin in Other Neurodegenerative Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hervey, G.R. The effects of lesions in the hypothalamus in parabiotic rats. J. Physiol. Lond. 1959, 145, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Huang, W.; Jaspan, J.B.; Maness, L.M. Leptin enters the brain by a saturable system independent of insulin. Peptides 1996, 17, 305–311. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Ihle, J.N. Cytokine receptor signalling. Nature 1995, 377, 591–594. [Google Scholar] [CrossRef]
- Kile, B.T.; Alexander, W.S. The suppressors of cytokine signalling (SOCS). Cell. Mol. Life Sci. 2001, 58, 1627–1635. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Leptin regulation of synaptic function at hippocampal TA-CA1 and SC-CA1 synapses: Implications for health and disease. Neurochem. Res. 2019, 44, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, J.K.; Bjørbaek, C.; Ahima, R.S.; Flier, J.S.; Saper, C.B. Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 1998, 395, 535–547. [Google Scholar] [CrossRef]
- Burguera, B.; Couce, M.E.; Long, J.; Lamsam, J.; Laakso, K.; Jensen, M.D.; Parisi, J.E.; Lloyd, R.V. The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology 2000, 71, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Hâkansson, M.L.; Brown, H.; Ghilardi, N.; Skoda, R.C.; Meister, B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J. Neurosci. 1998, 18, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahima, R.S.; Bjorbaek, C.; Osei, S.; Flier, J.S. Regulation of neuronal and glial proteins by leptin: Implications for brain development. Endocrinology 1999, 140, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Prabakaran, D.; Flier, J.S. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J. Clin. Investig. 1998, 101, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouret, S.G.; Draper, S.J.; Simerly, R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 2004, 304, 108–110. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.; Shanley, L.; O’Malley, D.; Irving, A. Leptin: A potential cognitive enhancer? Biochem. Soc. Trans. 2005, 33, 1029. [Google Scholar] [CrossRef] [Green Version]
- Irving, A.J.; Harvey, J. Leptin regulation of hippocampal synaptic function in health and disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130155. [Google Scholar] [CrossRef] [Green Version]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef]
- Winocur, G.; Greenwood, C.E.; Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Reagan, L.P.; McEwen, B.S. Memory impairment in obese Zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 2005, 119, 1389–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanley, L.; O’Malley, D.; Irving, A.; Ashford, M.; Harvey, J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J. Physiol. 2002, 545, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Rensing, N.; Yang, X.F.; Zhang, H.X.; Thio, L.L.; Rothman, S.M.; Weisenfeld, A.E.; Wong, M.; Yamada, K.A. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J. Clin. Investig. 2008, 118, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Moult, P.R.; Harvey, J. NMDA receptor subunit composition determines the polarity of leptin-induced synaptic plasticity. Neuropharmacology 2011, 61, 924–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moult, P.R.; Cross, A.; Santos, S.D.; Carvalho, A.L.; Lindsay, Y.; Connolly, C.N.; Irving, A.J.; Leslie, N.R.; Harvey, J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 2010, 30, 4088–4101. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, T.E.; Bannister, N.J.; Collett, V.J.; Dargan, S.L.; Massey, P.V.; Bortolotto, Z.A.; Fitzjohn, S.M.; Bashir, Z.I.; Collingridge, G.L.; Lodge, D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 2007, 52, 60–70. [Google Scholar] [CrossRef]
- Otmakhova, N.; Lisman, J.E. Dopamine selectively inhibits the direct cortical pathway to the CA1 hippocampal region. J. Neurosci. 1999, 19, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; McGregor, G.; Irving, A.J.; Harvey, J. Leptin induces a novel form of NMDA receptor-dependent LTP at hippocampal temporoammonic-CA1 synapses. eNeuro 2015, 2, 1–17. [Google Scholar] [CrossRef] [Green Version]
- McGregor, G.; Clements, L.; Farah, A.; Irving, A.J.; Harvey, J. Age-dependent regulation of excitatory synaptic transmission at hippocampal temporoammonic-CA1 synapses by leptin. Neurobiol. Aging 2018, 69, 76–93. [Google Scholar] [CrossRef]
- Nicolas, C.; Peineau, S.; Amici, M.; Csaba, Z.; Fafouri, A.; Javalet, C.; Collett, V.J.; Hildebrandt, L.; Seaton, G.; Choi, S.L.; et al. The JAK/STAT pathway is involved in synaptic plasticity. Neuron 2012, 73, 374–390. [Google Scholar] [CrossRef] [Green Version]
- Collingridge, G.L.; Isaac, J.T.; Wang, Y.T. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Morita, D.; Rah, J.C.; Isaac, J.T. Incorporation of inwardly rectifying AMPA receptors at silent synapses during hippocampal long-term potentiation. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranahan, A.M.; Mattson, M.P. Bidirectional metabolic regulation of neurocognitive function. Neurobiol. Learn. Mem. 2011, 96, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Hassing, L.B.; Dahl, A.K.; Thorvaldsson, V.; Berg, S.; Gatz, M.; Pedersen, N.L.; Johansson, B. Overweight in midlife and risk of dementia: A 40-year follow-up study. Int. J. Obes. 2009, 33, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.L.; Atti, A.R.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Midlife overweight and obesity increase late-life dementia risk: A population-based twin study. Neurology 2011, 76, 1568–1574. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, D.R.; Bäckman, K.; Joas, E.; Waern, M.; Östling, S.; Guo, X.; Skoog, I. 37 years of body mass index and dementia: Observations from the prospective population study of women in Gothenburg, Sweden. J. Alzheimers Dis. 2012, 28, 163–171. [Google Scholar] [CrossRef]
- Alhurani, R.E.; Vassilaki, M.; Aakre, J.A.; Mielke, M.M.; Kremers, W.K.; Machulda, M.M.; Geda, Y.E.; Knopman, D.S.; Petersen, R.C.; Roberts, R.O. Decline in Weight and Incident Mild Cognitive Impairment: Mayo Clinic Study of Aging. JAMA Neurol. 2016, 73, 439–446. [Google Scholar] [CrossRef]
- Power, D.A.; Noel, J.; Collins, R.; O’Neill, D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 2001, 12, 167–170. [Google Scholar] [CrossRef]
- Gao, S.; Nguyen, J.T.; Hendrie, H.C.; Unverzagt, F.W.; Hake, A.; Smith-Gamble, V.; Hall, K. Accelerated weight loss and incident dementia in an elderly African-American cohort. J. Am. Geriatr. Soc. 2011, 59, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, S.H.; Yun, S.H.; Kang, D.W.; Hahn, C.T.; Lim, H.K.; Lee, C.U. Body Mass Index in Mild Cognitive Impairment According to Age, Sex, Cognitive Intervention, and Hypertension and Risk of Progression to Alzheimer’s Disease. Front. Psychiatry 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, V.; Wark, P.A.; Jenab, M.; Pearce, N.; Brayne, C.; Vermeulen, R.; Andersen, P.M.; Hallmans, G.; Kyrozis, A.; Vanacore, N.; et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: The EPIC cohort. Neurology 2013, 80, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef]
- Bigalke, B.; Schreitmüller, B.; Sopova, K.; Paul, A.; Stransky, E.; Gawaz, M.; Stellos, K.; Laske, C. Adipocytokines and CD34 progenitor cells in Alzheimer’s disease. PLoS ONE 2011, 6, e20286. [Google Scholar] [CrossRef]
- Lieb, W.; Beiser, A.S.; Vasan, R.S.; Tan, Z.S.; Au, R.; Harris, T.B.; Roubenoff, R.; Auerbach, S.; DeCarli, C.; Wolf, P.A.; et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 2009, 302, 2565–2572. [Google Scholar] [CrossRef] [Green Version]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Greco, S.J.; Bryan, K.J.; Sarkar, S.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N.; Casadesus, G.J. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, C.E.; van de Flier, W.M.; Scheltens, P.; Duits, A.; Wijnstok, N.; Nijpels, G.; Dekker, J.M.; Blankenstein, R.M.; Heijboer, A.C. Serum leptin is not altered nor related to cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oania, R.; McEvoy, L.K. Plasma leptin levels are not predictive of dementia in patients with mild cognitive impairment. Age Ageing 2015, 44, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 632262–632272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaia, A.; Piantanelli, L. Insulin receptors in the brain cortex of aging mice. Mech. Ageing Dev. 2000, 113, 227–232. [Google Scholar] [CrossRef]
- Shek, E.W.; Scarpace, P.J. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regul. Pept. 2000, 92, 65–71. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Matheny, M.; Tümer, N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience 2001, 104, 1111–1117. [Google Scholar] [CrossRef]
- Doherty, G.H.; Oldreive, C.; Harvey, J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience 2008, 154, 1297–1307. [Google Scholar] [CrossRef]
- Davis, C.; Mudd, J.; Hawkins, M. Neuroprotective effects of leptin in the context of obesity and metabolic disorders. Neurobiol. Dis. 2014, 72, 61–71. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Oldreive, C.E.; Harvey, J.; Doherty, G.H. Neurotrophic effects of leptin on cerebellar Purkinje but not granule neurons in vitro. Neurosci. Lett. 2008, 438, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, J. Leptin protects hippocampal CA1 neurons against ischemic injury. J. Neurochem. 2008, 107, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.F.; Jin, Y.C.; Li, X.M.; Yang, Z.; Wang, D.; Cui, J.J. Protective effects of leptin against cerebral ischemia/reperfusin injury. Exp. Ther. Med. 2019, 17, 3282–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantea, D.; Tassorelli, C.; Russo, R.; Petrelli, F.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Neuroprotection by leptin in a rat model of permanent cerebral ischemia: Effects on STAT3 phosphorylation in discrete cells of the brain. Cell Death Dis. 2011, 2, e238. [Google Scholar] [CrossRef] [Green Version]
- Busch, H.J.; Schirmer, S.H.; Jost, M.; van Stijn, S.; Peters, S.L.M.; Piek, J.J.; Bode, C.; Buschmann, I.R.; Mies, G. Leptin augments cerebral hemodynamic reserve after three-vessel occlusion: Distinct effects on cerebrovascular tone and proliferation in a nonlethal model of hypoperfused rat brain. J. Cereb. Blood Flow. Metab. 2011, 31, 1085–1092. [Google Scholar] [CrossRef]
- Weng, Z.; Signore, A.P.; Gao, Y.; Wang, S.; Zhang, F.; Hastings, T.; Yin, X.M.; Chen, J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2008, 282, 34479–34491. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Park, C.S.; Lee, S.K.; Shin, D.W.; Kang, J.H. Leptin inhibits 1-methyl-4-phenylpyridinium-induced cell death in SH-SY5Y cells. Neurosci. Lett. 2006, 407, 240–243. [Google Scholar] [CrossRef]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef]
- Tong, J.Q.; Zhang, J.; Hao, M.; Yang, J.; Han, Y.F.; Liu, X.J.; Shi, H.; Wu, M.N.; Liu, Q.S.; Qi, J.S. Leptin attenuates the detrimental effects of β-amyloid on spatial memory and hippocampal later-phase long term potentiation in rats. Horm. Behav. 2015, 73, 125–130. [Google Scholar] [CrossRef]
- Marwarha, G.; Dasari, B.; Prasanthi, J.R.; Schommer, J.; Ghribi, O. Leptin reduces the accumulation of Aβ and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J. Alzheimers Dis. 2010, 19, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1587–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedowicz, D.M.; Studzinski, C.M.; Weidner, A.M.; Platt, T.L.; Kingry, K.N.; Beckett, T.L.; Bruce-Keller, A.J.; Keller, J.N.; Murphy, M.P. Leptin regulates amyloid β production via the γ-secretase complex. Biochim. Biophys. Acta 2013, 1832, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, S.J.; Sarkar, S.; Casadesus, G.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N. Leptin inhibits glycogen synthase kinase-3β to prevent tau phosphorylation in neuronal cells. Neurosci. Lett. 2019, 455, 191–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-González, R.; Antequera, D.; Vargas, T.; Spuch, C.; Bolós, M.; Carro, E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 24 (Suppl. 2), 17–25. [Google Scholar] [CrossRef]
- Perez-Gonzalez, R.O.; Alvira-Botero, M.X.; Robayo, O.; Antequera, D.; Garzon, M.; Martín-Moreno, A.M.; Brera, B.; de Ceballos, M.L.; Carro, E. Leptin gene therapy attenuates neuronal damages evoked by amyloid-β and rescues memory deficits in APP/PS1 mice. Gene Ther. 2014, 21, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Guo, M.; Zhang, J.; Du, C.; Xing, Y. Leptin Regulates Tau Phosphorylation through Wnt Signaling Pathway in PC12 Cells. Neurosignals 2016, 24, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.H.; Beccano-Kelly, D.; Yan, S.D.; Gunn-Moore, F.J.; Harvey, J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid β. Neurobiol. Aging 2013, 34, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Platt, T.L.; Beckett, T.L.; Kohler, K.; Niedowicz, D.M.; Murphy, M.P. Obesity, diabetes and leptin resistance promote tau pathology in a mouse model of disease. Neuroscience 2016, 315, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; et al. Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.L.; et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef] [PubMed]
- Zanini, P.; Arbo, B.D.; Niches, G.; Czarnabay, D.; Benetti, F.; Ribeiro, M.F.; Cecconello, A.L. Diet-induced obesity alters memory consolidation in female rats. Physiol. Behav. 2017, 180, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Malekizadeh, Y.; Holiday, A.; Redfearn, D.; Ainge, J.A.; Doherty, G.; Harvey, J. A leptin fragment mirrors the cognitive enhancing and neuroprotective actions of leptin. Cereb. Cortex. 2017, 27, 4769–4782. [Google Scholar] [CrossRef] [Green Version]
- Matochik, J.A.; London, E.D.; Yildiz, B.O.; Ozata, M.; Caglayan, S.; DePaoli, A.M.; Wong, M.L.; Licinio, J. Effect of leptin replacement on brain structure in genetically leptin-deficient adults. J. Clin. Endocrinol. Metab. 2005, 90, 2851–2854. [Google Scholar] [CrossRef] [Green Version]
- Paz-Filho, G.J.; Babikian, T.; Asarnow, R.; Esposito, K.; Erol, H.K.; Wong, M.L.; Licinio, J. Leptin replacement improves cognitive development. PLoS ONE 2008, 8, e3098. [Google Scholar] [CrossRef]
- Grasso, P.; Leinung, M.C.; Ingher, S.P.; Lee, D.W. In vivo effects of leptin-related synthetic peptides on body weight and food intake in female ob/ob mice: Localization of leptin activity to domains between amino acid residues 106–140. Endocrinology 1997, 138, 1413–1418. [Google Scholar] [CrossRef]
- Rozhavskaya-Arena, M.; Lee, D.W.; Leinung, M.C.; Grasso, P. Design of a synthetic leptin agonist: Effects on energy balance, glucose homeostasis and thermoregulation. Endocrinology 2000, 141, 2501–2507. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Nelson, J.S.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Popper, J.S.; Petrovitch, H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology 2002, 59, 1051–1057. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; de Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metab. Clin. Exp. 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Gaba, A.M.; Zhang, K.; Marder, K.; Moskowitz, C.B.; Werner, P.; Boozer, C.N. Energy balance in early-stage Huntington disease. Am. J. Clin. Nutr. 2005, 81, 1335–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, K.L.; Chitnis, T.; Ascherio, A. Body size and risk of MS in two cohorts of US women. Neurology 2009, 73, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratley, R.E.; Salbe, A.D.; Ravussin, E.; Caviness, J.N. Higher sedentary energy expenditure in patients with Huntington’s. Ann. Neurol. 2000, 47, 64–70. [Google Scholar] [CrossRef]
- Popovic, V.; Svetel, M.; Djurovic, M.; Petrovic, S.; Doknic, M.; Pekic, S.; Miljic, D.; Milic, N.; Glodic, J.; Dieguez, C.; et al. Circulating and cerebrospinal fluid ghrelin and leptin: Potential role in altered body weight in Huntington’s disease. Eur. J. Endocrinol. 2004, 151, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evidente, V.G.; Caviness, J.N.; Adler, C.H.; Gwinn-Hardy, K.A.; Pratley, R.E. Serum leptin concentrations and satiety in Parkinson’s disease patients with and without weight loss. Mov. Disord. 2001, 16, 924–927. [Google Scholar] [CrossRef]
- Rothman, S.M.; Griffioen, K.J.; Fishbein, K.W.; Spencer, R.G.; Makrogiannis, S.; Cong, W.N.; Martin, B.; Mattson, M.P. Metabolic abnormalities and hypoleptinemia in α-synuclein A53T mutant mice. Neurobiol. Aging 2014, 35, 1153–1161. [Google Scholar] [CrossRef] [Green Version]
- Frisullo, G.; Mirabella, M.; Angelucci, F.; Caggiula, M.; Morosetti, R.; Sancricca, C.; Patanella, A.K.; Nociti, V.; Iorio, R.; Bianco, A.; et al. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signaling-3 expression in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2007, 192, 174–183. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Phan, K.; Highton-Williamson, E.; Strikwerda-Brown, C.; Caga, J.; Ramsey, E.; Zoing, M.; Devenney, E.; Kim, W.S.; Hodges, J.R.; et al. Eating peptides: Biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Kölbel, H.; Hauffa, B.P.; Wudy, S.A.; Bouikidis, A.; Della Marina, A.; Schara, U. Hyperleptinemia in children with autosomal recessive spinal muscular atrophy type I-III. PLoS ONE 2017, 12, e0173144. [Google Scholar] [CrossRef]
- Nagel, G.; Peter, R.S.; Rosenbohm, A.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. Adipokines, C-reactive protein and Amyotrophic Lateral Sclerosis—Results from a population-based ALS registry in Germany. Sci. Rep. 2017, 7, 4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.A.; Bence, K.K.; Sandesara, I.; Andreux, P.; Auwerx, J.; Ishibashi, J.; Seale, P.; Kalb, R.G. Genetically altering organismal metabolism by leptin-deficiency benefits a mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 4995–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamilton, K.; Harvey, J. The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease. Pharmaceuticals 2021, 14, 52. https://doi.org/10.3390/ph14010052
Hamilton K, Harvey J. The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease. Pharmaceuticals. 2021; 14(1):52. https://doi.org/10.3390/ph14010052
Chicago/Turabian StyleHamilton, Kirsty, and Jenni Harvey. 2021. "The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease" Pharmaceuticals 14, no. 1: 52. https://doi.org/10.3390/ph14010052
APA StyleHamilton, K., & Harvey, J. (2021). The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease. Pharmaceuticals, 14(1), 52. https://doi.org/10.3390/ph14010052