Targeting the Proline–Glutamine–Asparagine–Arginine Metabolic Axis in Amino Acid Starvation Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
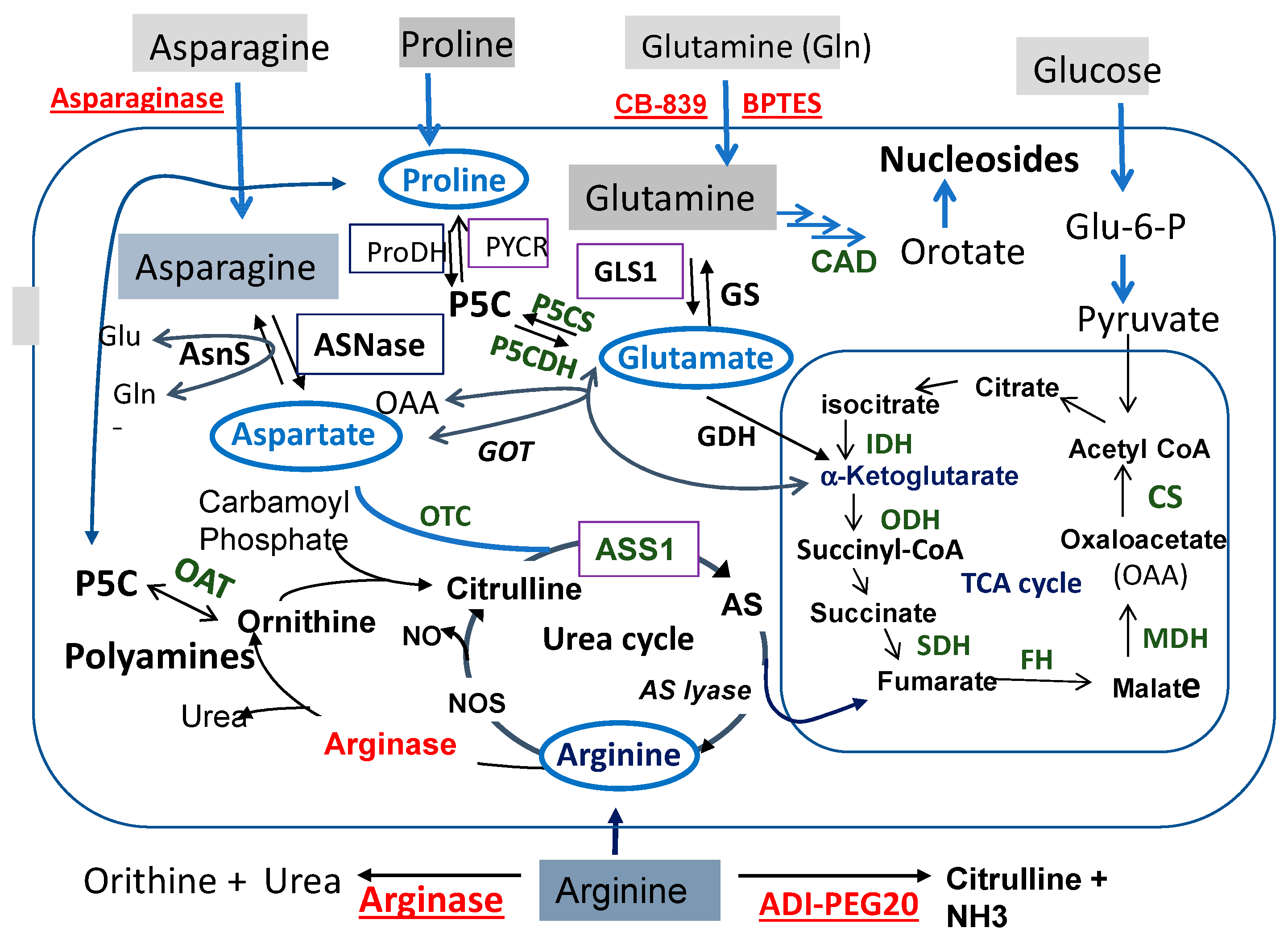
2. The Interconnecting Proline–Glutamine–Asparagine–Arginine Metabolic Wiring in Cancer Cells
3. Targeting Specific Amino Acid Starvation in Cancer Therapy
3.1. Proline Starvation
3.2. Glutamine Starvation
3.3. Asparagine Starvation
3.4. Arginine Starvation
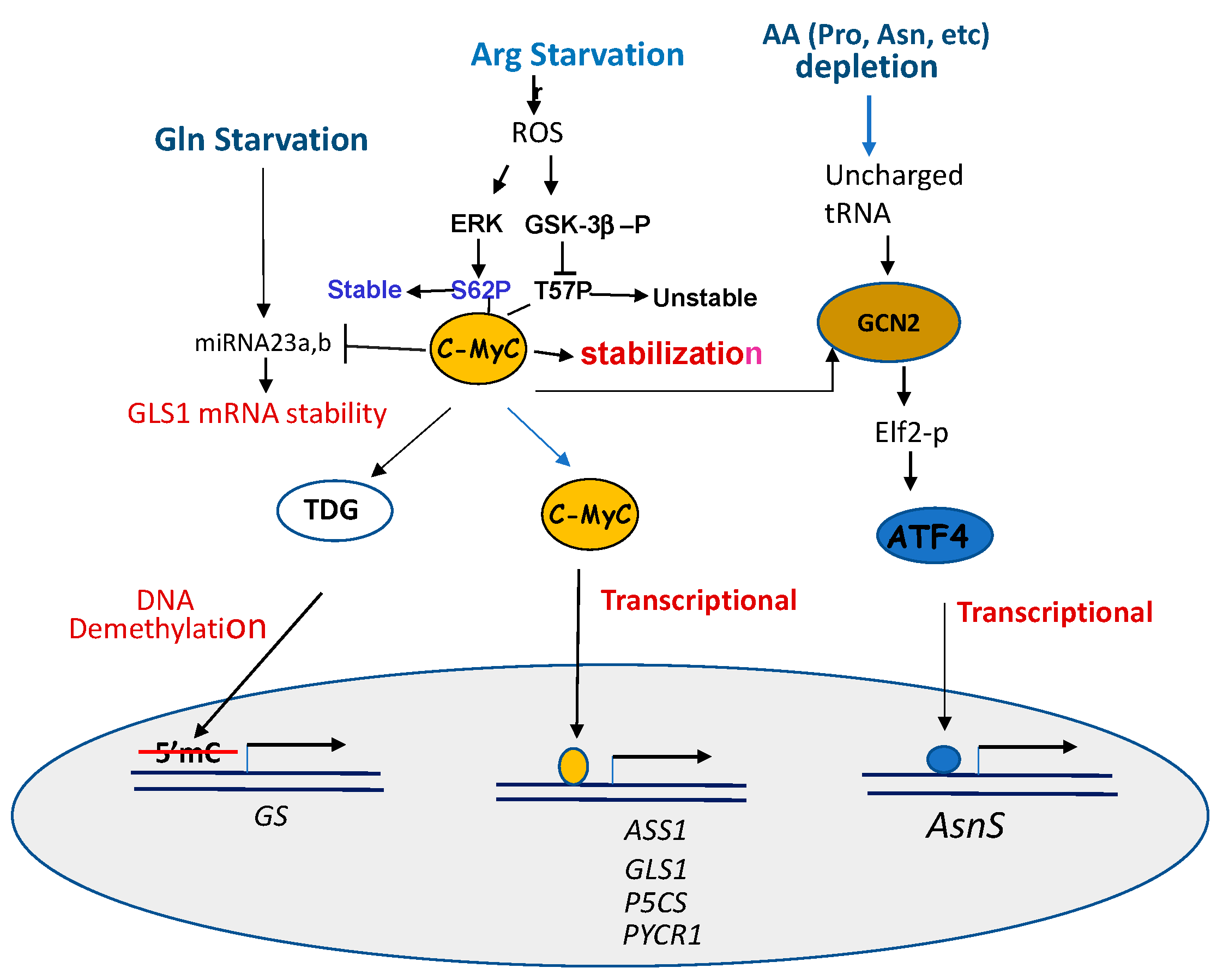
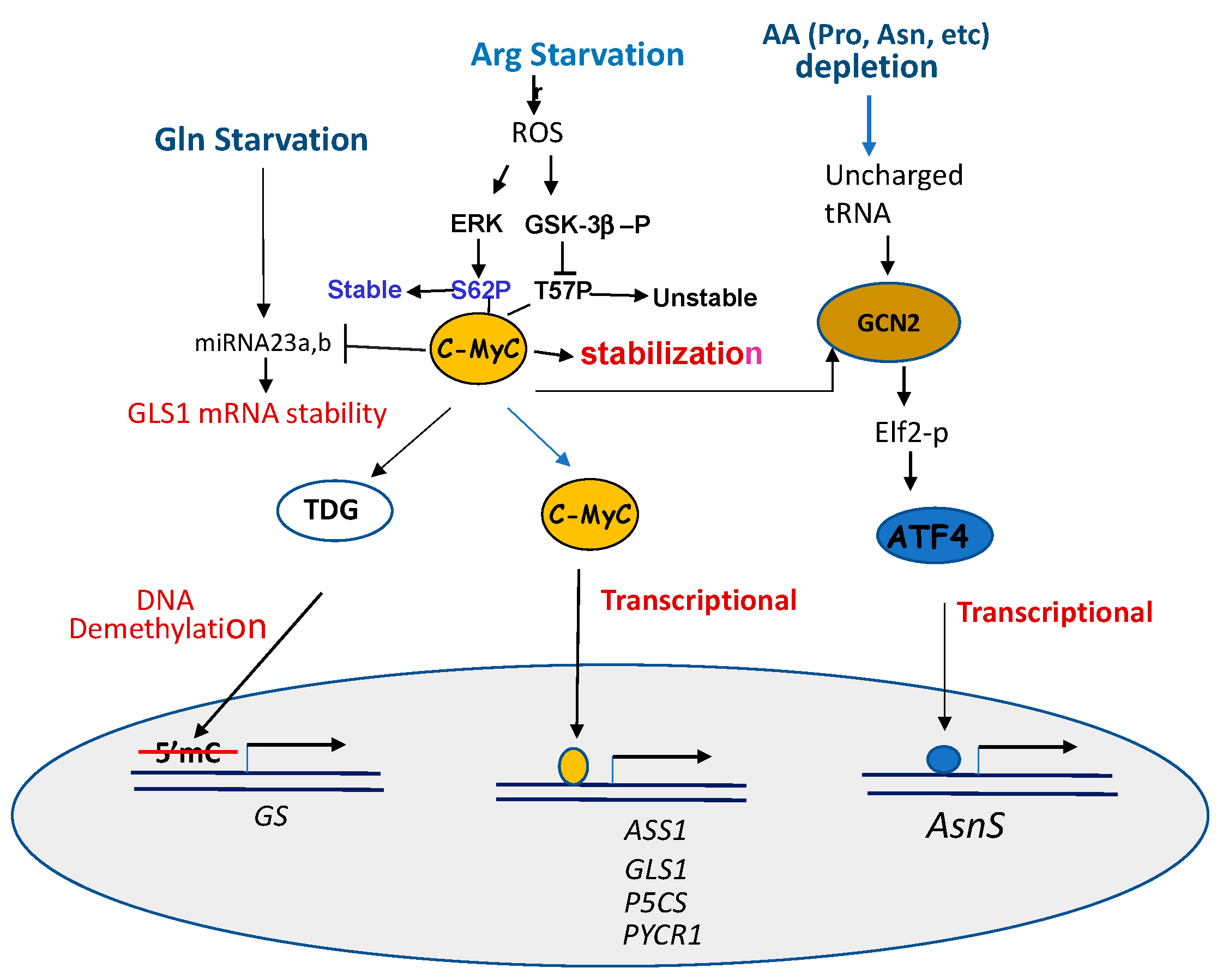
4. Common Mechanisms Associated with Pro- Gln- Asn- and Arg-Starvation Therapies
4.1. Production of ROS
4.2. Involvements of the Same Transcription Regulators
4.2.1. c-Myc
4.2.2. ATF4
5. Mechanisms of Drug Resistance
5.1. Reprogramming of Survival Amino Acid Metabolism
5.1.1. Re-Activation of the Silenced Genes
5.1.2. Compensatory Activation and Cross-Interference of Metabolic Pathways
5.2. Roles of Amino Acid Transporters
5.3. Induction of Immunogenic Reactions Associated with Using Microbial Enzymes
5.3.1. ADI-PEG20
5.3.2. Recombinant Asparaginases
6. Conclusions and Perspective
Funding
Conflicts of Interest
References
- Yahyaoui, R.; Pérez-Frías, J. Amino Acid Transport Defects in Human Inherited Metabolic Disorders. Int. J. Mol. Sci. 2019, 21, 119. [Google Scholar] [CrossRef] [Green Version]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase) in cancer. BioFactors 2012, 38, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Fendt, S.-M.; Becker, D.F. The Proline Cycle as a Potential Cancer Therapy Target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef] [PubMed]
- Loayza-Puch, F.; Rooijers, K.; Buil, L.C.M.; Zijlstra, J.; Vrielink, J.F.O.; Lopes, R.; Ugalde, A.P.; Van Breugel, P.; Hofland, I.; Wesseling, J.; et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nat. Cell Biol. 2016, 530, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Luo, L.; Xie, Y.; Zhao, Y.; Yao, J.; Liu, X.-L. PYCR1 knockdown inhibits the proliferation, migration, and invasion by affecting JAK/STAT signaling pathway in lung adenocarcinoma. Mol. Carcinog. 2020, 59, 503–511. [Google Scholar] [CrossRef]
- Zhuang, J.; Song, Y.; Ye, Y.; He, S.; Ma, X.; Zhang, M.; Ni, J.; Wang, J.; Xia, W. PYCR1 interference inhibits cell growth and survival via c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathway in hepatocellular cancer. J. Transl. Med. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nat. Cell Biol. 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline Oxidase Promotes Tumor Cell Survival in Hypoxic Tumor Microenvironments. Cancer Res. 2012, 72, 3677–3686. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.-M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Ericksen, R.E.; Escande-Beillard, N.; Lee, Q.Y.; Loh, A.; Haines, R.L.; Steckel, M.; Haegebarth, A.; Ho, T.S.W.; Chow, P.K.H.; et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J. Hepatol. 2020, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Yau, C.; Becker, B.C.; Khateeb, S.; Mahoney, S.; Jensen, M.B.; Hann, B.C.; Cowen, B.J.; Pegan, S.D.; Benz, C.C. Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition. Mol. Cancer Ther. 2019, 18, 1374–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, E.M.; Bogner, A.N.; Vandekeere, A.; Tam, G.S.; Patel, S.M.; Becker, D.F.; Fendt, S.-M.; Tanner, J.J. In crystallo screening for proline analog inhibitors of the proline cycle enzyme PYCR. J. Biol. Chem. 2020, 295, 18316–18327. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Dickman, M.B.; Becker, D.F. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med. 2008, 44, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine Plays a Critical Role in Regulating Cellular Adaptation to Glutamine Depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Chen, S. Metabolic Signaling Cascades Prompted by Glutaminolysis in Cancer. Cancers 2020, 12, 2624. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.C.; Dechaume, A.-L.; Murray, P.M.; Katt, W.P.; Baguley, B.C.; Leung, E. Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer. BMC Cancer 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampa, M.; Arlt, H.; Christopher, W.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomme, J.; Su, Y.; Lavie, A. Elucidation of the Specific Function of the Conserved Threonine Triad Responsible for Human l-Asparaginase Autocleavage and Substrate Hydrolysis. J. Mol. Biol. 2014, 426, 2471–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baruchel, A.; Brown, P.; Rizzari, C.; Silverman, L.; Van Der Sluis, I.; Wolthers, B.O.; Schmiegelow, K. Increasing completion of asparaginase treatment in childhood acute lymphoblastic leukaemia (ALL): Summary of an expert panel discussion. ESMO Open 2020, 5, e000977. [Google Scholar] [CrossRef]
- Chan, W.K.; Horvath, T.D.; Tan, L.; Link, T.; Harutyunyan, K.G.; Pontikos, M.A.; Anishkin, A.; Du, D.; Martin, L.A.; Yin, E.; et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity against Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2019, 18, 1587–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, D.; Oh, A.; Mahmud, D.L.; et al. A Novel l-Asparaginase with low l-Glutaminase Coactivity Is Highly Efficacious against Both T- and B-cell Acute Lymphoblastic Leukemias In Vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Radadiya, A.; Zhu, W.; Coricello, A.; Alcaro, S.; Richards, N.G.J. Improving the Treatment of Acute Lymphoblastic Leukemia. Biochemistry 2020, 59, 3193–3200. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Srivastava, S.; Seim, G.; Pavlova, N.N.; King, B.; Zou, L.; Zhang, C.; Zhong, M.; Feng, H.; Kapur, R.; et al. Promoter demethylation of the asparagine synthetase gene is required for ATF4-dependent adaptation to asparagine depletion. J. Biol. Chem. 2019, 294, 18674–18684. [Google Scholar] [CrossRef] [PubMed]
- Touzart, A.; Lengliné, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic Silencing Affects l-Asparaginase Sensitivity and Predicts Outcome in T-ALL. Clin. Cancer Res. 2019, 25, 2483–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 9, 1480. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, L.-W.; Tan, Y.-X.; Zhang, J.; Pan, Y.-F.; Yang, C.; Li, M.-H.; Ding, Z.-W.; Liu, L.-J.; Jiang, T.-Y.; et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br. J. Cancer 2013, 109, 14–23. [Google Scholar] [CrossRef]
- Dufour, E.; Gay, F.; Aguera, K.; Scoazec, J.-Y.; Horand, F.; Lorenzi, P.L.; Godfrin, Y. Pancreatic Tumor Sensitivity to Plasma L-Asparagine Starvation. Pancreas 2012, 41, 940–948. [Google Scholar] [CrossRef]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nat. Cell Biol. 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Aslanian, A.M.; Fletcher, B.S.; Kilberg, M.S. Asparagine synthetase expression alone is sufficient to induce l-asparaginase resistance in MOLT-4 human leukaemia cells. Biochem. J. 2001, 357, 321–328. [Google Scholar] [CrossRef]
- Su, N.; Pan, Y.-X.; Zhou, M.; Harvey, R.C.; Hunger, S.P.; Kilberg, M.S. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr. Blood Cancer 2007, 50, 274–279. [Google Scholar] [CrossRef]
- Fung, M.K.L.; Chan, G.C.F. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017, 10, 1–18. [Google Scholar] [CrossRef]
- Closs, E.I.; Simon, A.; Vékony, N.; Rotmann, A. Plasma Membrane Transporters for Arginine. J. Nutr. 2004, 134, 2752S–2759S. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Savaraj, N.; Feun, L.G. Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget 2010, 1, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Delage, B.; Luong, P.; Maharaj, L.; O’Riain, C.; Syed, N.; Crook, T.; Hatzimichael, E.; Papoudou-Bai, A.; Mitchell, T.J.; Whittaker, S.J.; et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. 2012, 3, e342. [Google Scholar] [CrossRef]
- Syed, N.; Langer, J.; Janczar, K.; Singh, P.K.; Nigro, C.L.; Lattanzio, L.; Coley, H.M.; Hatzimichael, E.; Bomalaski, J.S.; Szlosarek, P.W.; et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis. 2013, 4, e458. [Google Scholar] [CrossRef] [Green Version]
- Szlosarek, P.W.; Grimshaw, M.J.; Wilbanks, G.D.; Hagemann, T.; Wilson, J.L.; Burke, F.; Stamp, G.; Balkwill, F.R. Aberrant regulation of argininosuccinate synthetase by TNF-α in human epithelial ovarian cancer. Int. J. Cancer 2007, 121, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Wu, W.-R.; Wang, Y.-H.; Wang, J.-W.; Fang, F.-M.; Tsai, J.-W.; Li, S.-H.; Hung, H.-C.; Yu, S.-C.; Lan, J.; et al. ASS1 as a Novel Tumor Suppressor Gene in Myxofibrosarcomas: Aberrant Loss via Epigenetic DNA Methylation Confers Aggressive Phenotypes, Negative Prognostic Impact, and Therapeutic Relevance. Clin. Cancer Res. 2013, 19, 2861–2872. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberly, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef]
- Lan, J.; Tai, H.-C.; Lee, S.-W.; Chen, T.-J.; Huang, H.-Y.; Li, C.-F. Deficiency in expression and epigenetic DNA Methylation of ASS1 gene in nasopharyngeal carcinoma: Negative prognostic impact and therapeutic relevance. Tumor Biol. 2013, 35, 161–169. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Luong, P.; Phillips, M.M.; Baccarini, M.; Ellis, S.; Szyszko, T.; Sheaff, M.T.; Avril, N.; Stephen, E. Metabolic Response to Pegylated Arginine Deiminase in Mesothelioma With Promoter Methylation of Argininosuccinate Synthetase. J. Clin. Oncol. 2013, 31, e111–e113. [Google Scholar] [CrossRef]
- Allen, M.D.; Luong, P.; Hudson, C.; Leyton, J.; Delage, B.; Ghazaly, E.; Cutts, R.; Yuan, M.; Syed, N.; Nigro, C.L.; et al. Prognostic and Therapeutic Impact of Argininosuccinate Synthetase 1 Control in Bladder Cancer as Monitored Longitudinally by PET Imaging. Cancer Res. 2014, 74, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, T.; Strauss, M.; Hering, S.; Redanz, U.; William, D.; Rosche, Y.; Classen, C.F.; Kreikemeyer, B.; Linnebacher, M.; Maletzki, C. Arginine deprivation by arginine deiminase of Streptococcus pyogenes controls primary glioblastoma growth in vitro and in vivo. Cancer Biol. Ther. 2015, 16, 1047–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-B.; Aiba, I.; Lee, S.-Y.; Feun, L.; Savaraj, N.; Kuo, M.T. Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1α/Sp4. Mol. Cancer Ther. 2009, 8, 3223–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-B.; Long, Y.; Chang, J.T.; Savaraj, N.; Feun, L.G.; Jung, M.; Chen, H.H.W.; Kuo, M.T. Chromatin remodeling system p300-HDAC2-Sin3A is involved in Arginine Starvation-Induced HIF-1α Degradation at the ASS1 promoter for ASS1 Derepression. Sci. Rep. 2017, 7, 10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-B.; Long, Y.; Park, J.-R.; Chang, J.T.; Liu, H.; Rodriguez-Canales, J.; Savaraj, N.; Feun, L.G.; Davies, M.A.; Wistuba, I.; et al. Gas6/Axl is the sensor of arginine-auxotrophic response in targeted chemotherapy with arginine-depleting agents. Oncogene 2016, 35, 1632–1642. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-B.; Aiba, I.; Long, Y.; Lin, H.-K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK Pathway Induces c-Myc Stabilization to Upregulate Argininosuccinate Synthetase, Leading to Arginine Deiminase Resistance in Melanoma Cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.T.; Long, Y.; Tsai, W.B.; Li, Y.Y.; Chen, H.H.W.; Feun, L.G.; Savaraj, N. Collaboration Between RSK-EphA2 and Gas6-Axl RTK Signaling in Arginine Starvation Response That Confers Resistance to EGFR Inhibitors. Trans. Oncol. 2020, 13, 355–364. [Google Scholar] [CrossRef]
- Long, Y.; Tsai, W.-B.; Chang, J.T.; Estécio, M.R.; Wangpaichitr, M.; Savaraj, N.; Feun, L.G.; Chen, H.H.; Kuo, M.T. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1α, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016, 7, 82658–82670. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting Arginine-Dependent Cancers with Arginine-Degrading Enzymes: Opportunities and Challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef]
- Zhou, S.; Allard, P.-M.; Wolfrum, C.; Ke, C.; Tang, C.; Ye, Y.; Wolfender, J.-L. Identification of chemotypes in bitter melon by metabolomics: A plant with potential benefit for management of diabetes in traditional Chinese medicine. Metabolomics 2019, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.-Y.; Lu, S.-N.; Yen, C.-J.; Feng, Y.-H.; Lim, H.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.C.-C.; Cheng, P.N.; Chan, P.; Chen, L.; Yuen, J.; Pang, R.; Fan, S.T.; Wheatley, D.N.; Poon, R.T. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Investig. New Drugs 2015, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; Egan, S.A.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; Kin, P.U.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic Signaling Mediated by Oxidants in Ras-Transformed Fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craze, M.L.; Cheung, H.; Jewa, N.; Coimbra, N.D.M.; Soria, D.; El-Ansari, R.; Aleskandarany, M.A.; Cheng, K.W.; Diez-Rodriguez, M.; Nolan, C.C.; et al. MYC regulation of glutamine–proline regulatory axis is key in luminal B breast cancer. Br. J. Cancer 2017, 118, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, S.K.; Zhu, W.; Liang, X.; Zhang, L.; Demers, A.J.; Zimmerman, M.C.; Simpson, M.A.; Becker, D.F. Proline dehydrogenase is essential for proline protection against hydrogen peroxide-induced cell death. Free Radic. Biol. Med. 2012, 53, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Tołoczko-Iwaniuk, N.; Dziemianczyk-Pakiela, D.; Celińska-Janowicz, K.; Ościłowska, I.; Klupczynska, A.; Kokot, Z.; Nowaszewska, K.B.; Reszeć, J.; Borys, J.; Miltyk, W. Proline-Dependent Induction of Apoptosis in Oral Squamous Cell Carcinoma (OSCC)—The Effect of Celecoxib. Cancers 2020, 12, 136. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.Y.L.; Zareba, I.; Baszanowska, W.; Lewoniewska, S.; Pałka, J. Understanding the role of key amino acids in regulation of proline dehydrogenase/proline oxidase (prodh/pox)-dependent apoptosis/autophagy as an approach to targeted cancer therapy. Mol. Cell. Biochem. 2020, 466, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mick, E.; Titov, D.V.; Skinner, O.S.; Sharma, R.; Jourdain, A.; Mootha, V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. eLife 2020, 9, e49178. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol. Res. 2019, 52, 15. [Google Scholar] [CrossRef]
- Shen, Y.A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.C.; Rahmanto, Y.S.; Jung, J.G.; Chen, Y.W.; Yen, T.T.; Tomaszewski, A.; et al. Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res. 2020, 80, 4514–4526. [Google Scholar]
- Seitz, V.; Butzhammer, P.; Hirsch, B.; Hecht, J.; Gütgemann, I.; Ehlers, A.; Lenze, D.; Oker, E.; Sommerfeld, A.; Von Der Wall, E.; et al. Deep Sequencing of MYC DNA-Binding Sites in Burkitt Lymphoma. PLoS ONE 2011, 6, e26837. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Feun, L.; Thongkum, A.; Tu, C.-H.; Chen, S.-M.; Wangpaichitr, M.; Wu, C.; Kuo, M.T.; Savaraj, N. Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1297. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ye, L.; Fan, J.; Zhang, X.; Wang, H.; Liao, S.; Song, P.; Wang, Z.; Wang, S.; Li, Y.; et al. Autophagy suppression potentiates the anti-glioblastoma effect of asparaginase in vitro and in vivo. Oncotarget 2017, 8, 91052–91066. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, L.; Tao, Q.; Zhang, X.; Luan, J.; Zhao, S.; Liu, H.; Ju, D. Deprivation of asparagine triggers cytoprotective autophagy in laryngeal squamous cell carcinoma. Appl. Microbiol. Biotechnol. 2017, 101, 4951–4961. [Google Scholar] [CrossRef] [PubMed]
- Changou, C.A.; Chen, Y.-R.; Xing, L.; Yen, Y.; Chuang, F.Y.S.; Cheng, R.H.; Bold, R.J.; Ann, D.K.; Kung, H.-J. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 14147–14152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Wu, C.; Chen, S.-M.; Shah, S.S.; Wangpaichitr, M.; Feun, L.G.; Kuo, M.T.; Suarez, M.; Prince, J.; Savaraj, N. BRAF inhibitor resistance enhances vulnerability to arginine deprivation in melanoma. Oncotarget 2016, 7, 17665–17680. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Wu, C.; Shah, S.S.; Chen, S.-M.; Wangpaichitr, M.; Kuo, M.T.; Feun, L.G.L.G.; Han, X.; Suarez, M.; Prince, J.; et al. Degradation of AMPK-α1 sensitizes BRAF inhibitor-resistant melanoma cells to arginine deprivation. Mol. Oncol. 2017, 11, 1806–1825. [Google Scholar] [CrossRef] [Green Version]
- Brashears, C.B.; Barlin, M.; Ehrhardt, W.R.; Rathore, R.; Schultze, M.; Tzeng, S.-C.; Van Tine, B.A.; Held, J.M. Systems level profiling of arginine starvation reveals MYC and ERK adaptive metabolic reprogramming. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Tiziani, S.; Kang, Y.; Harjanto, R.; Axelrod, J.; Piermarocchi, C.; Roberts, W.; Paternostro, G. Metabolomics of the Tumor Microenvironment in Pediatric Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e82859. [Google Scholar] [CrossRef] [Green Version]
- Gaglio, D.; Bonanomi, M.; Valtorta, S.; Bharat, R.; Ripamonti, M.; Conte, F.; Fiscon, G.; Righi, N.; Napodano, E.; Papa, F.; et al. Disruption of redox homeostasis for combinatorial drug efficacy in K-Ras tumors as revealed by metabolic connectivity profiling. Cancer Metab. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Sahu, N.; Cruz, D.D.; Gao, M.; Sandoval, W.; Haverty, P.M.; Liu, J.; Stephan, J.-P.; Haley, B.; Classon, M.; Hatzivassiliou, G.; et al. Proline Starvation Induces Unresolved ER Stress and Hinders mTORC1-Dependent Tumorigenesis. Cell Metab. 2016, 24, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapproth, K.; Wirth, T. Advances in the understanding of MYC-induced lymphomagenesis. Br. J. Haematol. 2010, 149, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Marchingo, J.M.; Sinclair, L.V.; Howden, A.J.; Cantrell, D.A. Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. eLife 2020, 9, e53725. [Google Scholar] [CrossRef]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.T.; Tsai, W.-B.; Wangpaichitr, M.; Tsukamoto, T.; Savaraj, N.; Feun, L.G.; Kuo, M.T. Arginine Deiminase Resistance in Melanoma Cells Is Associated with Metabolic Reprogramming, Glucose Dependence, and Glutamine Addiction. Mol. Cancer Ther. 2013, 12, 2581–2590. [Google Scholar] [CrossRef] [Green Version]
- Le, A.-P.; Zhang, L.-L.; Liu, W.; Shi, Y.-F. Cantharidin inhibits cell proliferation and induces apoptosis through G2/M phase cell cycle arrest in hepatocellular carcinoma stem cells. Oncol. Rep. 2016, 35, 2970–2976. [Google Scholar] [CrossRef] [Green Version]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salinas, C.S.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Xu, X.; Watt, D.S.; Liu, C. Multifaceted roles for thymine DNA glycosylase in embryonic development and human carcinogenesis. Acta Biochim. Biophys. Sin. 2015, 48, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Bott, A.J.; Peng, I.-C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nat. Cell Biol. 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.-M.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardos, G.R.; Wastyk, H.C.; Robertson, G.P. Disruption of Proline Synthesis in Melanoma Inhibits Protein Production Mediated by the GCN2 Pathway. Mol. Cancer Res. 2015, 13, 1408–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Li, Y.; Xia, X.; Guo, W.; Zhai, T.; Jin, Y.; Che, Y.; Gao, H.; Duan, X.; Ma, H.; et al. Arginine inhibits the malignant transformation induced by interferon-gamma through the NF-kappaB-GCN2/eIF2alpha signaling pathway in mammary epithelial cells in vitro and in vivo. Exp. Cell Res. 2018, 368, 236–247. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829.e6. [Google Scholar] [CrossRef] [Green Version]
- Cherasse, Y.; Maurin, A.-C.; Chaveroux, C.; Jousse, C.; Carraro, V.; Parry, L.; Deval, C.; Chambon, C.; Fafournoux, P.; Bruhat, A. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007, 35, 5954–5965. [Google Scholar] [CrossRef]
- Long, Y.; Tsai, W.-B.; Wang, D.; Hawke, D.; Savaraj, N.; Feun, L.G.; Hung, M.-C.; Chen, H.H.; Kuo, M.T. Argininosuccinate synthetase 1 (ASS1) is a common metabolic marker of chemosensitivity for targeted arginine- and glutamine-starvation therapy. Cancer Lett. 2017, 388, 54–63. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5. [Google Scholar] [CrossRef] [Green Version]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Gabra, M.B.I.; Tran, T.Q.; Li, H.; Kong, M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019, 26, 3051–3060.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Bröer, S. The SLC38 family of sodium–amino acid co-transporters. Pflügers Archiv. 2013, 466, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.B.; Wang, C.; Huang, C.C.; Yaman, I.; Li, Y.; Chakravarty, K.; Johnson, P.F.; Chiang, C.-M.; Snider, M.D.; Wek, R.C.; et al. A feedback transcriptional mechanism controls the level of the arginine/lysine transporter cat-1 during amino acid starvation. Biochem. J. 2007, 402, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Hall, P.E.; Lewis, R.; Syed, N.; Shaffer, R.; Evanson, J.; Ellis, S.; Williams, M.; Feng, X.; Johnston, A.; Thomson, J.; et al. A Phase I Study of Pegylated Arginine Deiminase (Pegargiminase), Cisplatin, and Pemetrexed in Argininosuccinate Synthetase 1-Deficient Recurrent High-grade Glioma. Clin. Cancer Res. 2019, 25, 2708–2716. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-S.; Lu, S.-N.; Chao, Y.; Sheen, I.-S.; Lin, C.-C.; Wang, T.-E.; Chen, S.-C.; Wang, J.-H.; Liao, L.-Y.; Thomson, J.; et al. A randomised phase II study of pegylated arginine deiminase (ADI-PEG 20) in Asian advanced hepatocellular carcinoma patients. Br. J. Cancer 2010, 103, 954–960. [Google Scholar] [CrossRef]
- Hijiya, N.; Van Der Sluis, I. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 57, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Cecconello, D.K.; De Magalhães, M.R.; Werlang, I.C.R.; Lee, M.L.D.M.; Michalowski, M.B.; Daudt, L.E. Asparaginase: An old drug with new questions. Hematol. Transfus. Cell Ther. 2020, 42, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ko, R.H.; Jones, T.L.; Radvinsky, D.; Robison, N.; Gaynon, P.S.; Panosyan, E.H.; Avramis, I.A.; Avramis, V.I.; Rubin, J.; Ettinger, L.J.; et al. Allergic reactions and antiasparaginase antibodies in children with high-risk acute lymphoblastic leukemia: A children’s oncology group report. Cancer 2015, 12, 4205–4211. [Google Scholar] [CrossRef] [PubMed]
- Pession, A.; Valsecchi, M.G.; Masera, G.; Kamps, W.A.; Magyarosy, E.; Rizzari, C.; Van Wering, E.R.; Nigro, L.L.; Van Der Does, A.; Locatelli, F.; et al. Long-Term Results of a Randomized Trial on Extended Use of High Dose l-Asparaginase for Standard Risk Childhood Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2005, 23, 7161–7167. [Google Scholar] [CrossRef] [PubMed]
- Amylon, M.D.; Shuster, J.; Pullen, J.; Berard, C.; Link, M.P.; Wharam, M.; Katz, J.; Yu, A.; Laver, J.; Ravindranath, Y.; et al. Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: A Pediatric Oncology Group study. Leukemia 1999, 13, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, W. 50 Years Ago in The Journal of Pediatrics: L-Asparaginase Therapy in Pediatric Acute Lymphoblastic Leukemia: Optimizing Efficacy and Minimizing Toxicity. J. Pediatr. 2020, 224, 86. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Duvall, B.; Tsukamoto, T. Recent Progress in the Discovery of Allosteric Inhibitors of Kidney-Type Glutaminase. J. Med. Chem. 2019, 62, 46–59. [Google Scholar] [CrossRef]
- Huang, Q.; Stalnecker, C.; Zhang, C.; McDermott, L.A.; Iyer, P.; O’Neill, J.; Reimer, S.; Cerione, R.A.; Katt, W.P. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [Google Scholar] [CrossRef] [Green Version]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Ding, J.; Kuo, M.-L.; Su, L.; Xue, L.; Luh, F.; Zhang, H.; Wang, J.; Lin, T.G.; Zhang, K.; Chu, P.; et al. Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis 2017, 38, 519–531. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, M.T.; Chen, H.H.W.; Feun, L.G.; Savaraj, N. Targeting the Proline–Glutamine–Asparagine–Arginine Metabolic Axis in Amino Acid Starvation Cancer Therapy. Pharmaceuticals 2021, 14, 72. https://doi.org/10.3390/ph14010072
Kuo MT, Chen HHW, Feun LG, Savaraj N. Targeting the Proline–Glutamine–Asparagine–Arginine Metabolic Axis in Amino Acid Starvation Cancer Therapy. Pharmaceuticals. 2021; 14(1):72. https://doi.org/10.3390/ph14010072
Chicago/Turabian StyleKuo, Macus Tien, Helen H. W. Chen, Lynn G. Feun, and Niramol Savaraj. 2021. "Targeting the Proline–Glutamine–Asparagine–Arginine Metabolic Axis in Amino Acid Starvation Cancer Therapy" Pharmaceuticals 14, no. 1: 72. https://doi.org/10.3390/ph14010072
APA StyleKuo, M. T., Chen, H. H. W., Feun, L. G., & Savaraj, N. (2021). Targeting the Proline–Glutamine–Asparagine–Arginine Metabolic Axis in Amino Acid Starvation Cancer Therapy. Pharmaceuticals, 14(1), 72. https://doi.org/10.3390/ph14010072