
Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP

Abstract
1. Introduction
2. Results
2.1. Design of PT1.2
2.2. Spectrum of Activity and Specificity of PT.2
2.3. Speed of Kill
2.4. Kinetics of PT1.2:S. aureus Interaction
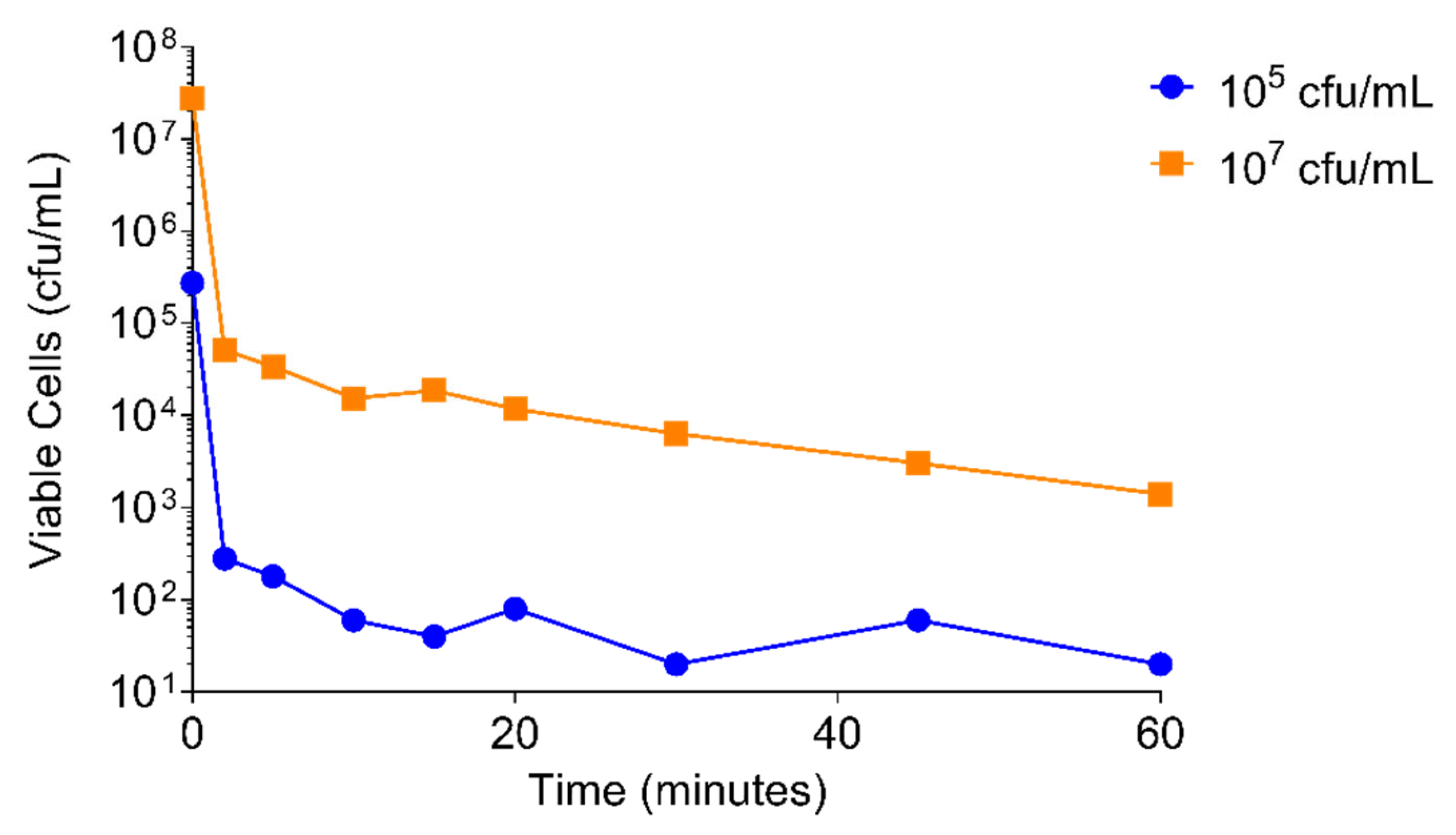
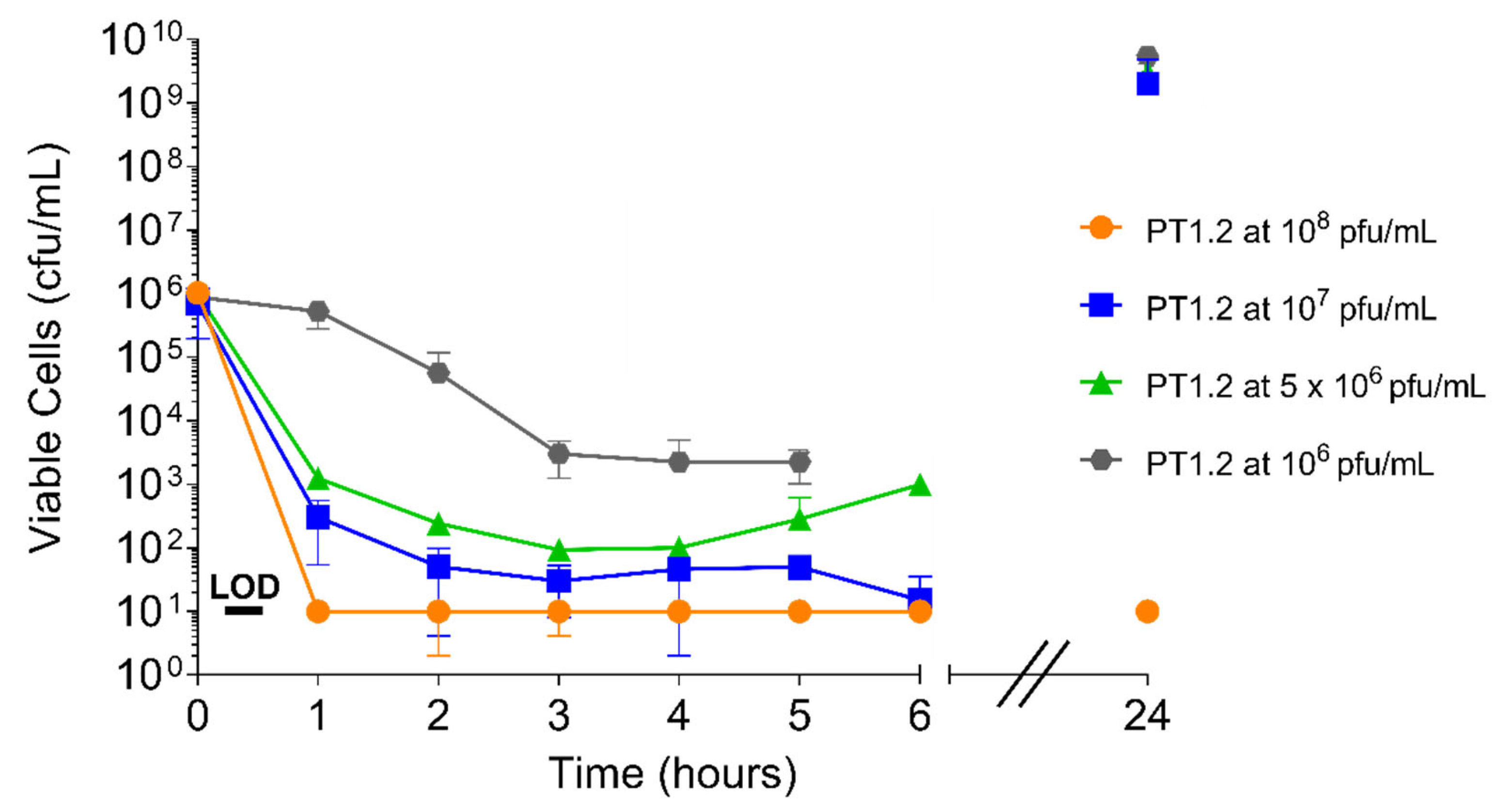
2.4.1. PT1.2 Concentration
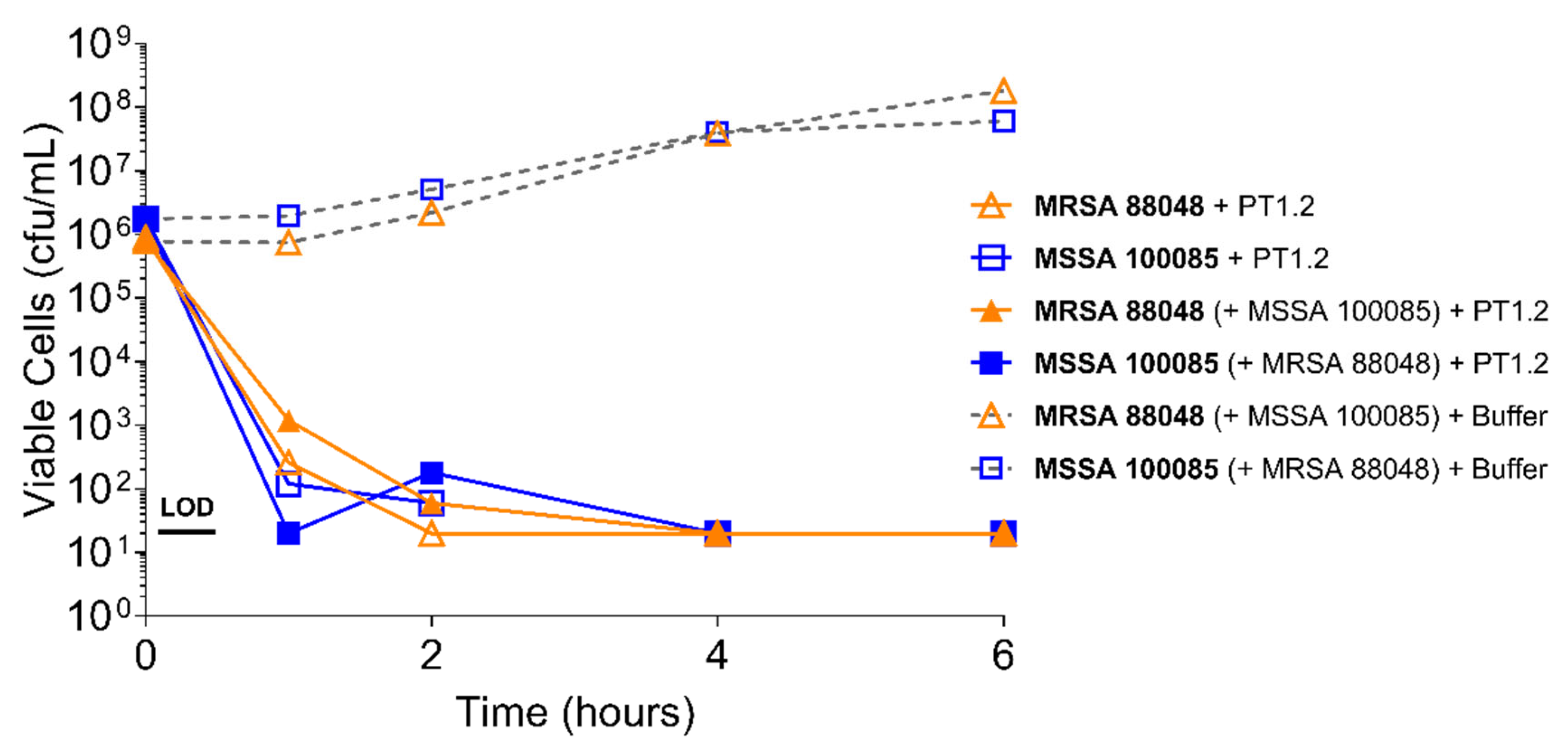
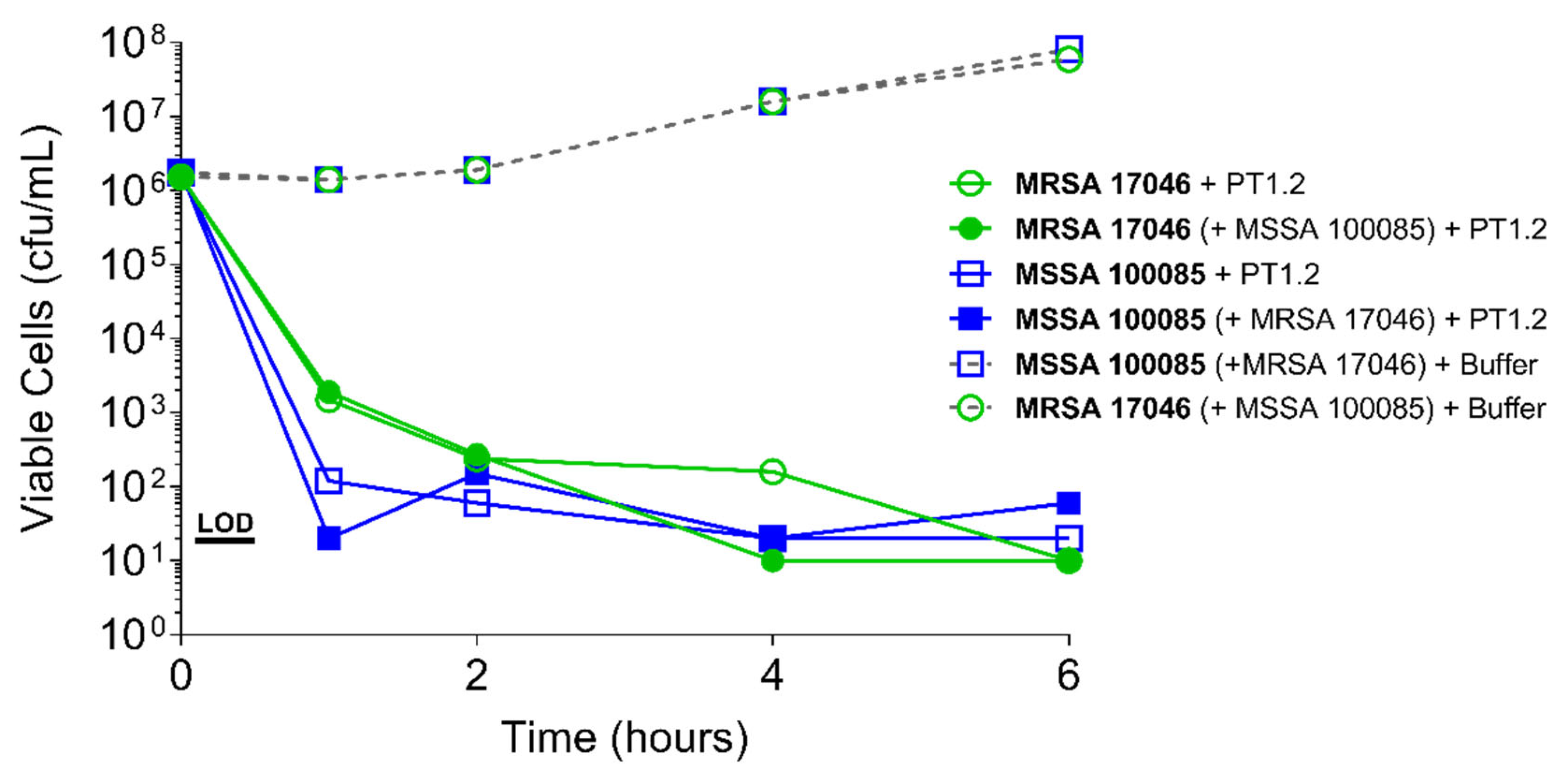
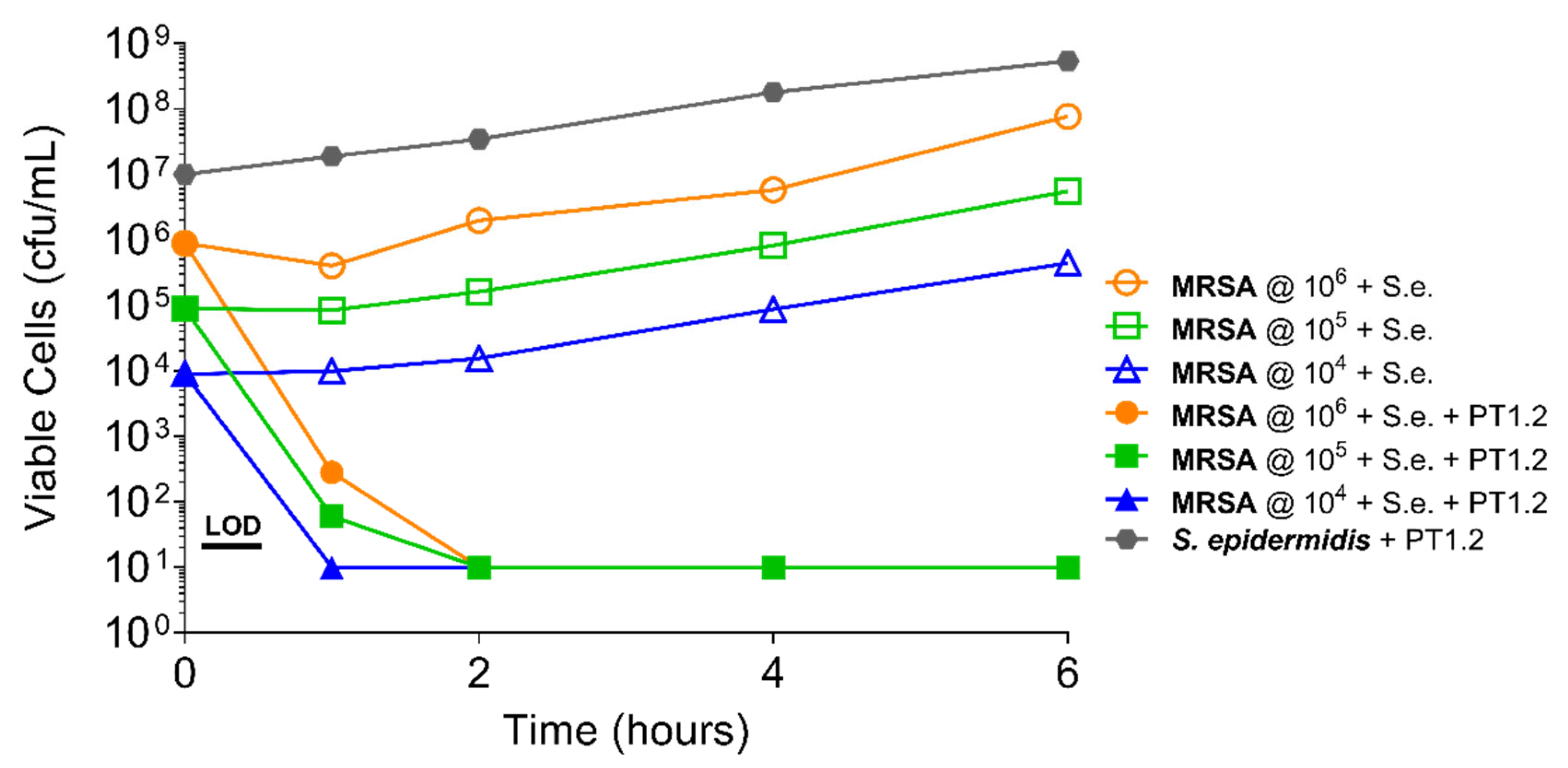
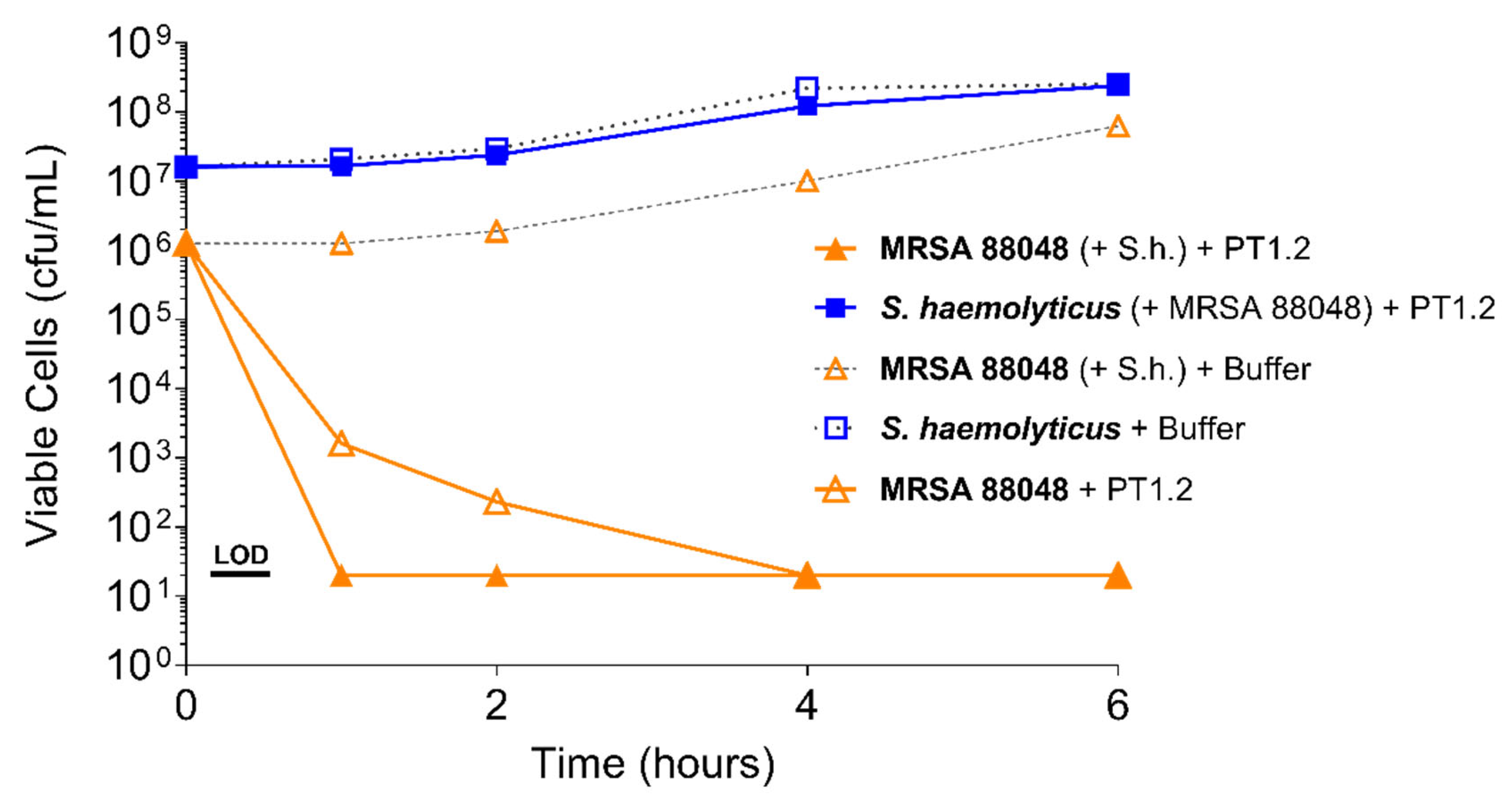
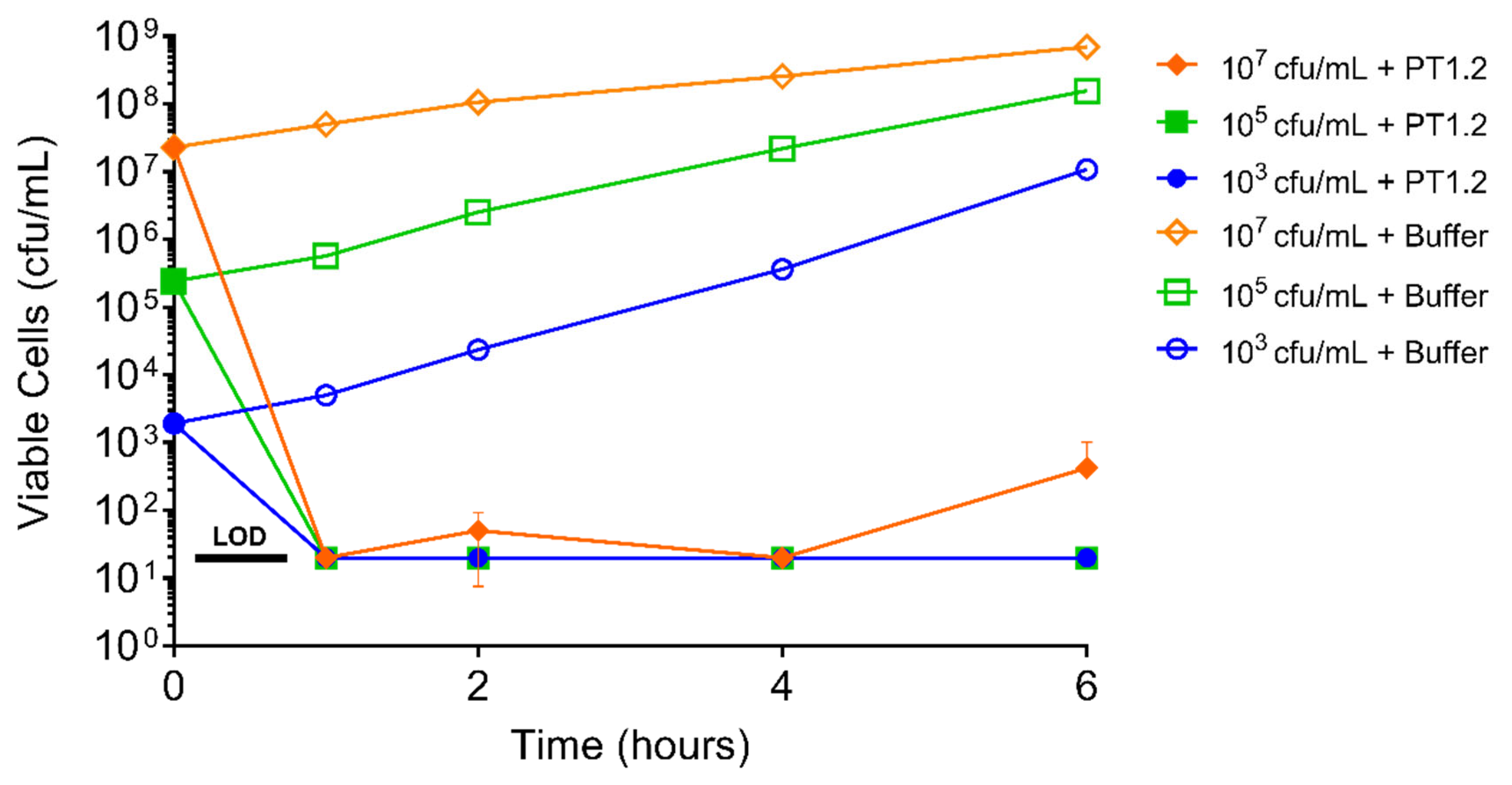
2.4.2. S. aureus Concentration
2.5. Effect of Human Serum Albumin on PT1.2 Activity
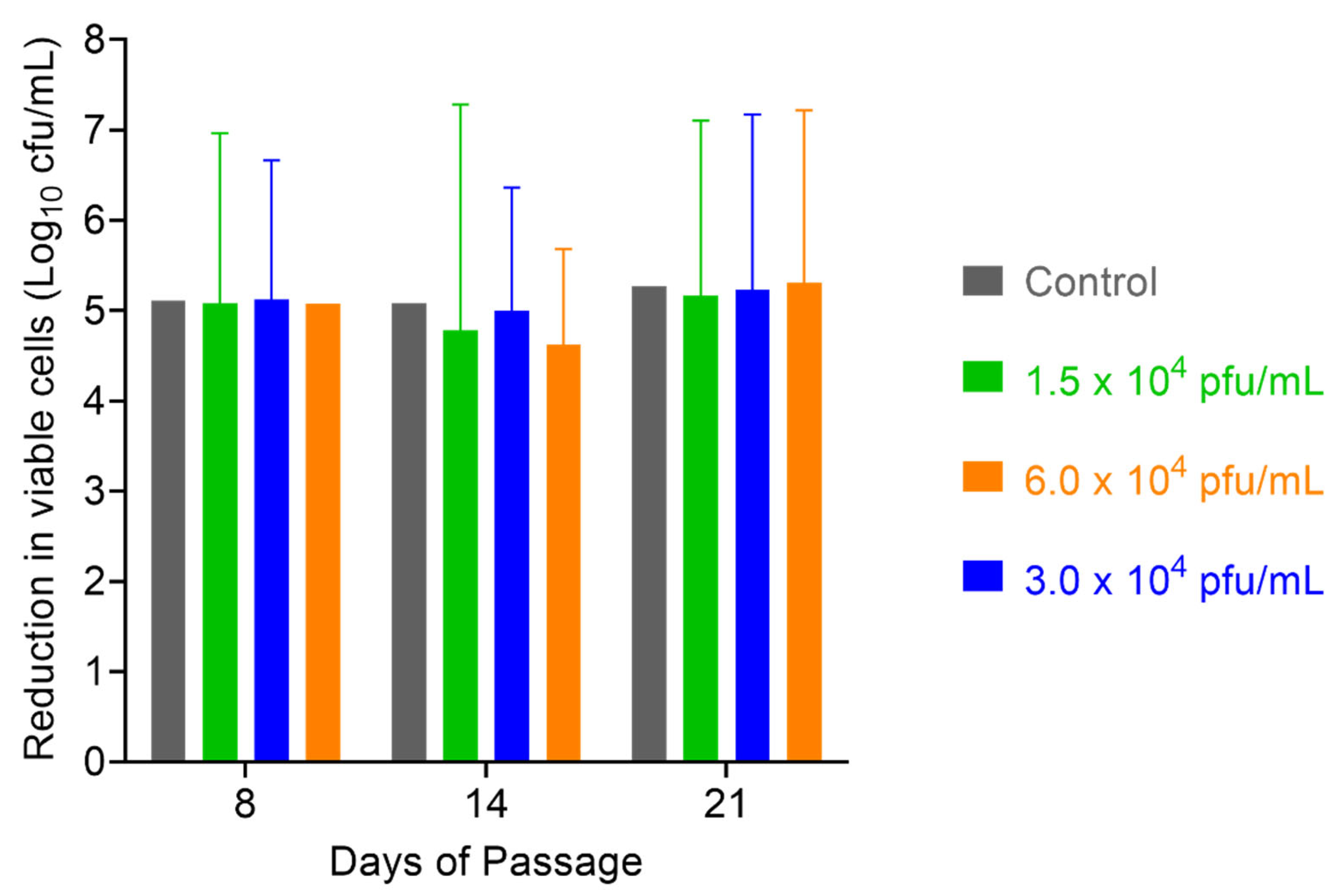
2.6. Assessment of S. aureus Resistance to PT1.2
3. Discussion
4. Materials and Methods
4.1. The Strains, Media and Chemicals
4.2. Spectrum of Activity of PT1.2
4.2.1. 3 h Kill Assay
4.2.2. Time-Kill Curves
4.3. Specificity of PT1.2
4.3.1. Activity of PT1.2 against Coagulase Negative Staphylococci
4.3.2. Activity of PT1.2 in Mixed Bacterial Cultures
4.4. Effect of PT1.2 on Bacterial Viability
4.4.1. Speed of Kill
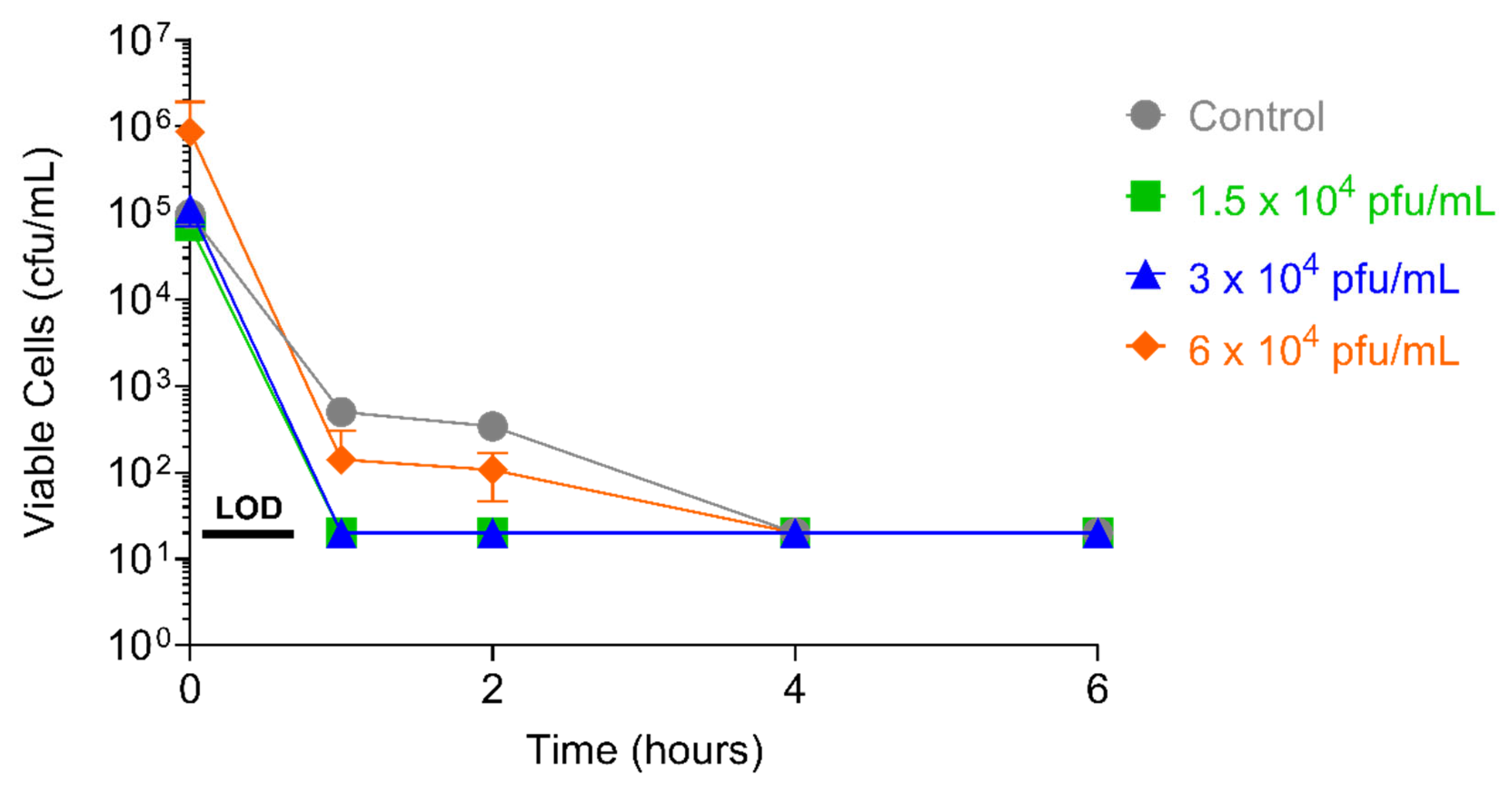
4.4.2. Concentration of PT1.2
4.4.3. Concentration of Bacterial Culture
4.5. Effect of Human Serum Albumin (HSA) on PT1.2 Activity
4.6. Assessment of S. aureus Resistance to SASPject PT1.2
4.6.1. Passaging Studies
4.6.2. Single Dosing Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dheman, N.; Mahoney, N.; Cox, E.M.; Farley, J.F.; Amini, T.; Lanthier, M.L. An Analysis of Antibacterial Drug Development Trends in the United States, 1980–2019. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. Antibiotics alter the gut microbiome and host health. Nat. Res. 2019, 15, S19. [Google Scholar]
- Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 225–280. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef]
- Brüssow, H.; Kutter, E. Phage Ecology. In Bacteriophages: Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2005; Volume 70, pp. 129–163. [Google Scholar]
- Chanishvili, N. Phage Therapy—History from Twort and d’Herelle through Soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [PubMed]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.R.; Oliveira, H.; Pires, D.P.; Dąbrowska, K.; Azeredo, J. Phage therapy efficacy: A review of the last 10 years of preclinical studies. Crit. Rev. Microbiol. 2020, 46, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef]
- Fairhead, H.; Wilkinson, A.; Barnard, A.; Severi, E.; Anderson, N.; Pitts, K. Modifying Bacteriophage. European Patent No. EP3201323, 6 November 2019. [Google Scholar]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, H. SASP gene delivery: A novel antibacterial approach. Drug News Perspect. 2009, 22, 197–203. [Google Scholar] [CrossRef]
- Sanchez-Salas, J.-L.; Santiago-Lara, M.L.; Setlow, B.; Sussman, M.D.; Setlow, P. Properties of Bacillus megaterium and Bacillus subtilis Mutants Which Lack the Protease That Degrades Small, Acid-Soluble Proteins during Spore Germination. J. Bacteriol. 1992, 174, 807–814. [Google Scholar] [CrossRef]
- Fairhead, H.; Setlow, B.; Setlow, P. Prevention of DNA damage in spores and in vitro by small, acid-soluble proteins from Bacillus species. J. Bacteriol. 1993, 175, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, H.; Setlow, P. Binding of DNA to ac/d-Type Small, Acid-Soluble Proteins from Spores of Bacillus or Clostnidium Species Prevents Formation of Cytosine Dimers, Cytosine-Thymine Dimers, and Bipyrimidine Photoadducts after UV Irradiation. J. Bacteriol. 1992, 174, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Setlow, P. I will survive: DNA protection in bacterial spores. Trends Microbiol. 2007, 15, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Bumbaca, D.; Kosman, J.; Setlow, P.; Jedrzejas, M.J. Structure of a protein–DNA complex essential for DNA protection in spores of Bacillus species. Proc. Natl. Acad. Sci. USA 2008, 105, 2806–2811. [Google Scholar] [CrossRef]
- Setlow, B.; Hand, A.R.; Setlow, P. Synthesis of a Bacillus subtilis small, acid-soluble spore protein in Escherichia coli causes cell DNA to assume some characteristics of spore DNA. J. Bacteriol. 1991, 173, 1642–1653. [Google Scholar] [CrossRef]
- Hayes, C.S.; Setlow, P. An alpha/beta-type small, acid-soluble spore protein which has a very high affinity for DNA prevents outgrowth of Bacillus subtilis spores. J. Bacteriol. 2001, 183, 3982–3990. [Google Scholar] [CrossRef]
- Hatzixanthis, K.; Wilkinson, A.; Fairhead, H. Double-Blind, Placebo-Controlled Phase I study of PT1.2, a Novel Antibacterial Protein (SASP) delivery vector. In Proceedings of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Boston, MA, USA, 12–15 September 2010. Poster F1-2086b. [Google Scholar]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Jenks, P.J.; Laurent, M.; McQuarry, S.; Watkins, R. Clinical and economic burden of surgical site infection (SSI) and predicted financial consequences of elimination of SSI from an English hospital. J. Hosp. Infect. 2014, 86, 24–33. [Google Scholar] [CrossRef]
- NICE Guideline Updates Team (UK). Evidence Review for Effectiveness of Nasal Decolonisation in Prevention of Surgical Site Infection: Surgical Site Infections: Prevention and Treatment; NICE guideline NG125; National Institute for Health and Care Excellence: London, UK, 2019. [Google Scholar]
- Dadashi, M.; Hajikhani, B.; Darban-Sarokhalil, D.; van Belkum, A.; Goudarzi, M. Mupirocin resistance in Staphylococcus aureus: A systematic review and meta-analysis. J. Glob. Antimicrob. Resist. 2020, 20, 238–247. [Google Scholar] [CrossRef]
- Bisognano, C.; Vaudaux, P.; Rohner, P.; Lew, D.P.; Hooper, D.C. Induction of fibronectin-binding proteins and increased adhesion of quinolone-resistant Staphylococcus aureus by subinhibitory levels of ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Harbarth, S.; Liassine, N.; Dharan, S.; Herrault, P.; Auckenthaler, R.; Pittet, D. Risk Factors for Persistent Carriage of Methicillin-Resistant Staphylococcus aureus. Clin. Infect. Dis. 2000, 31, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Pitts, K.; Brown, D.F.; Barnard, A.; Wilkinson, A.; Fairhead, H. SASP: Rapid Bactericidal Activity against MRSA and Stationary Phase Staphylococcus aureus. In Proceedings of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Poster F1-2131. [Google Scholar]
- Bowker, K.E.; Noel, A.R.; MacGowan, A.P.; Pitts, K.; Wilkinson, A.; Fairhead, H. SASP: Kill Kinetics against Diverse Antibiotic Resistant Staphylococcus aureus. Proceeding of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Poster F1-2131. [Google Scholar]
- Barnard, A.; Pitts, K.; Brown, D.F.; Wilkinson, A.; Fairhead, H. SASP: Rapid Bactericidal Activity against USA strains of Methicillin Resistant Staphylococcus aureus (MRSA). In Proceedings of the European Congress on Clinical Microbiology and Infectious Diseases, Barcelona, Spain, 19–22 April 2008. Poster P-561. [Google Scholar]
- Pitts, K.; Barnard, A.; Brown, D.F.J.; Wilkinson, A.; Fairhead, H. The Efficacy of SASP Targeted to Methicillin Resistant Staphylococcus aureus (MRSA) in Mixed Staphylococcal Cultures. In Proceedings of the European Congress on Clinical Microbiology and Infectious Diseases, Barcelona, Spain, 19–22 April 2008. Poster P-561. [Google Scholar]
- National Committee for Clinical Laboratory Standards; Barry, A.L. Methods for Determining Bactericidal Activity of Antimicrobial Agents: Approved Guideline; M26-A. CLSI; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2009; Volume 19. [Google Scholar]
- Loshon, C.A.; Setlow, P. Bacillus megaterium spore protease: Purification, radioimmunoassay, and analysis of antigen level and localization during growth, sporulation and spore germination. J. Bacteriol. 1982, 150, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Loshon, C.A.; Setlow, B.M.; Setlow, P. Bacillus megaterium spore protease: Synthesis and processing of precursor forms during sporulation and germination. J. Biol. Chem. 1982, 257, 10838–10845. [Google Scholar] [CrossRef]
- Setlow, P. Small acid-soluble, spore proteins of Bacillus species: Structure, synthesis, genetics, function and degradation. Annu. Rev. Microbiol. 1988, 42, 319–338. [Google Scholar] [CrossRef]
- Illades-Aguiar, B.; Setlow, P. Autoprocessing of the protease that degrades small, acid-soluble proteins of spores of Bacillus species is triggered by low pH, dehydration, and dipicolinic acid. J. Bacteriol. 1994, 176, 7032–7037. [Google Scholar] [CrossRef]
- Verhoef, J.; van Boven, C.P.A.; Holtrigter, B. Host controlled modification and restriction of phages in coagulase negative staphylococci. J. Gen. Microbiol. 1972, 71, 231–239. [Google Scholar] [CrossRef]
- Coyette, J.; Ghuysen, J.M. Structure of the cell wall of Staphylococcus aureus, strain Copenhagen. IX. Teichoic acid and phage adsorption. Biochemistry 1968, 7, 2385–2389. [Google Scholar] [CrossRef]
- Holme, S.; Math, V.; Pitts, K.; Barnard, A.; Wang, H.; Wilkinson, A.; Fairhead, H. Low Propensity for S. aureus to Develop Resistance to SASP or its Delivery Vector. In Proceedings of the Interscience Conference Antimicrobe Agents Chemotherapy, Washington, DC, USA, 25–28 October 2008. Poster F1-4001. [Google Scholar]
- Duplessis, M.; Moineau, S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 2001, 41, 325–336. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Pluckthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, H.; Wilkinson, A. Modified bacteriophage. European Patent No. EP3340200, 24 February 2021. [Google Scholar]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- McCallin, S.; Sacher, J.C.; Zheng, J.; Chan, B.K. Current State of Compassionate Phage Therapy. Viruses 2019, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails to Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Character | Detail (Total No. of Isolates, n = 225) (Total No. of Isolates in the 3 h (3 h) or Time-Kill (tk) Assays) | No. of Isolates | |||||
|---|---|---|---|---|---|---|---|
| 3 h Kill Assay log10 Drop at 3 h | Time-Kill Curves log10 Drop at 4 h | ||||||
| ≥2 | ≥3 | ≥4 | ≥2 | ≥3 | ≥4 | ||
| mecA | mecA− (38) (3 h = 34; tk = 4) | 26 | 26 | 23 | 3 | 3 | 2 |
| mecA+ (187) (3 h = 163; tk = 24) | 158 | 156 | 153 | 22 | 21 | 15 | |
| Panton Valentine Leukocidin (PVL) | PVL− (3 h = 10) | 10 | 10 | 10 | |||
| PVL+ (3 h = 12) | 12 | 12 | 12 | ||||
| SCCmec | I (3 h = 22) | 21 | 21 | 20 | |||
| II (3 h = 31) | 31 | 31 | 31 | ||||
| III (3 h = 23) | 23 | 23 | 23 | ||||
| IV (3 h = 38) | 37 | 36 | 36 | ||||
| V (3 h = 1) | 1 | 1 | 1 | ||||
| Untypeable (3 h = 3) | 3 | 3 | 3 | ||||
| Sequence type | 1 (3 h = 3) | 3 | 3 | 3 | |||
| 22 (3 h = 15) | 15 | 15 | 15 | ||||
| 30 (3 h = 1) | 1 | 1 | 1 | ||||
| 36 (3 h = 12) | 12 | 12 | 12 | ||||
| 80 (3 h = 2) | 2 | 2 | 2 | ||||
| 239 (3 h = 6) | 6 | 6 | 6 | ||||
| 240 (3 h = 2) | 2 | 2 | 2 | ||||
| 247 (3 h = 4) | 4 | 4 | 4 | ||||
| 5 (3 h = 6) | 6 | 5 | 5 | ||||
| 8 (3 h = 16) | 16 | 15 | 15 | ||||
| USA type | ORSA-I (3 h = 7) | 7 | 7 | 7 | |||
| ORSA-II (3 h = 13) | 13 | 12 | 12 | ||||
| ORSA-III (3 h = 7) | 7 | 7 | 7 | ||||
| ORSA-IV (3 h = 14) | 14 | 14 | 14 | ||||
| ORSA-I/III/IV (3 h = 11) | 11 | 10 | 10 | ||||
| USA-100 (3 h = 1; tk = 1) | 1 | 0 | 0 | 1 | 1 | 1 | |
| USA-200 (13), -300 (1), -400 (1), -500 (1), -700 (1), -800 (1), -1000 (1), -1100 (1) (3 h = 20) | 12 | 12 | 12 | ||||
| VISA | VISA (3 h = 3; tk = 3) | 2 | 0 | 0 | 2 | 2 | 0 |
| hVISA | hVISA (3 h = 9; tk = 2) | 9 | 9 | 0 | 1 | 0 | 0 |
| Historic Clones | Archaic/Iberian (3 h = 15) | 15 | 14 | 14 | |||
| Brazilian/Hungarian (3 h = 28) | 28 | 28 | 28 | ||||
| Pediatric (3 h = 6) | 6 | 6 | 6 | ||||
| UK epidemic clones | EMRSA-1 (1), -2 (1), -3 (3), -4 (3), -5 (2), -6 (3), -7 (2), -8 (2), -9 (2), -10 (2), -11 (2), -12 (3) -13 (2), -14 (1), -15 (14), -16 (11), -17 (3) (3 h = 57) | 54 | 54 | 54 | |||
| EMRSA-6 (3 h = 3) | 3 | 3 | 2 | ||||
| Community MRSA | (3 h = 10; tk = 5) | 10 | 10 | 10 | 5 | 5 | 5 |
| Geographic location | Australia (3 h = 6) | 6 | 6 | 6 | |||
| Japan (3 h = 4) | 4 | 3 | 3 | ||||
| USA (3 h = 16) | 15 | 15 | 15 | ||||
| Denmark (3 h = 1) | 1 | 1 | 1 | ||||
| France (3 h = 2) | 2 | 2 | 2 | ||||
| Ireland (3 h = 5) | 5 | 5 | 5 | ||||
| Staphylococcal Species | Cambridge Identity Codes (CC) | Log10 Reduction in Viable Cells |
|---|---|---|
| S. epidermidis | 72003 | 0 |
| 72037 | 0 | |
| 72025 | 0 | |
| 72004 | 0 | |
| 72029 | 0 | |
| 72020 | 0 | |
| 72030 | 0 | |
| S. haemolyticus | 133072 | 0 |
| 133034 | 0 | |
| 133068 | 0 | |
| S. warneri | 133029 | 0 |
| 133019 | 0 | |
| S. hominis | 133075 | 0 |
| 133097 | 0 | |
| S. cohnii | 133089 | 0 |
| 133041 | 0 | |
| S. capitis | 133095 | 0 |
| 133092 | 0 | |
| S. simulans | 133080 | 0 |
| 133053 | 0 | |
| S. lugdunensis | 133074 | 0 |
| 133082 | 0 | |
| S. saprophyticus | 133002 | 0 |
| 133091 | 0 |
| Staphylococcal Species | Cambridge Identity Codes (CC) | Methicillin Sensitivity Profile |
|---|---|---|
| S. epidermidis | 72003 | S |
| 72037 | S | |
| 72025 | S | |
| 72004 | R | |
| 72029 | R | |
| 72020 | R | |
| 72030 | R | |
| S. haemolyticus | 133072 | S |
| 133034 | R | |
| 133068 | R | |
| S. warneri | 133029 | S |
| 133019 | R | |
| S. hominis | 133075 | S |
| 133097 | R | |
| S. cohnii | 133089 | S |
| 133041 | R | |
| S. capitis | 133095 | S |
| 133092 | R | |
| S. simulans | 133080 | S |
| 133053 | R | |
| S. lugdunensis | 133074 | S |
| 133082 | R | |
| S. saprophyticus | 133002 | S |
| 133091 | Unknown |
| Staphylococcal Species | Cambridge Identity Codes (CC) | Methicillin Sensitivity Profile |
|---|---|---|
| S. aureus | 88048 | R |
| 17046 | R | |
| 100085 | S | |
| S. epidermidis | 72020 | R |
| S. haemolyticus | 133034 | R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cass, J.; Barnard, A.; Fairhead, H. Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP. Pharmaceuticals 2021, 14, 1038. https://doi.org/10.3390/ph14101038
Cass J, Barnard A, Fairhead H. Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP. Pharmaceuticals. 2021; 14(10):1038. https://doi.org/10.3390/ph14101038
Chicago/Turabian StyleCass, James, Anne Barnard, and Heather Fairhead. 2021. "Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP" Pharmaceuticals 14, no. 10: 1038. https://doi.org/10.3390/ph14101038
APA StyleCass, J., Barnard, A., & Fairhead, H. (2021). Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP. Pharmaceuticals, 14(10), 1038. https://doi.org/10.3390/ph14101038