Author Contributions
Conceptualization, J.Y., M.H. and A.C.; methodology, J.Y., M.H. and A.C.; software, J.Y.; validation, J.Y., M.H. and A.C.; formal analysis, J.Y., M.H. and A.C.; investigation, J.Y., M.H. and A.C.; resources, J.Y., M.H. and A.C.; data curation, J.Y., M.H. and A.C.; writing—original draft preparation, J.Y.; writing—review and editing, J.Y., M.H. and A.C.; visualization, J.Y.; supervision, M.H. and A.C.; project administration, A.C.; funding acquisition, A.C. All authors have read and agreed to the published version of the manuscript.
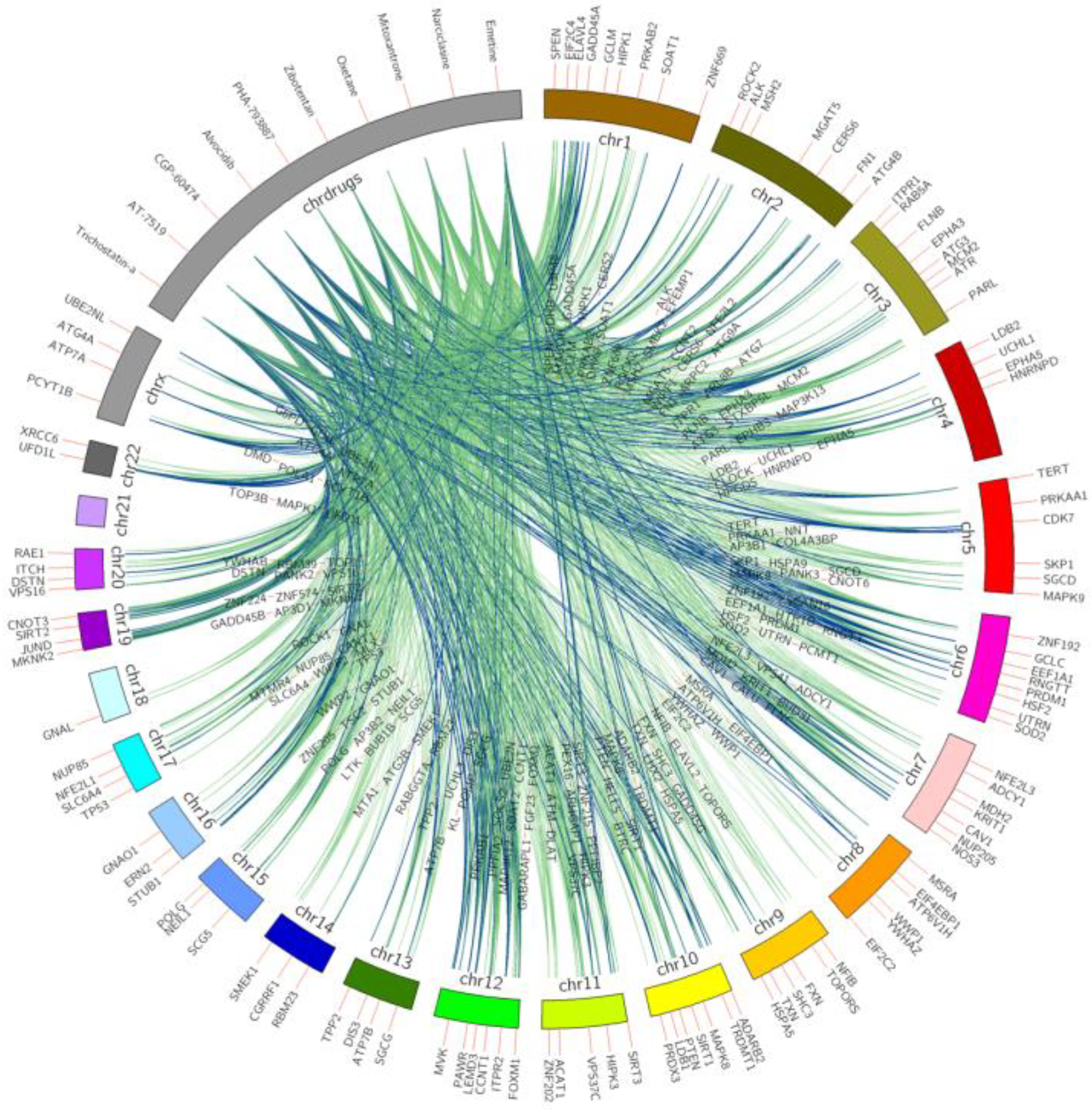
Figure 1.
Top 10 small molecules that upregulate pro-longevity genes across all LINCS cell lines. Pro-longevity genes are shown on chromosomes bands, repurposed chemicals are shown on the drug band. Colors of interactions indicate the relationship occurrence on different cell lines. Green D–G (pro-longevity genes) links indicate the interactions were captured in less than six cell lines; pairs found in above five cell lines are labeled in blue.
Figure 1.
Top 10 small molecules that upregulate pro-longevity genes across all LINCS cell lines. Pro-longevity genes are shown on chromosomes bands, repurposed chemicals are shown on the drug band. Colors of interactions indicate the relationship occurrence on different cell lines. Green D–G (pro-longevity genes) links indicate the interactions were captured in less than six cell lines; pairs found in above five cell lines are labeled in blue.
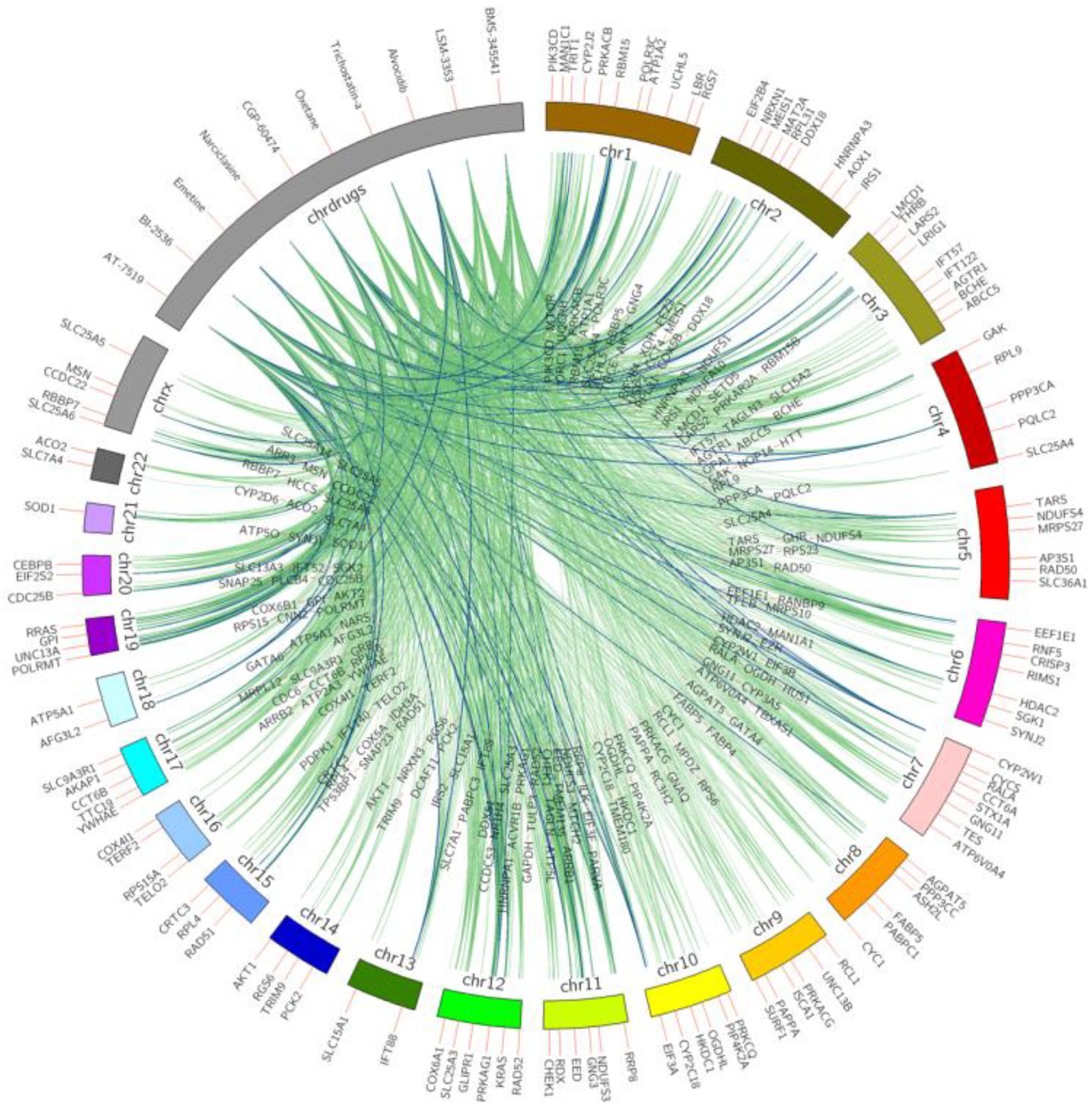
Figure 2.
Top 10 small molecules that downregulate anti-longevity genes across all LINCS cell lines. Anti-longevity genes are shown on chromosomes bands, repurposed chemicals are shown on the drug band. Colors of interactions indicate the relationship occurrence on different cell lines. Green D–G (anti-longevity genes) links indicate the interactions were captured in less than six cell lines; pairs found in above five are labeled in blue.
Figure 2.
Top 10 small molecules that downregulate anti-longevity genes across all LINCS cell lines. Anti-longevity genes are shown on chromosomes bands, repurposed chemicals are shown on the drug band. Colors of interactions indicate the relationship occurrence on different cell lines. Green D–G (anti-longevity genes) links indicate the interactions were captured in less than six cell lines; pairs found in above five are labeled in blue.
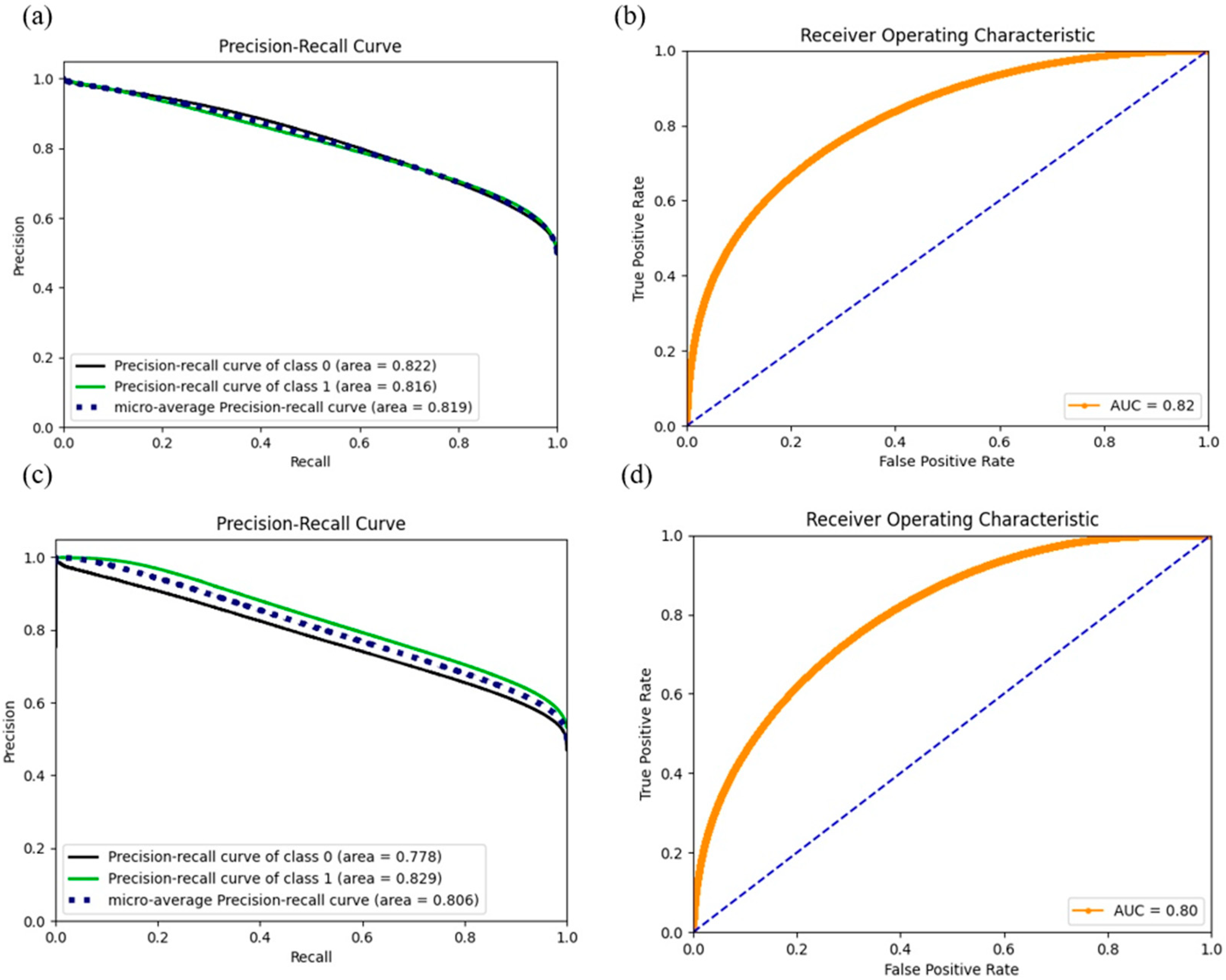
Figure 3.
ROC and precision curves for model 1 ((a): PRC, (b): ROC) and model 2 ((c): PRC, (d): ROC). While the AUC score dropped compared with models 3–8, the dramatic increase in the APR score (positive class) gained confidence in predicting the positives in further exploration.
Figure 3.
ROC and precision curves for model 1 ((a): PRC, (b): ROC) and model 2 ((c): PRC, (d): ROC). While the AUC score dropped compared with models 3–8, the dramatic increase in the APR score (positive class) gained confidence in predicting the positives in further exploration.
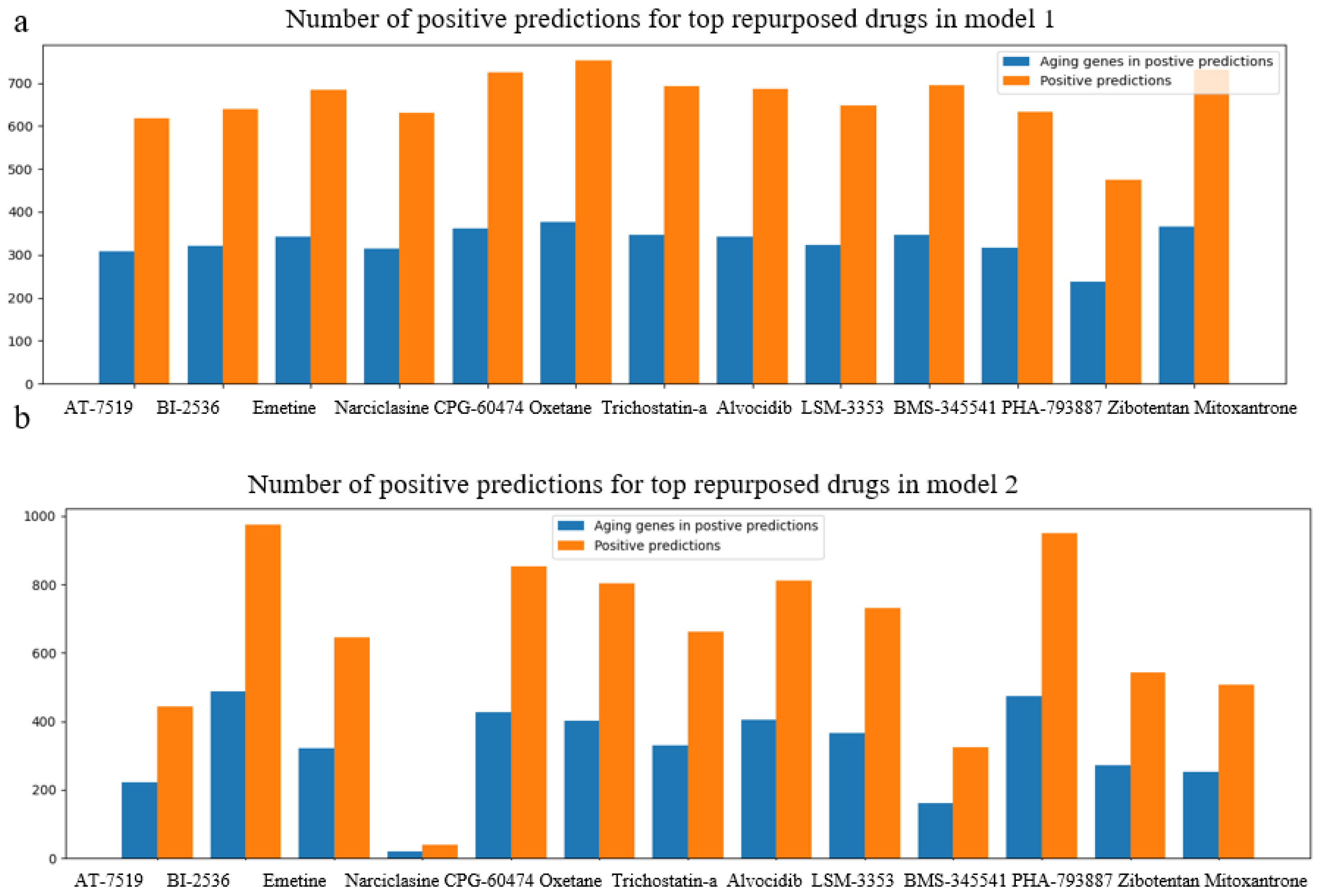
Figure 4.
Bar charts on positive predictions for repurposed drugs in CMAP LINCS dataset, for normal cell lines, NHBEC and HGEC6B, using model 1(a) and model 2(b). Orange bars demonstrate the total number of positive predictions for each drug candidate, and blue bars illustrate the number of unique aging genes among positive predictions.
Figure 4.
Bar charts on positive predictions for repurposed drugs in CMAP LINCS dataset, for normal cell lines, NHBEC and HGEC6B, using model 1(a) and model 2(b). Orange bars demonstrate the total number of positive predictions for each drug candidate, and blue bars illustrate the number of unique aging genes among positive predictions.
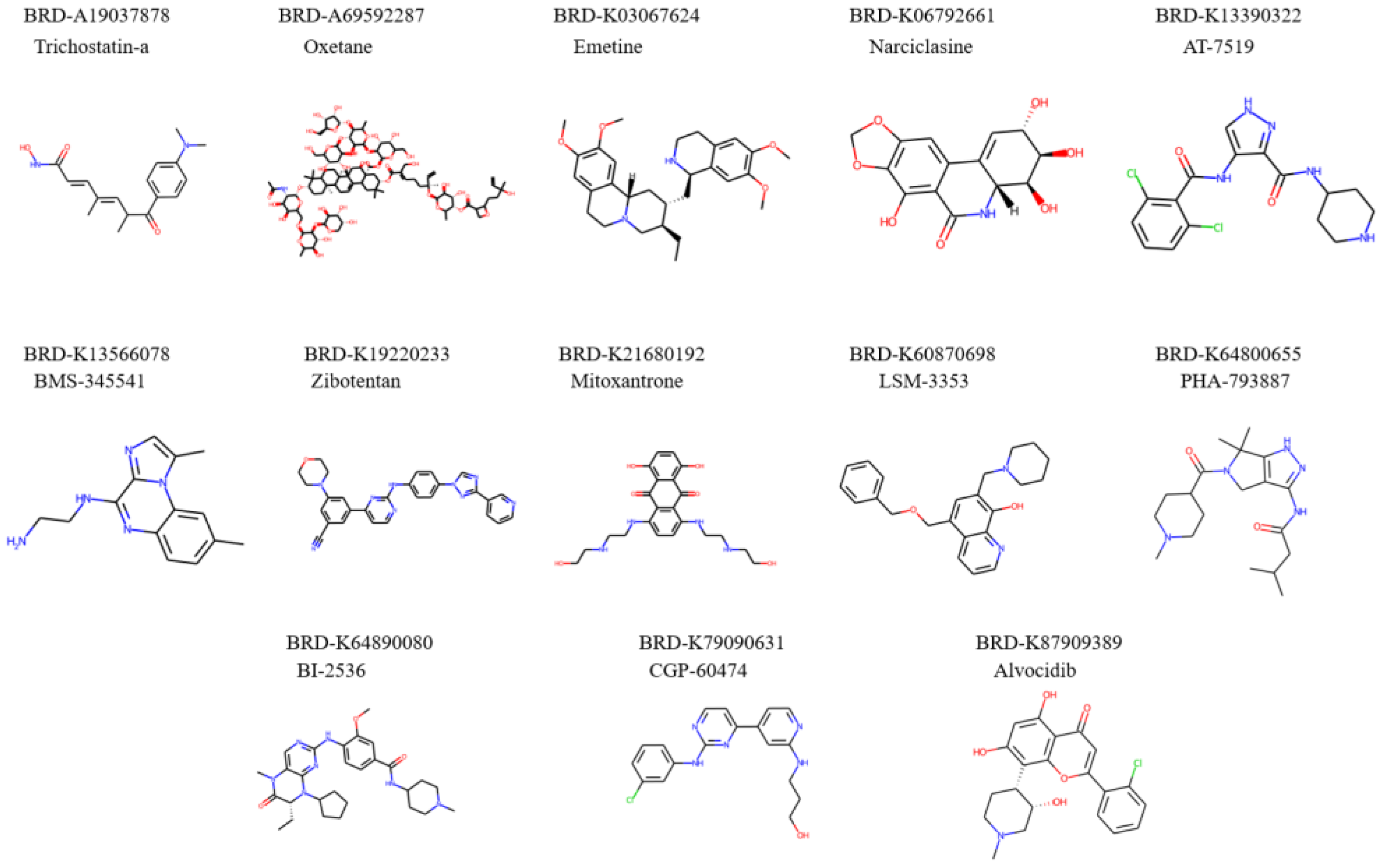
Figure 5.
Molecular structures of repurposed drug candidates for longevity purpose.
Figure 5.
Molecular structures of repurposed drug candidates for longevity purpose.
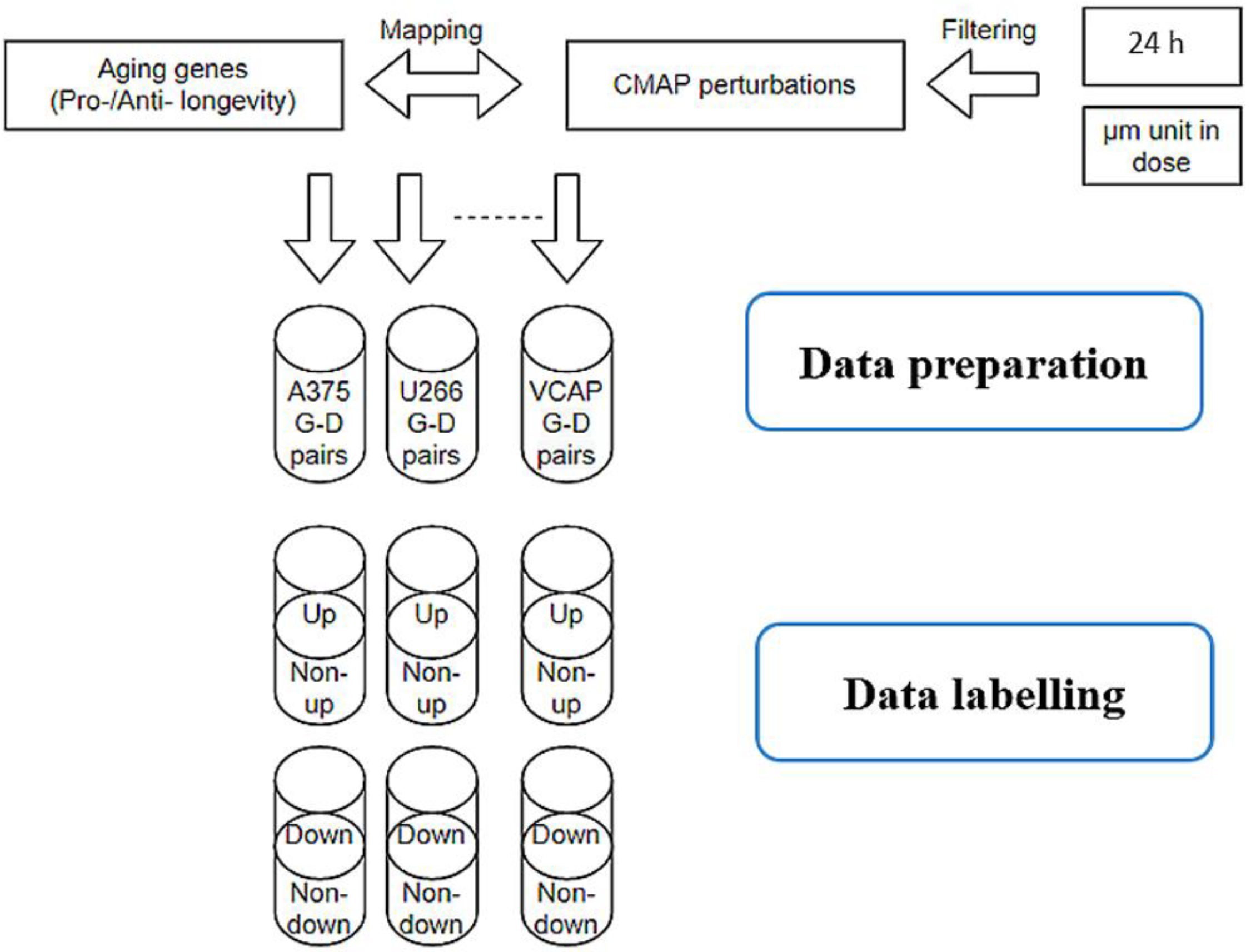
Figure 6.
Data pipeline in collecting and labelling LINCS perturbations. Only perturbations include aging genes were kept for further analysis. The left–right percentile method was applied to label upregulation and downregulation effects with 5% threshold on the Z-score for each cell line.
Figure 6.
Data pipeline in collecting and labelling LINCS perturbations. Only perturbations include aging genes were kept for further analysis. The left–right percentile method was applied to label upregulation and downregulation effects with 5% threshold on the Z-score for each cell line.
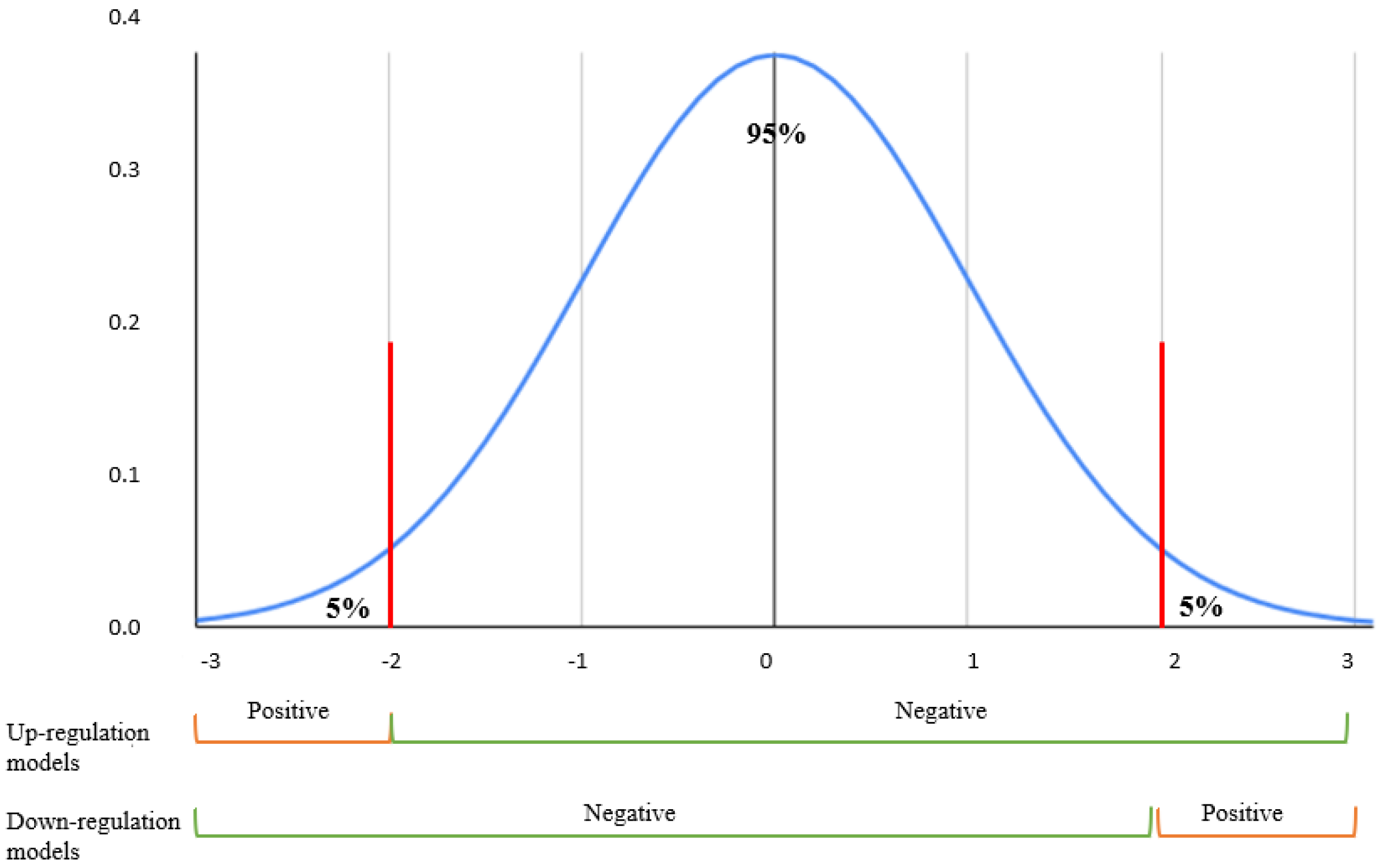
Figure 7.
Z-score normal distribution. Positive samples were selected from the top 5% gene expressions perturbations for each cell lines in models predicting up/non-upregulation effects. Similarly, the bottom 5% perturbations were identified as the positives in models predicting down/non-downregulation effects.
Figure 7.
Z-score normal distribution. Positive samples were selected from the top 5% gene expressions perturbations for each cell lines in models predicting up/non-upregulation effects. Similarly, the bottom 5% perturbations were identified as the positives in models predicting down/non-downregulation effects.
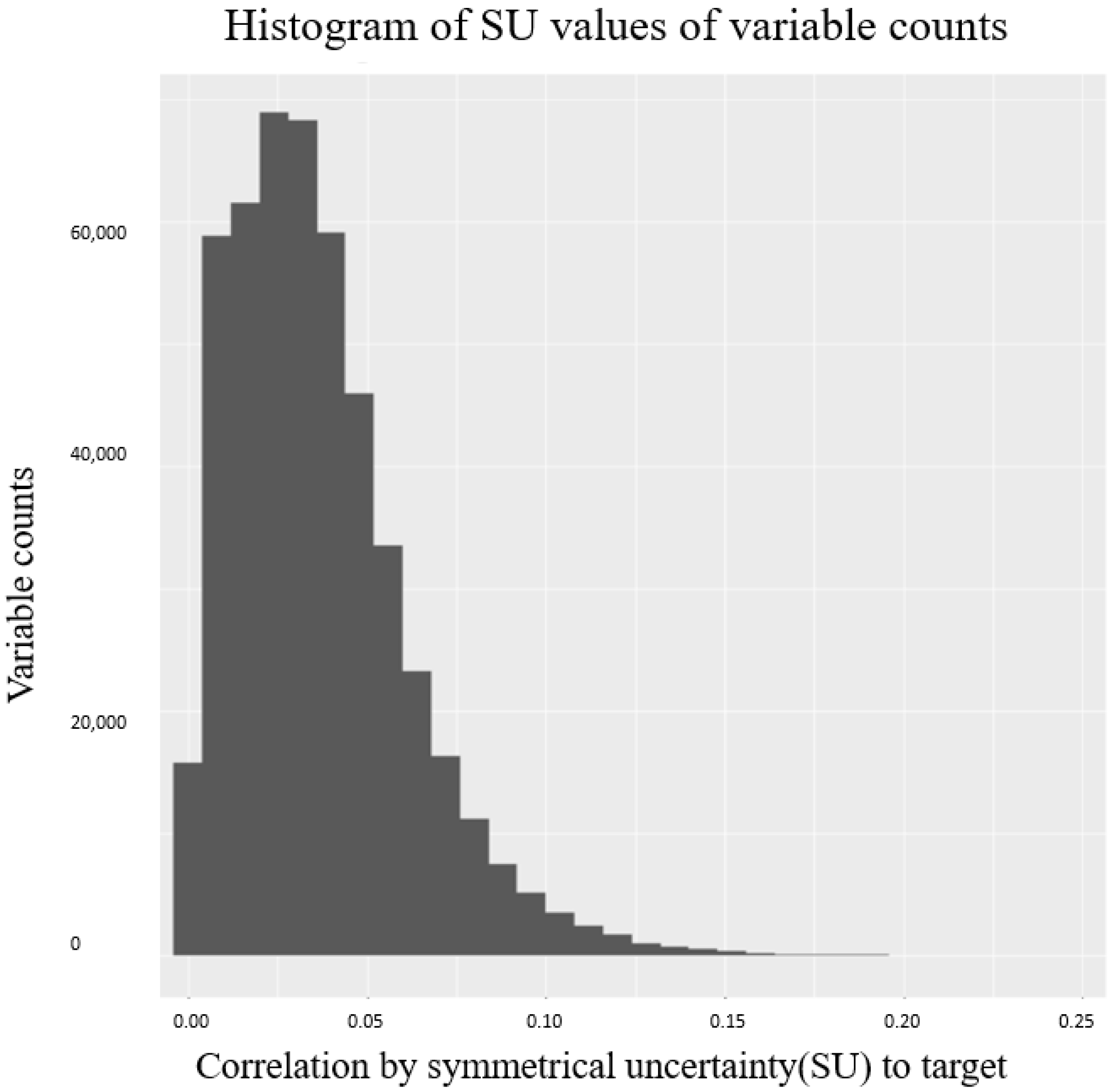
Figure 8.
Histogram of correlation values and the corresponding variable counts using FCBF. Correlations were calculated between CpG sites and cancer histology categories.
Figure 8.
Histogram of correlation values and the corresponding variable counts using FCBF. Correlations were calculated between CpG sites and cancer histology categories.
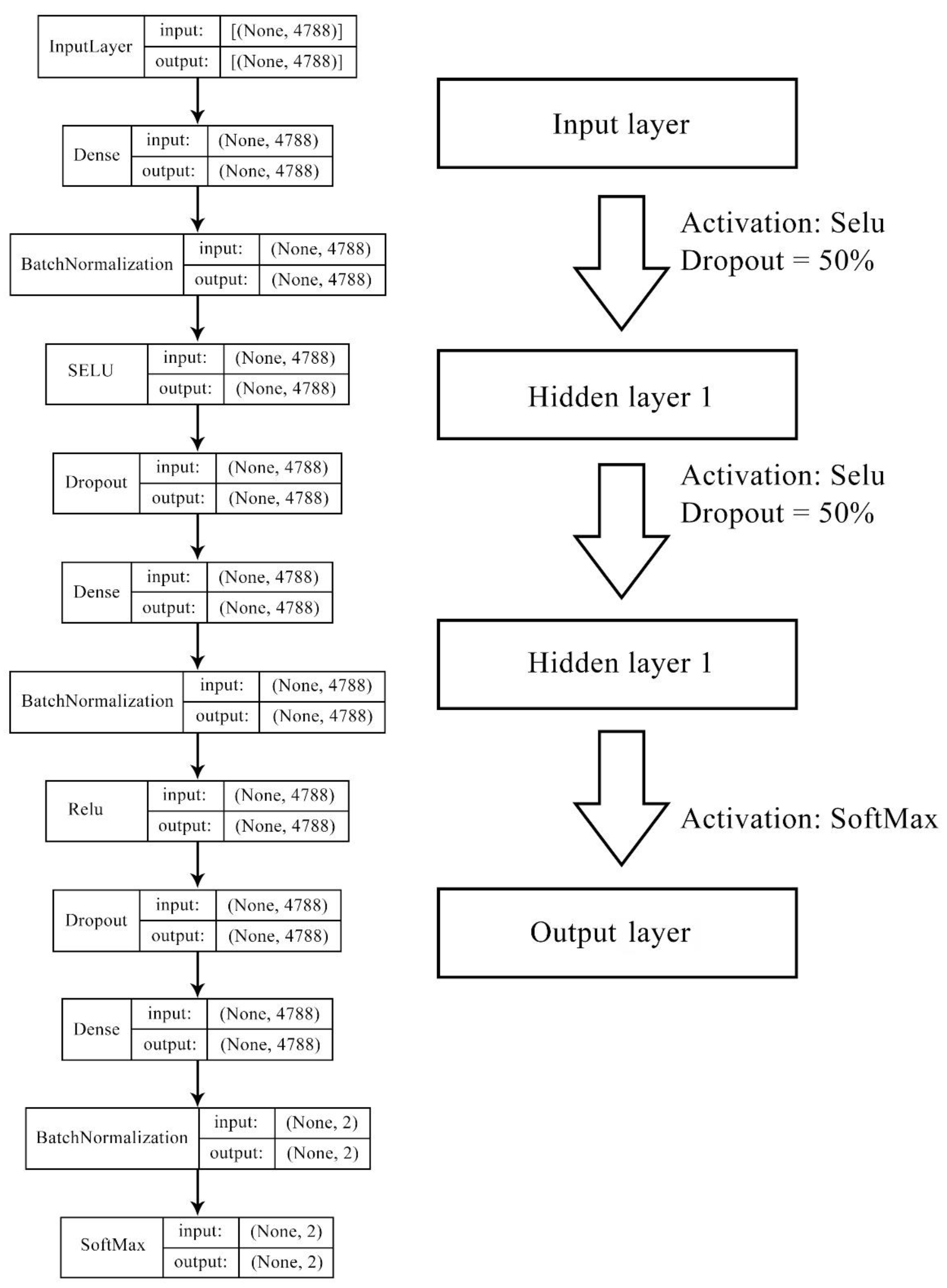
Figure 9.
Deep neural network structure for model 1 and model 2. Features of models 1–2 were contributed by gene ontology terms (946 bits), molecular fingerprints (2048 bits), cell line mutation status (735 bits), and cell line methylation beta level (1183 bits), forming the length of 4788 bits in total.
Figure 9.
Deep neural network structure for model 1 and model 2. Features of models 1–2 were contributed by gene ontology terms (946 bits), molecular fingerprints (2048 bits), cell line mutation status (735 bits), and cell line methylation beta level (1183 bits), forming the length of 4788 bits in total.
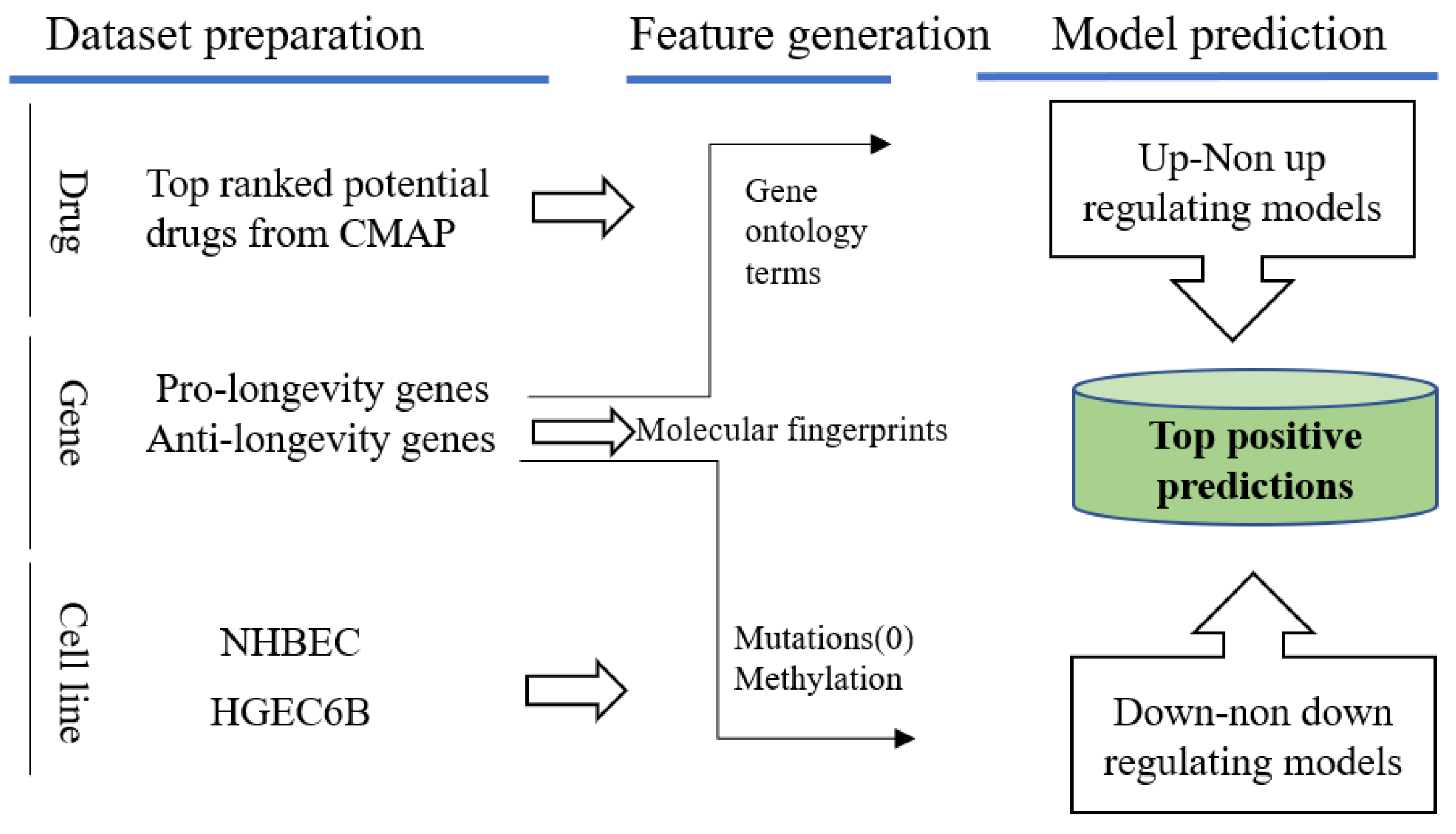
Figure 10.
Predictions on normal cell lines with aging related genes and promising drug candidates queried from LINCS. Positive predictions will be of interest with desired regulating effects towards pro-/anti-longevity genes.
Figure 10.
Predictions on normal cell lines with aging related genes and promising drug candidates queried from LINCS. Positive predictions will be of interest with desired regulating effects towards pro-/anti-longevity genes.
Table 1.
Sample distribution for each cell line in upregulated models; positive samples are defined as the top 5% gene signatures, while negative samples are the remaining 95%. Unique numbers of drugs and aging genes are summarized below. For example, cell line A375 contains 73,610 unique D–G–Cs as positive samples with the top 5% threshold, while the remaining 1.2 million D–G–Cs with unknown regulating effects form the negatives. (#: Counts.)
Table 1.
Sample distribution for each cell line in upregulated models; positive samples are defined as the top 5% gene signatures, while negative samples are the remaining 95%. Unique numbers of drugs and aging genes are summarized below. For example, cell line A375 contains 73,610 unique D–G–Cs as positive samples with the top 5% threshold, while the remaining 1.2 million D–G–Cs with unknown regulating effects form the negatives. (#: Counts.)
Table 2.
Top-ranked 10 small molecules that upregulate the most pro-longevity genes across all cell lines in LINCS. (#: Counts.)
Table 2.
Top-ranked 10 small molecules that upregulate the most pro-longevity genes across all cell lines in LINCS. (#: Counts.)
| Rank | Drug | # Interactions | # Unique Pro-Longevity Genes | # Unique Cell Lines |
|---|
| 1 | Trichostatin-a | 926 | 218 | 10 |
| 2 | AT 7519 | 871 | 226 | 7 |
| 3 | CGP-60474 | 843 | 200 | 6 |
| 4 | Alvocidib | 743 | 218 | 6 |
| 5 | PHA-793887 | 730 | 244 | 7 |
| 6 | Emetine | 714 | 225 | 6 |
| 7 | Narciclasine | 711 | 239 | 6 |
| 8 | Zibotentan | 700 | 218 | 7 |
| 9 | Oxetane | 673 | 214 | 6 |
| 10 | Mitoxantrone | 632 | 201 | 6 |
Table 3.
Top-ranked 10 small molecules that downregulate the most anti-longevity genes across all cell lines in LINCS. (#: Counts.)
Table 3.
Top-ranked 10 small molecules that downregulate the most anti-longevity genes across all cell lines in LINCS. (#: Counts.)
| Rank | Drug | # Interactions | # Unique Anti-Longevity Genes | # Unique Cell Lines |
|---|
| 1 | AT-7519 | 585 | 201 | 7 |
| 2 | BI-2536 | 536 | 207 | 9 |
| 3 | Emetine | 520 | 205 | 6 |
| 4 | Narciclasine | 510 | 212 | 6 |
| 5 | CGP-60474 | 497 | 177 | 6 |
| 6 | Oxetane | 494 | 177 | 7 |
| 7 | Trichostatin-a | 454 | 198 | 6 |
| 8 | Alvocidib | 445 | 174 | 6 |
| 9 | LSM-3353 | 433 | 215 | 6 |
| 10 | BMS-345541 | 426 | 176 | 6 |
Table 4.
Model performance on overall accuracy, Area under the ROC curve (AUC), and area under the precision-recall curve (PRC) for the positive class on deep neural network(DNN), random forest, naïve bayes and logistic regression models.
Table 4.
Model performance on overall accuracy, Area under the ROC curve (AUC), and area under the precision-recall curve (PRC) for the positive class on deep neural network(DNN), random forest, naïve bayes and logistic regression models.
| Model | Evaluation | DNN | Random Forest | Naïve Bayes | Logistic Regression |
|---|
| 1 | Accuracy | 0.73 | 0.71 | 0.54 | 0.62 |
| AUC | 0.82 | 0.71 | 0.54 | 0.62 |
| APR | 0.82 | 0.78 | 0.55 | 0.68 |
| 2 | Accuracy | 0.73 | 0.66 | 0.54 | 0.59 |
| AUC | 0.8 | 0.66 | 0.54 | 0.59 |
| APR | 0.78 | 0.71 | 0.54 | 0.64 |
| 3 | Accuracy | 0.95 | 0.94 | 0.6 | 0.95 |
| AUC | 0.89 | 0.64 | 0.76 | 0.63 |
| APR | 0.46 | 0.39 | 0.12 | 0.44 |
| 4 | Accuracy | 0.95 | 0.95 | 0.59 | 0.95 |
| AUC | 0.84 | 0.58 | 0.74 | 0.56 |
| APR | 0.4 | 0.29 | 0.1 | 0.33 |
| 5 | Accuracy | 0.95 | 0.94 | 0.68 | 0.95 |
| AUC | 0.85 | 0.64 | 0.78 | 0.65 |
| APR | 0.46 | 0.4 | 0.14 | 0.46 |
| 6 | Accuracy | 0.95 | 0.95 | 0.54 | 0.95 |
| AUC | 0.81 | 0.58 | 0.72 | 0.57 |
| APR | 0.39 | 0.29 | 0.09 | 0.35 |
| 7 | Accuracy | 0.95 | 0.95 | 0.58 | 0.95 |
| AUC | 0.88 | 0.64 | 0.75 | 0.64 |
| APR | 0.46 | 0.4 | 0.11 | 0.45 |
| 8 | Accuracy | 0.95 | 0.95 | 0.58 | 0.95 |
| AUC | 0.84 | 0.58 | 0.74 | 0.56 |
| APR | 0.4 | 0.3 | 0.1 | 0.54 |
Table 5.
Percentage of positively predicted D (drug candidates)–G (aging genes)–C (normal cell line) pairs for each promising drug candidate in model 1 and model 2. Highlighted repurposed drugs showed great potential in regulating aging gene expressions on normal cell lines in both models. Drugs in bold achieved high positive rate (above 80%) on both models.
Table 5.
Percentage of positively predicted D (drug candidates)–G (aging genes)–C (normal cell line) pairs for each promising drug candidate in model 1 and model 2. Highlighted repurposed drugs showed great potential in regulating aging gene expressions on normal cell lines in both models. Drugs in bold achieved high positive rate (above 80%) on both models.
| Drug | Model 1 Positive Rate | Model 2 Positive Rate |
|---|
| AT 7519 | 78% | 45% |
| BI-2536 | 81% | 100% |
| Emetine | 87% | 66% |
| Narciclasine | 80% | 4% |
| CGP-60474 | 92% | 87% |
| Oxetane | 95% | 82% |
| Trichostatin-a | 88% | 68% |
| Alvocidib | 87% | 83% |
| LSM-3353 | 82% | 75% |
| BMS-345541 | 88% | 33% |
| PHA-793887 | 80% | 97% |
| Zibotentan | 60% | 56% |
| Mitoxantrone | 92% | 52% |
Table 6.
Summary of previous research findings for repurposed pro-longevity drugs.
Table 6.
Summary of previous research findings for repurposed pro-longevity drugs.
| Repurposed Drug | Traits | Evidence |
|---|
| AT 7519 | Inhibitor of CDKs | / |
| BI-2536 | Inhibits tumor growth | [16] |
| Emetine | Increases lifespan of leukemic mice | [17] |
| Narciclasine | Attenuates diet-induced obesity | [18] |
| CGP-60474 | Inhibitor of CDKs | / |
| Oxetane | / | / |
| Trichostatin-a | Increases lifespan by promoting hsp22 gene expression | [19] |
| Alvocidib | Inhibits metastasis of human osteosarcoma cells | [23] |
| LSM-3353 | / | / |
| BMS-345541 | Inhibitor of kB-kinase (IKK) | / |
| PHA-793887 | Inhibitor of pan-CDK | / |
| Zibotentan | Inhibits blood vessel growth | [21] |
| Mitoxantrone | Treatment of advanced breast cancer | [22] |
Table 7.
Detailed model layouts on predictive labels, sample cell lines, used feature set, and whether the negative class is being downsampled. For example, model 3 was trained with gene ontology term, drug descriptors, both cell line mutation status, and methylation values on perturbation responses on cell lines “U266” and “NOMO1”. The positive samples from model 3 were D–G–C interactions with the top 5% upregulated gene expression signatures, whereas the negative samples were the remaining 95% perturbation data.
Table 7.
Detailed model layouts on predictive labels, sample cell lines, used feature set, and whether the negative class is being downsampled. For example, model 3 was trained with gene ontology term, drug descriptors, both cell line mutation status, and methylation values on perturbation responses on cell lines “U266” and “NOMO1”. The positive samples from model 3 were D–G–C interactions with the top 5% upregulated gene expression signatures, whereas the negative samples were the remaining 95% perturbation data.
| Model Predictive Direction | Cultured Cell Lines | If Balanced Sample Feature |
|---|
| 1 Up/Non-upregulation | Includes all cell lines | True, Type 1 |
| 2 Down/Non-downregulation | Includes all cell lines | True, Type 1 |
| 3 Up/Non-upregulation | U266, NOMO1 | False, Type 1 |
| 4 Down/Non-downregulation | U266, NOMO1 | False, Type 1 |
| 5 Up/Non-upregulation | U266, NOMO1 | False, Type 2 |
| 6 Down/Non-downregulation | U266, NOMO1 | False, Type 2 |
| 7 Up/Non-upregulation | U266, NOMO1 | False, Type 3 |
| 8 Down/Non-downregulation | U266, NOMO1 | False, Type 3 |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}