
GC-MS- and NMR-Based Metabolomics and Molecular Docking Reveal the Potential Alpha-Glucosidase Inhibitors from Psychotria malayana Jack Leaves

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. α-Glucosidase Inhibition Assay
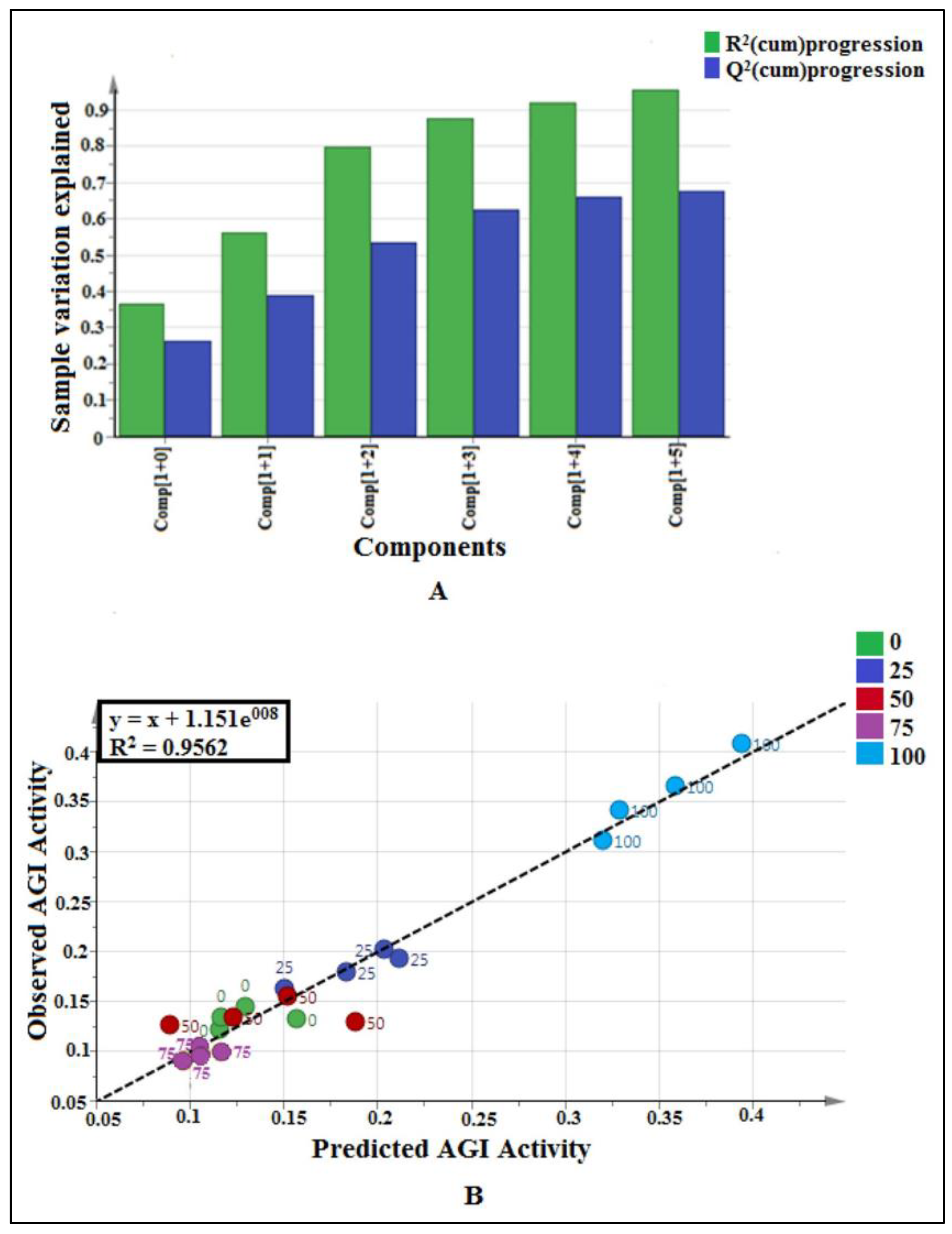
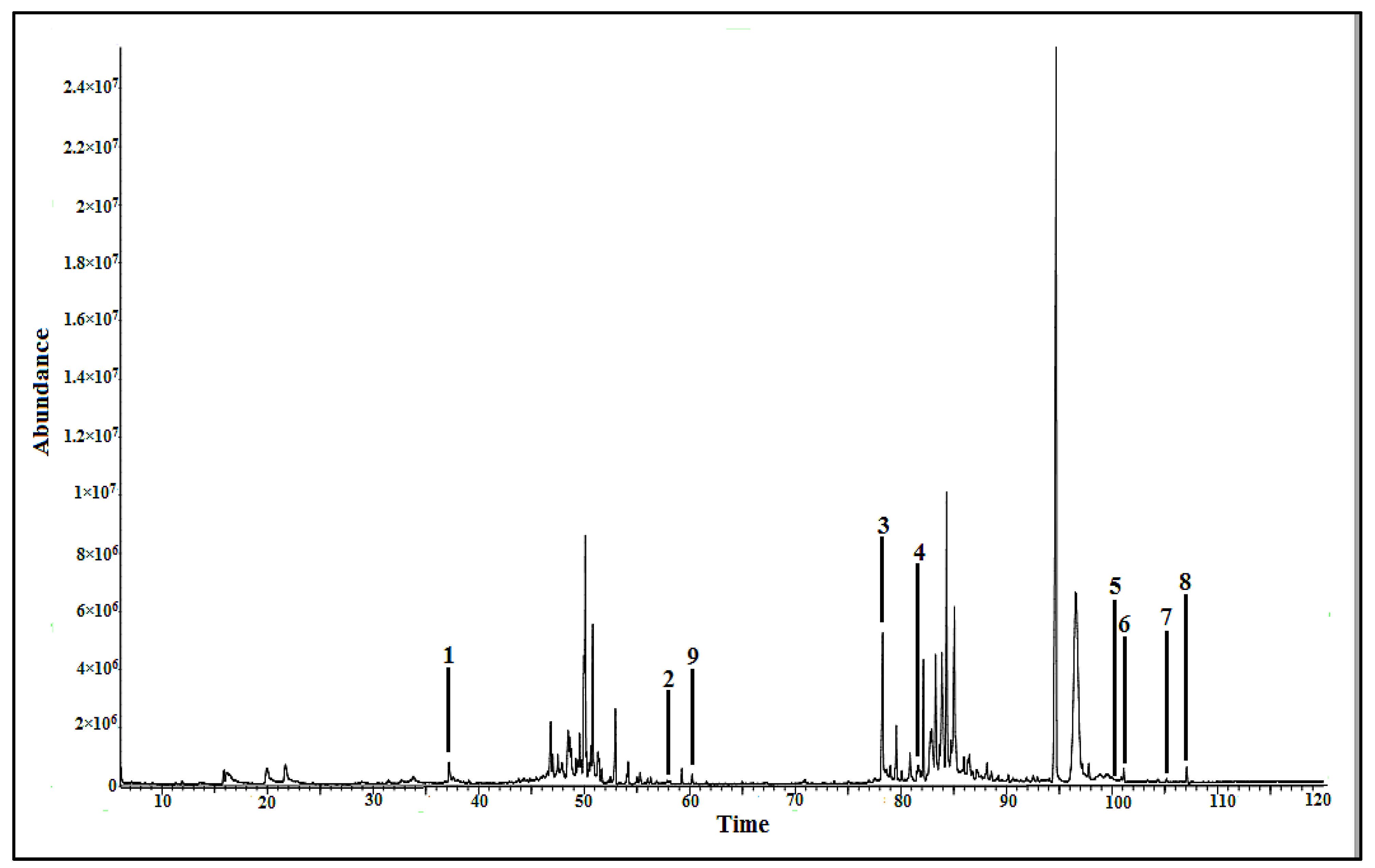
2.2. GC-MS-Based Metabolomics
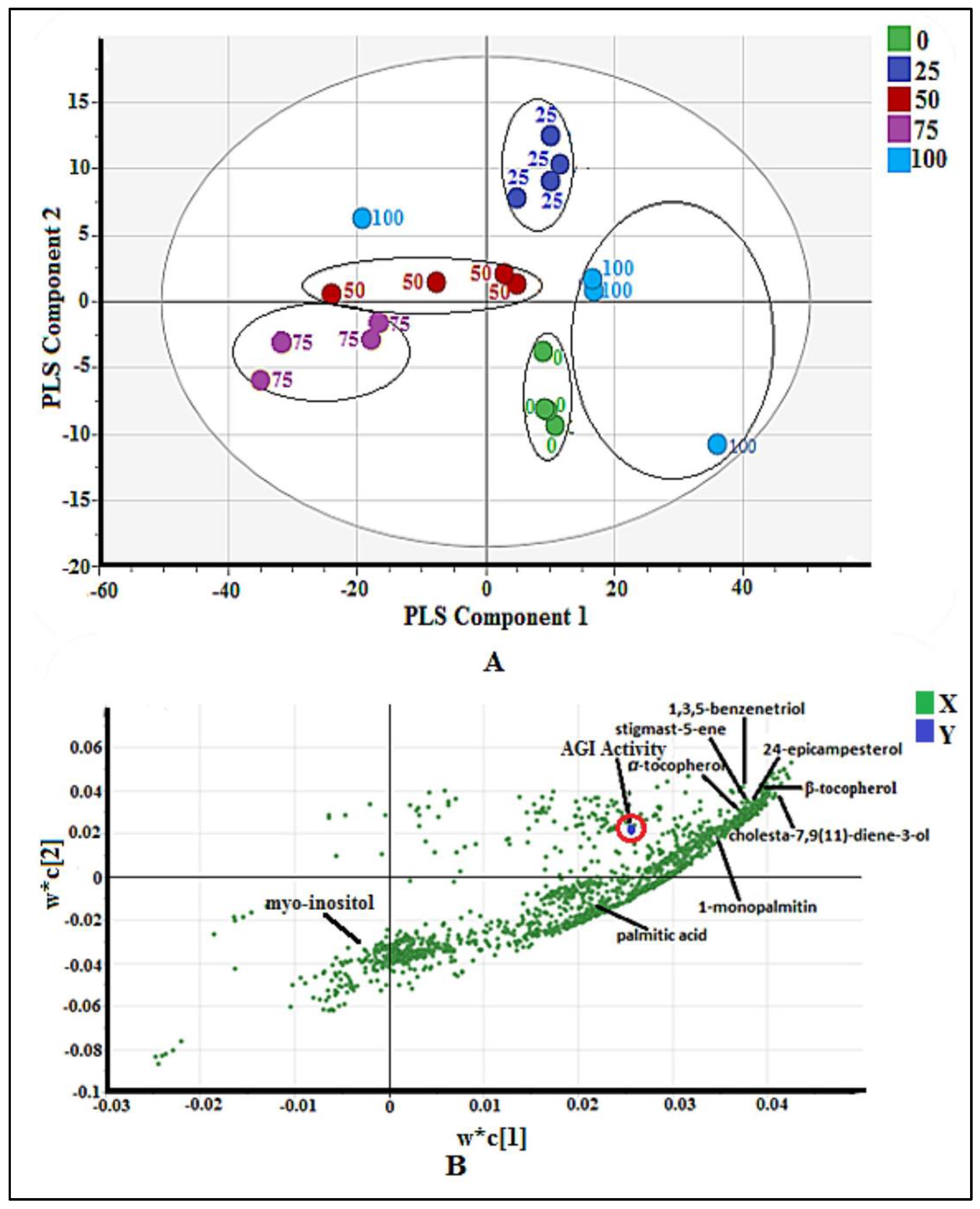
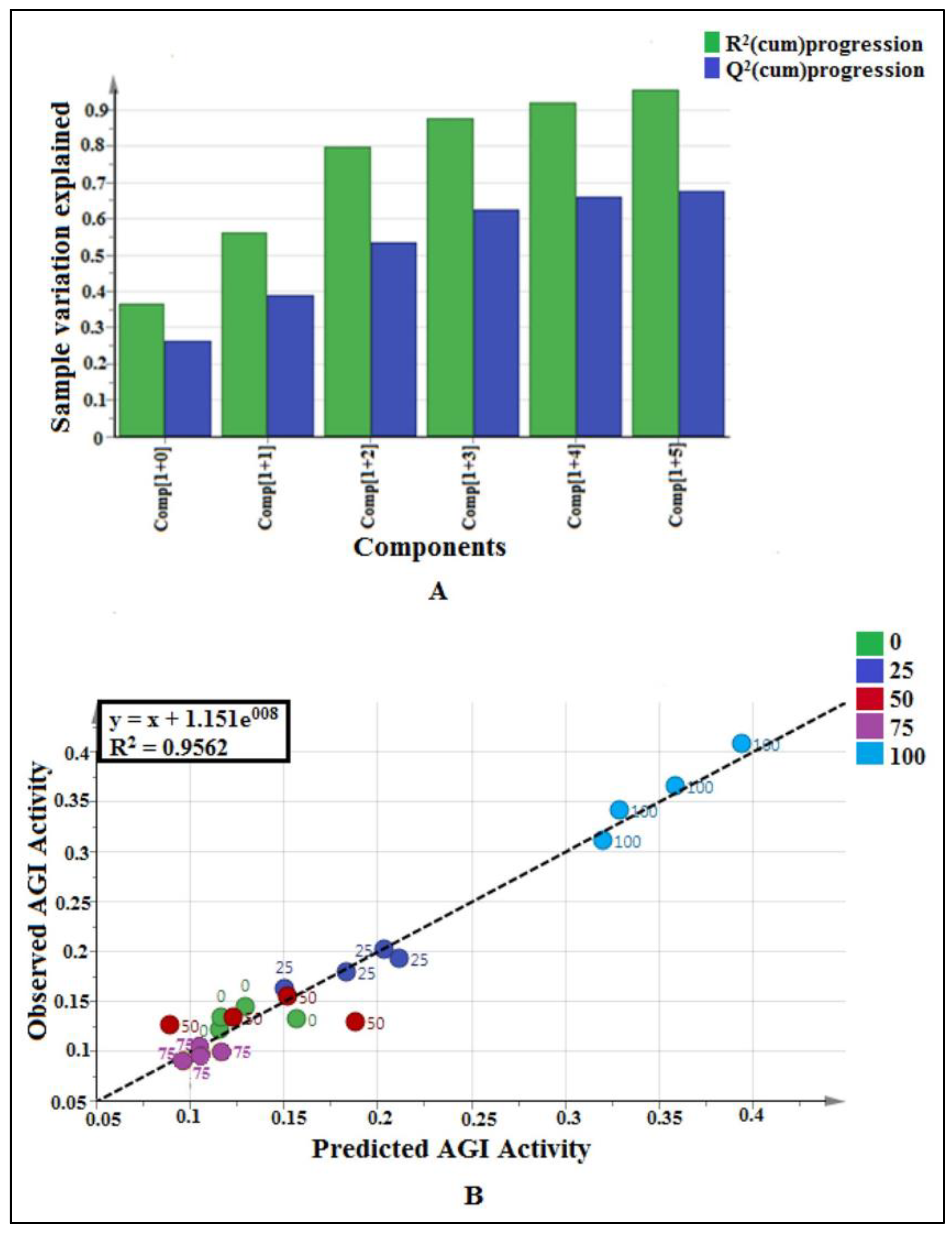
2.2.1. Multivariate Data Analysis
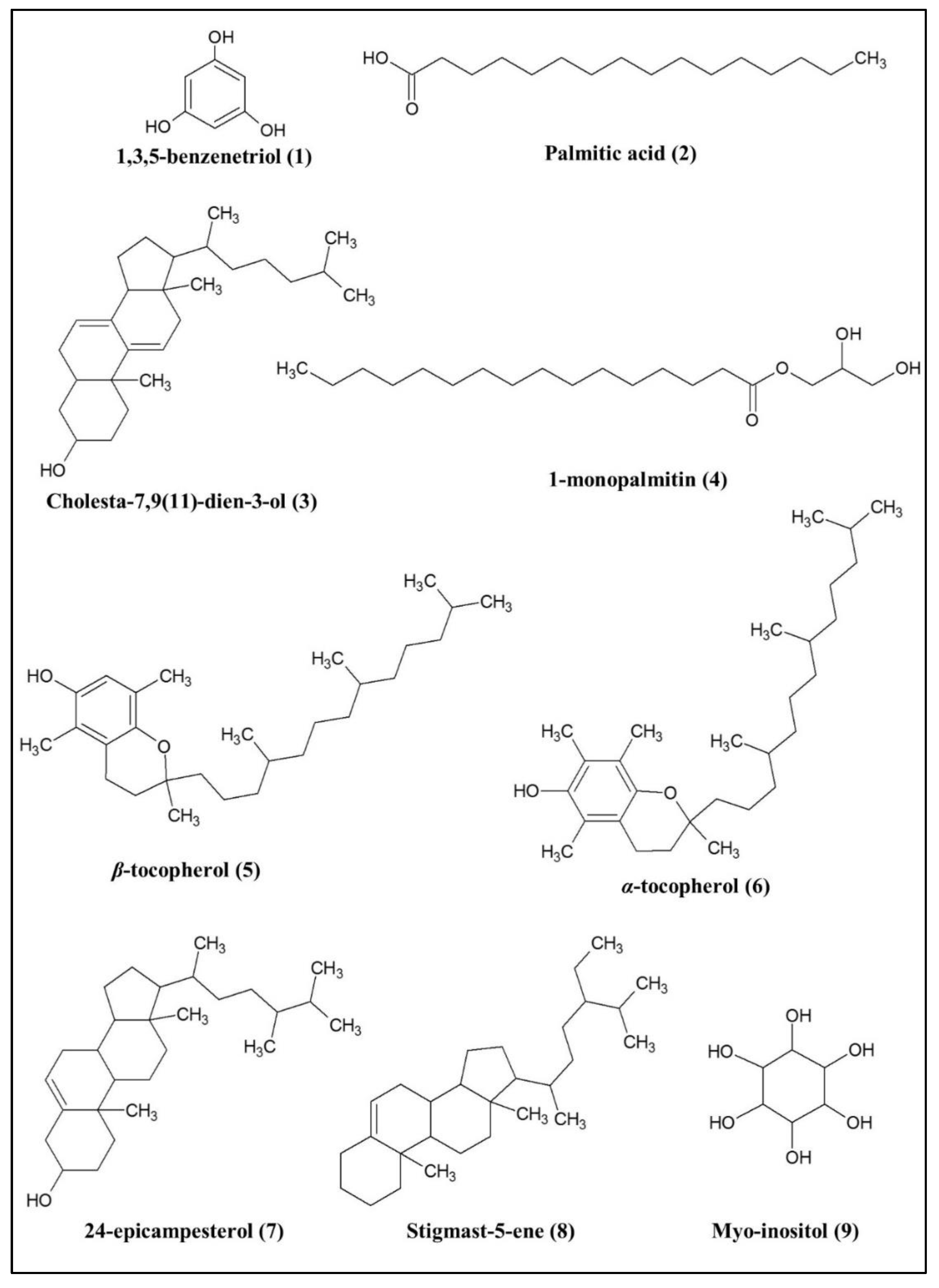
2.2.2. Bioactive Metabolites Profiling of P. malayana Extracts by GC-MS
2.2.3. Bioactive Confirmation of Three Pure Bioactive Compounds
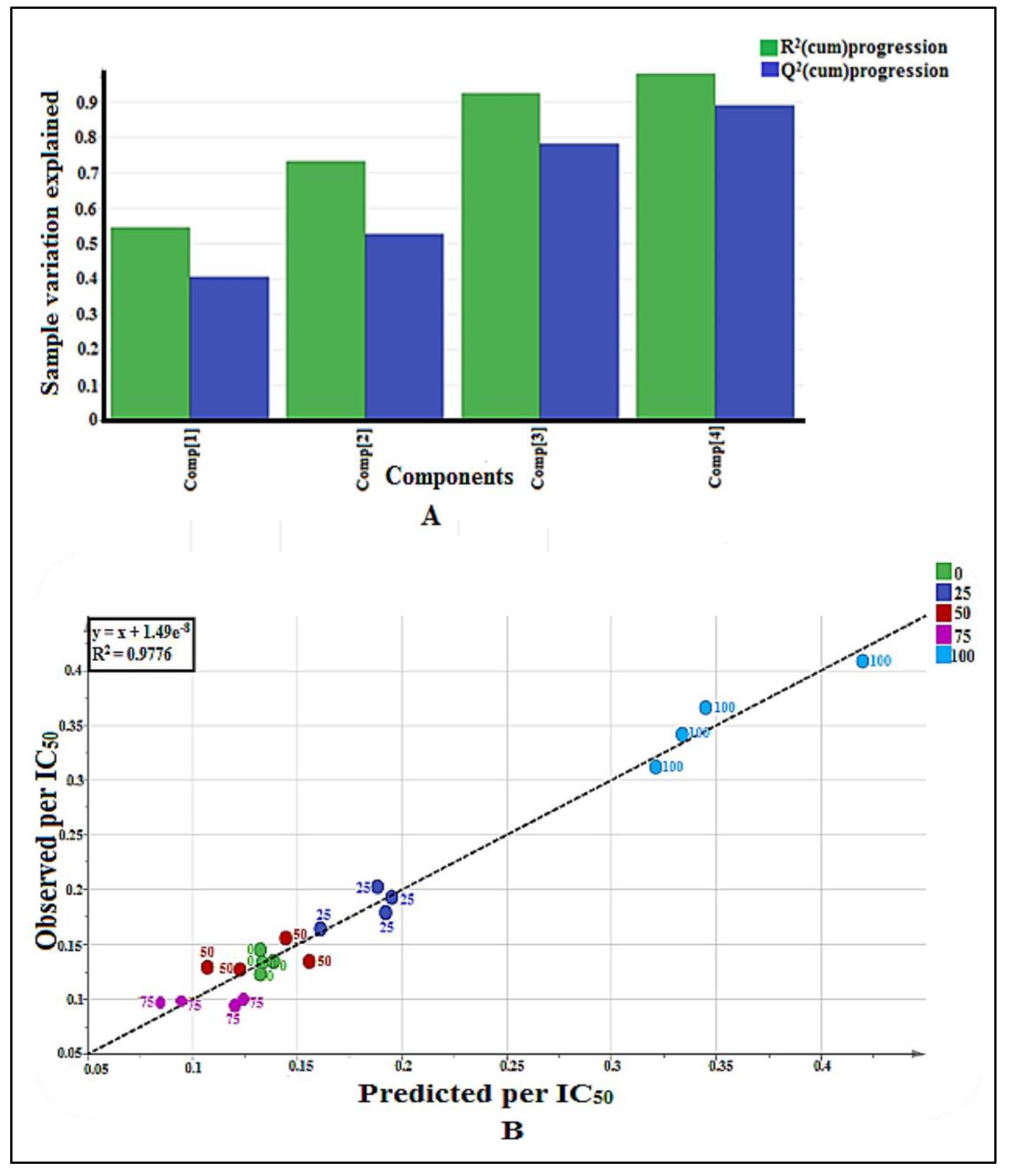
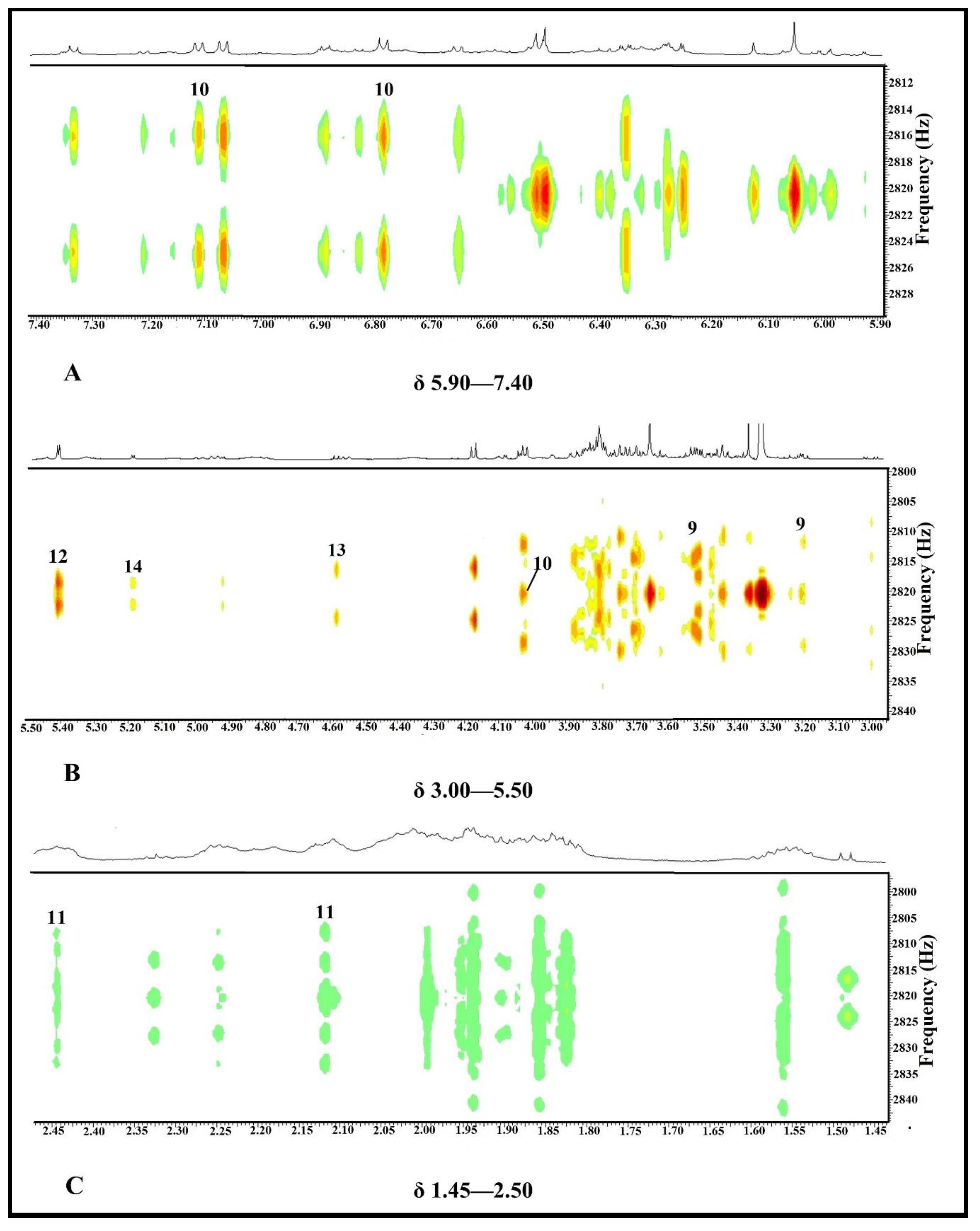
2.3. NMR-Based Metabolomics
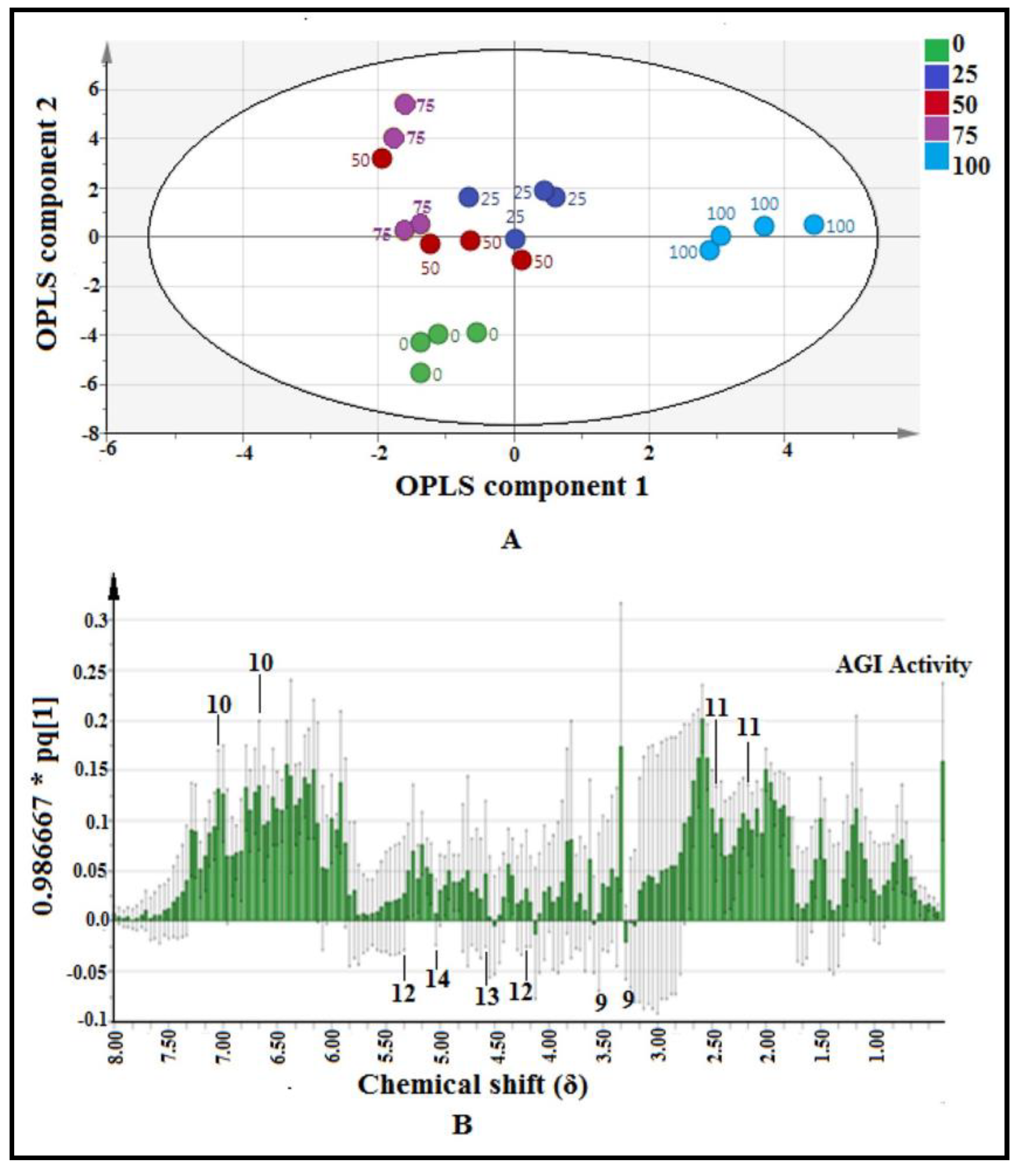
2.3.1. Multivariate Data Analysis
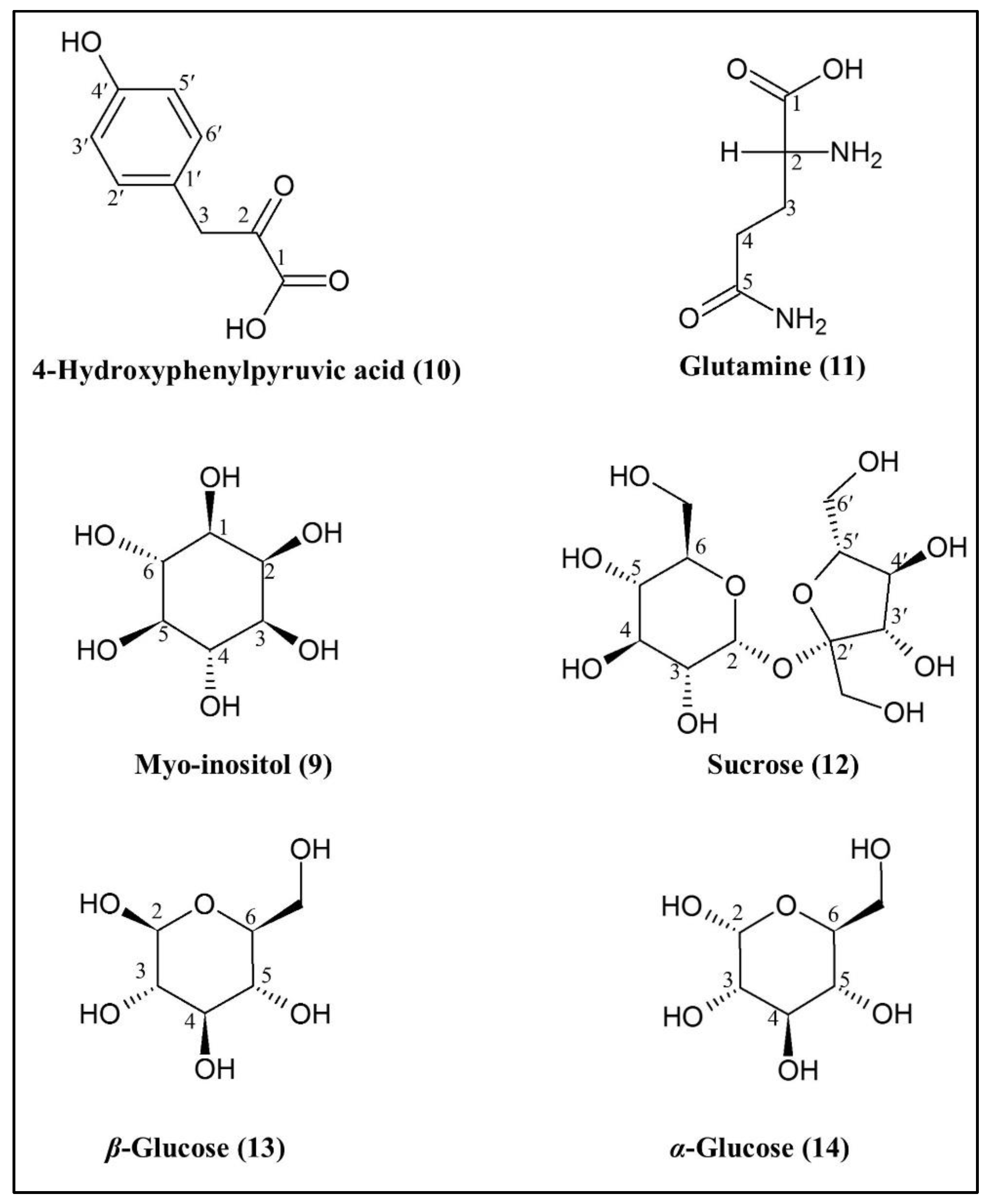
2.3.2. Identification of Putative Bioactive and Other Metabolites
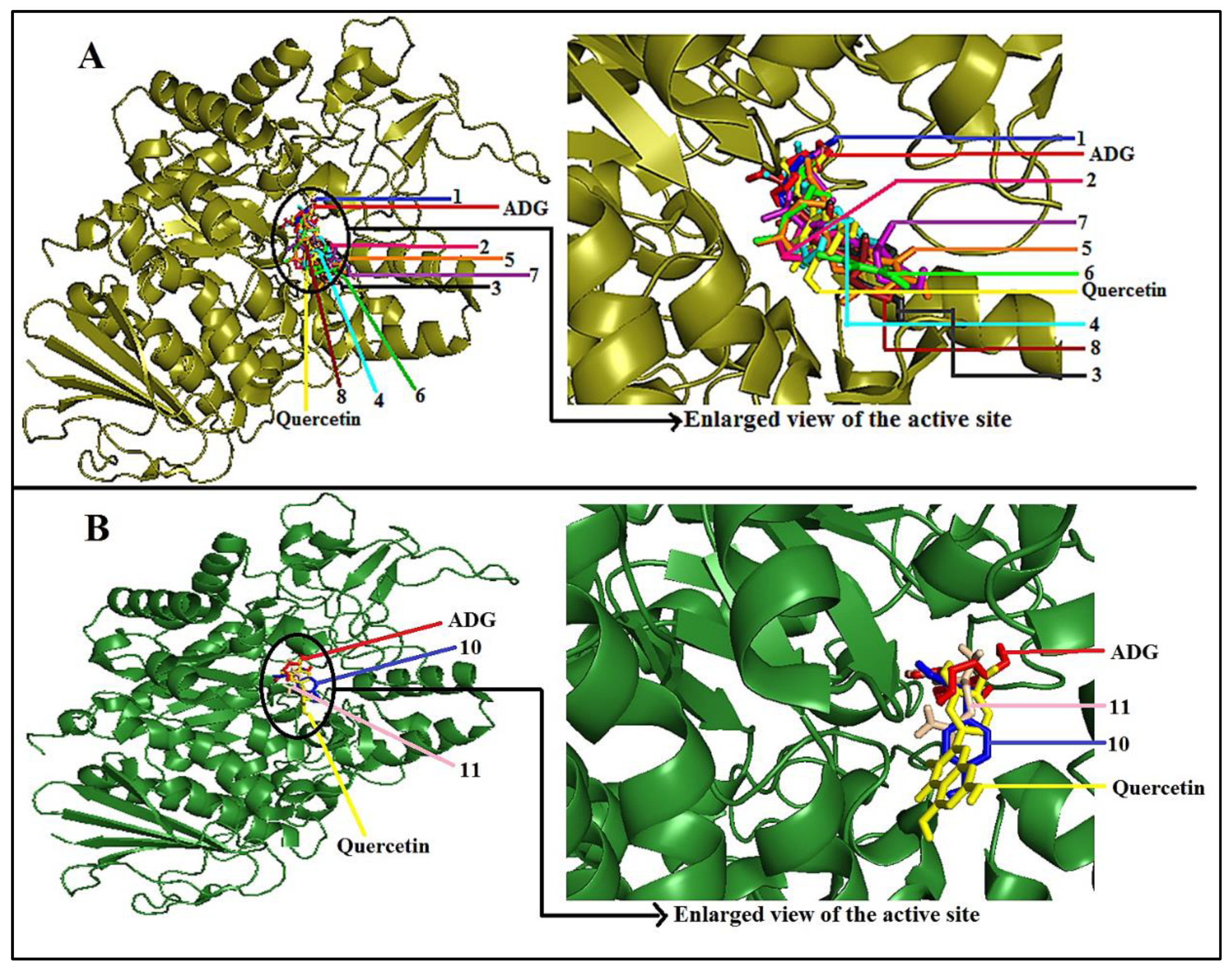
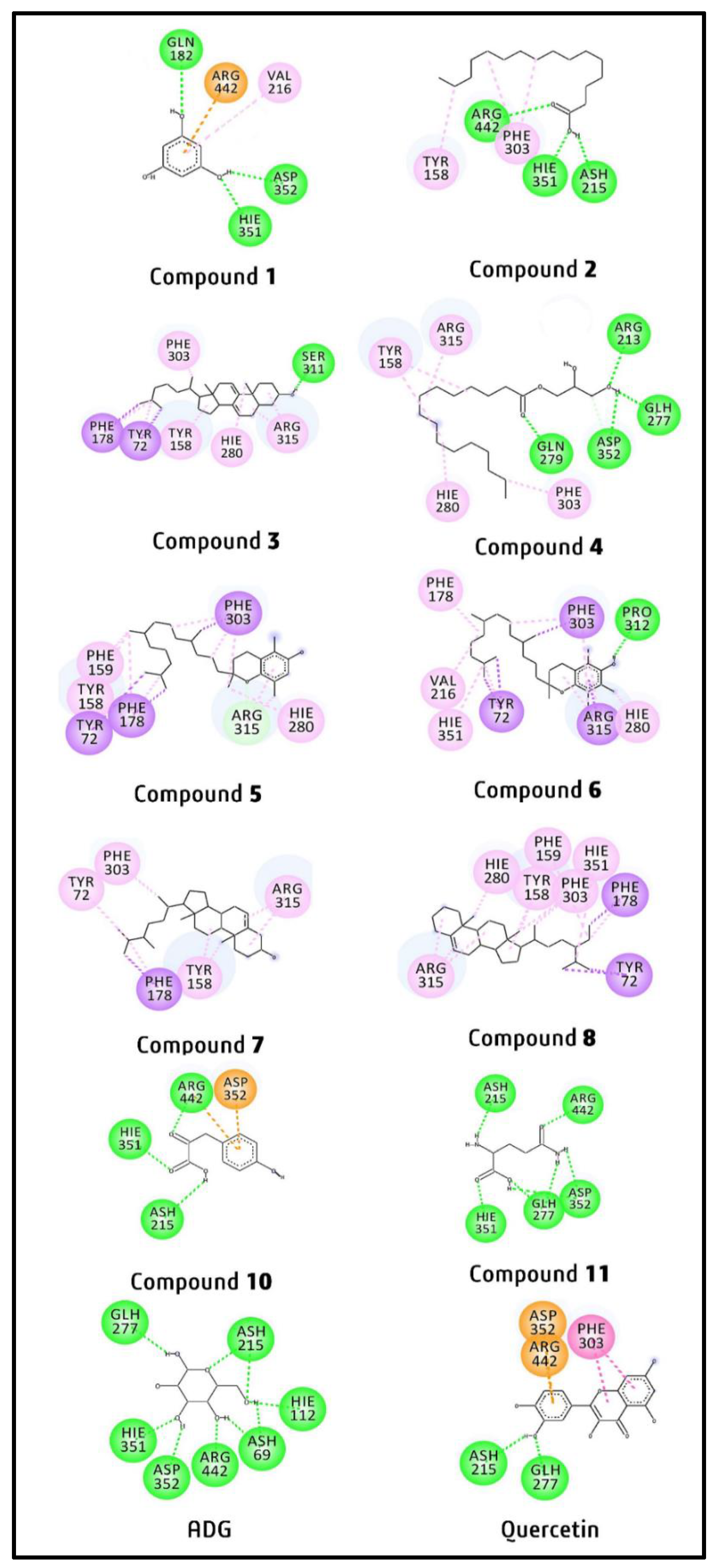
2.4. Molecular Docking Study of Putative Compounds Identified by GC-MS and NMR Analysis
3. Discussion
3.1. AGI Activity
3.2. Putative Compounds Identified by GC-MS
3.3. Putative Compounds Identified by NMR
3.4. In-Silico Study
4. Materials and Methods
4.1. Materials
4.2. Plant Material
4.3. Plant Extract Preparation
4.4. Assay of α-Glucosidase Inhibitory Activity
4.5. Derivatization Procedure for GC-MS
4.6. GC-MS Analysis
4.7. Bioactivity Confirmation of Pure Compounds
4.8. H-NMR Sample Preparation
4.9. H-NMR Data Acquisition and Data Processing
4.10. Metabolite Assignment
4.11. In-Silico Study
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.-U.; Yusoff, N.-I.N.; Uzir, B.-F.; Perumal, V.; Abas, F.; Saari, K.; El-Seedi, H.; Khatib, A. Characterization of α-glucosidase inhibitors from Clinacanthus nutans lindau leaves by gas chromatography-mass spectrometry-based metabolomics and molecular docking simulation. Molecules 2018, 23, 2402. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Kavrekar, V.; Mishra, A. In vitro studies on alpha amylase and alpha glucosidase inhibitory activities of selected plant extracts. Eur. J. Exp. Biol. 2013, 3, 128–132. [Google Scholar]
- Graham, J.E.; Stoebner-may, D.G.; Ostir, G.V.; Al, S.; Peek, M.K.; Markides, K.; Ottenbacher, K.J. Health related quality of life in older Mexican Americans with diabetes: A cross-sectional study. Health Qual. Life Outcomes 2007, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, N.; Salma, U.; Nurunnabi, T.R.; Uddin, M.J.; Jahangir, M.F.K.; Islam, S.M.Z.; Kamruzzaman, M. Management of Type 2 Diabetes mellitus by lifestyle, diet and medicinal plants. Pakistan J. Biol. Sci. 2011, 14, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Gro, J.K.; Yada, S.; Vats, V. Medicinal Plants of India with Anti-Diabetic Potential. J. Ethnopharmacol. 2002, 81, 81–100. [Google Scholar]
- Situmorang, R.O.P.; Harianja, A.H.; Silalahi, J. Karo’s Local Wisdom: The use of woody plants for traditional diabetic medicines. Indones. J. For. Res. 2015, 2, 121–130. [Google Scholar] [CrossRef]
- Hadi, S.; Asnawati, D.; Ersalena, V.F.; Azwari, A.; Rahmawati, K.P. Characterization of alkaloids from the leaves of Psychotria malayana jack of Lombok island on the basis of gas chromatography-mass spectroscopy. J. Pure Appl. Chem. Res. 2014, 3, 108–113. [Google Scholar] [CrossRef]
- Matsuura, H.N.; Porto, D.D.; Fett-Neto, A.G. Bioactive alkaloids from south american Psychotria and related rubiaceae. In Natural Products; Springer: Berlin/Heidelberg, Germany, 2013; pp. 119–147. [Google Scholar] [CrossRef]
- Chebib, M.; Duke, R.K.; Duke, C.C.; Connor, M.; Mewett, K.N.; Johnston, G.A.R. Convulsant actions of calycanthine. Toxicol. Appl. Pharmacol. 2003, 190, 58–64. [Google Scholar] [CrossRef]
- Hadi, S. Bioactive Alkaloids from Medicinal Plants of Lombok; University of Wollongong: Wollongong, Australia, 2002. [Google Scholar]
- Nokhala, A.; Siddiqui, M.J.; Ahmed, Q.U.; Bustamam, M.S.A.; Zakaria, Z.A. Investigation of α-glucosidase inhibitory metabolites from Tetracera scandens leaves by GC–MS metabolite profiling and docking studies. Biomolecules 2020, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Byrne, T.; Johansson, E.; Trygg, J.; Vikström, C. Multi- and Megavariate Data Analysis Basic Principles and Applications, 2nd ed.; Umetrics Academy: Umea, Sweden, 2006. [Google Scholar]
- Javadi, N.; Abas, F.; Hamid, A.A.; Simoh, S.; Shaari, K.; Ismail, I.S.; Mediani, A.; Khatib, A. GC-MS-based metabolite profiling of Cosmos caudatus leaves possessing alpha-glucosidase inhibitory activity. J. Food Sci. 2014, 79, C1130–C1136. [Google Scholar] [CrossRef]
- Das, S.; Dutta, M.; Chaudhury, K.; De, B. Metabolomic and Chemometric Study of Achras Sapota L. Fruit extracts for identification of metabolites contributing to the inhibition of α-amylase and α-glucosidase. Eur. Food Res. Technol. 2016, 242, 733–743. [Google Scholar] [CrossRef]
- Tan, D.C.; Kassim, N.K.; Ismail, I.S.; Hamid, M.; Ahamad Bustamam, M.S. Identification of antidiabetic metabolites from Paederia foetida l. twigs by gas chromatography-mass spectrometry-based metabolomics and molecular docking study. Biomed. Res. Int. 2019, 2019, 7603125. [Google Scholar] [CrossRef]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Zolkeflee, N.K.Z.; Isamail, N.A.; Maulidiani, M.; Abdul Hamid, N.A.; Ramli, N.S.; Azlan, A.; Abas, F. Metabolite variations and antioxidant activity of Muntingia calabura leaves in response to different drying methods and ethanol ratios elucidated by NMR-based metabolomics. Phytochem. Anal. 2020, 32, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Sussulini, A. (Ed.) Metabolomics: From Fundamentals to Clinical Applications, 1st ed.; Springer: Gewerbestrasse, Switzerland, 2017. [Google Scholar] [CrossRef]
- Mediani, A.; Abas, F.; Maulidiani, M.; Khatib, A.; Tan, C.P.; Safinar Ismail, I.; Shaari, K.; Ismail, A. Characterization of metabolite profile in Phyllanthus niruri and correlation with bioactivity elucidated by nuclear magnetic resonance based metabolomics. Molecules 2017, 22, 902. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, R.K.; Dubey, A.K. Phytochemical Profiling, GC-MS analysis and α-amylase inhibitory potential of ethanolic extract of Cocos Nucifera Linn. Endocarp. Endocr. Metab. Immune Disord.-Drug Targets 2019, 19, 419–442. [Google Scholar] [CrossRef]
- Jardim, C.M.; Jham, G.N.; Dhingra, O.D.; Freire, M.M. Composition and antifungal activity of the essential oil of the brazilian Chenopodium Ambrosioides L. J. Chem. Ecol. 2008, 34, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Efron, B.; Gong, G. A Leisurely Look at the Bootstrap, the Jackknife, and Cross-Validation. Am. Stat. 1983, 37, 36–48. [Google Scholar]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomics analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, H.K.; Linthorst, H.J.M.; Hollander, J.G.; Lefeber, A.W.M.; Erkelens, C.; Nuzillard, J.M.; Verpoorte, R. NMR metabolomics to revisit the Tobacco Mosaic virus infection in Nicotiana tabacum leaves. J. Nat. Prod. 2006, 69, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.J.; Hom, K.; Bai, G.; Shapiro, M. NMR metabolomics analysis of caco-2 cell differentiation. J. Proteome Res. 2009, 8, 4104–4108. [Google Scholar] [CrossRef] [PubMed]
- Jindra, A.; Šípal, Z.; Hudecová, V. Biosynthesis of Alkaloids. On the occurrence of keto acids in Papaver Somniferum L. plants. Experientia 1964, 20, 371–372. [Google Scholar] [CrossRef]
- Hou, Z.; Li, Y.; Su, F.; Chen, J.; Zhang, X.; Xu, L.; Yang, D.; Liang, Z. Application of 1H-NMR Combined with QRT-PCR technology in the exploration of rosmarinic acid biosynthesis in hair roots of Salvia miltiorrhiza Bunge and Salvia castanea f. Tomentosa Stib. Planta 2021, 253, 2. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Ikegami, F.; Lambein, F. Neuroactive and other free amino acids in seed and young plants of Panax ginseng. Phytochemistry 2003, 62, 1087–1091. [Google Scholar] [CrossRef]
- Peksa, A.; Miedzianka, J.; Nems, A.; Rytel, E. The free-amino-acid content in six potatoes cultivars through storage. Molecules 2021, 26, 1322. [Google Scholar] [CrossRef]
- Khoo, L.W.; Mediani, A.; Zolkeflee, N.K.Z.; Leong, S.W.; Ismail, I.S.; Khatib, A.; Shaari, K.; Abas, F. Phytochemical diversity of Clinacanthus nutans extracts and their bioactivity correlations elucidated by NMR based metabolomics. Phytochem. Lett. 2015, 14, 123–133. [Google Scholar] [CrossRef]
- Maroyi, A. Hilliardiella Elaeagnoides: Review of its medicinal uses, phytochemistry and pharmacological properties. Res. J. Pharm. Technol. 2020, 13, 5539–5545. [Google Scholar] [CrossRef]
- Nowakowski, P.; Naliwajko, S.K.; Markiewicz-Żukowska, R.; Borawska, M.H.; Socha, K. The two faces of Coprinus Comatus—functional properties and potential hazards. Phyther. Res. 2020, 34, 2932–2944. [Google Scholar] [CrossRef]
- Maulidiani, H.; Khatib, A.; Shaari, K.; Abas, F.; Shitan, M.; Kneer, R.; Neto, V.; Lajis, N.H. Discrimination of three pegaga (centella) varieties and determination of growth-lighting effects on metabolites content based on the chemometry of 1h nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 2012, 60, 410–417. [Google Scholar] [CrossRef]
- Mediani, A.; Abas, F.; Khatib, A.; Tan, C.P.; Ismail, I.S.; Shaari, K.; Ismail, A.; Lajis, N.H. Phytochemical and biological features of Phyllanthus niruri and Phyllanthus urinaria harvested at different growth stages revealed by 1h NMR-based metabolomics. Ind. Crops Prod. 2015, 77, 602–613. [Google Scholar] [CrossRef]
- Wei, S.; Abas, F.; Wai, K.; Yusoff, K. In Vitro and In Silico Evaluations of diarylpentanoid series as α-glucosidase inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 302–309. [Google Scholar] [CrossRef]
- Kidane, Y.; Bokrezion, T.; Mebrahtu, J.; Mehari, M.; Gebreab, Y.B.; Fessehaye, N.; Achila, O.O. In vitro inhibition of α-amylase and α-glucosidase by extracts from Psiadia punctulata and Meriandra bengalensis. Evid.-Based Complement Altern. Med. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, A.; Singh, B.; Arora, R.; Arora, S. In Vitro Evaluation of the α-glucosidase inhibitory potential of methanolic extracts of traditionally used antidiabetic plants. BMC Complement Altern. Med. 2019, 19, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, M.F.; Bennett, M.T. The Effect of different methods and solvents on the extraction of polyphenols in ginger (Zingiber officinale). J. Teknol. 2016, 78, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Nipun, T.S.; Khatib, A.; Ahmed, Q.U.; Redzwan, I.E.; Ibrahim, Z.; Khan, A.Y.F.; Primaharinastiti, R.; Khalifa, S.A.M.; El-Seedi, H.R. Alpha-glucosidase inhibitory effect of Psychotria malayana jack leaf: A rapid analysis using infrared fingerprinting. Molecules 2020, 25, 4161. [Google Scholar] [CrossRef]
- Prihantini, A.I.; Tachibana, S.; Itoh, K. Evaluation of Antioxidant and α-Glucosidase Inhibitory Activities of Some Subtropical Plants. Pak. J. Biol. Sci. 2014, 17, 1106–1114. [Google Scholar] [CrossRef]
- Indrianingsih, A.W.; Tachibana, S.; Itoh, K. In Vitro Evaluation of Antioxidant and α-Glucosidase Inhibitory Assay of Several Tropical and Subtropical Plants. Procedia Environ. Sci. 2015, 28, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A Review of Alpha-Glucosidase Inhibitors from Plants as Potential Candidates for the Treatment of Type-2 Diabetes. Phytochem. Rev. 2021. [Google Scholar] [CrossRef]
- Mauldina, M.G.; Sauriasari, R.; Elya, B. α-Glucosidase Inhibitory Activity from Ethyl Acetate Extract of Antidesma Bunius (L.) Spreng Stem Bark Containing Triterpenoids. Pharmacogn. Mag. 2017, 13, 590–594. [Google Scholar] [CrossRef]
- Ankita Bachhawat, J.; Shihabudeen, M.S.; Thirumurugan, K. Screening of Fifteen Indian Ayurvedic Plants for Alpha-Glucosidase Inhibitory Activity and Enzyme Kinetics. Int. J. Pharm. Pharm. Sci. 2011, 3, 267–274. [Google Scholar]
- Alam, M.A.; Zaidul, I.S.M.; Ghafoor, K.; Sahena, F.; Hakim, M.A.; Rafii, M.Y.; Abir, H.M.; Bostanudin, M.F.; Perumal, V.; Khatib, A. In vitro antioxidant and, α-glucosidase inhibitory activities and comprehensive metabolite profiling of methanol extract and its fractions from Clinacanthus nutans. BMC Complement Altern. Med. 2017, 17, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadi, N.; Abas, F.; Mediani, A.; Abd Hamid, A.; Khatib, A.; Simoh, S.; Shaari, K. Effect of Storage Time on metabolite profile and alpha-glucosidase inhibitory activity of Cosmos caudatus leaves-GCMS based metabolomics approach. J. Food Drug Anal. 2015, 23, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Yumna, M.; Angelina; Abdullah; Arbianti, R.; Utami, T.S.; Hermansyah, H. Effect of mother-in-law’s tongue leaves (Sansevieria trifasciata) extract’s solvent polarity on anti-diabetic activity through in vitro α-glucosidase enzyme inhibition test. In E3S Web of Conferences; Auffret-Babak, I., Ed.; EDP Sciences: Paris, Franch, 2018; Volume 67, pp. 1–5. [Google Scholar] [CrossRef] [Green Version]
- Srilatha, B.R.; Ananda, S. Antidiabetic Effects of Mukia Maderaspatana and Its Phenolics: An in vitro study on gluconeogenesis and glucose uptake in rat tissues. Pharm. Biol. 2014, 52, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.Y.; Choi, H.; Jun, H.S. The Effect of phloroglucinol, a component of Ecklonia cava extract, on hepatic glucose production. Mar. Drugs 2017, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Freeman, C.W.; Rahimtula, A.D.; Wilton, D.C. A Novel olefinic rearrangement the enzymic conversion of cholesta-7,9-dien-3β-ol into cholesta-8,14-dien-3β-ol. Biochem. J. 1972, 129, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Jamaluddin, F.; Mohamed, S.; Lajis, M.N. Hypoglycaemic effect of Parkia speciosa seeds due to the synergistic action of β-sitosterol and stigmasterol. Food Chem. 1994, 49, 339–345. [Google Scholar] [CrossRef]
- Kuhlisch, C.; Pohnert, G. Metabolomics in chemical ecology. Nat. Prod. Rep. 2015, 32, 937–955. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.A.; De Souza, L.P.; Serag, A.; Fernie, A.R.; Farag, M.A.; Ezzat, S.M.; Alseekh, S. Metabolomics in the context of plant natural products research: From sample preparation to metabolite analysis. Metabolites 2020, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Huang, F.; Deng, C.; Wang, Y.; Kai, G. Bioactivities, biosynthesis and biotechnological production of phenolic acids in Salvia miltiorrhiza. Crit. Rev. Food Sci. Nutr. 2018, 59, 953–964. [Google Scholar] [CrossRef]
- Białecka-Florjańczyk, E.; Fabiszewska, A.; Zieniuk, B. Phenolic acids derivatives-biotechnological methods of synthesis and bioactivity. Curr. Pharm. Biotechnol. 2018, 19, 1098–1113. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, Y.; Yue, Y.; Chen, W.; Zhang, Y.; Shi, G.; Wu, Z. Glutamine enhances the hypoglycemic effect of insulin in l6 cells via phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT)/glucose transporter 4 (GLUT4) signaling pathway. Med. Sci. Monit. 2018, 24, 1241–1250. [Google Scholar] [CrossRef]
- Jafari-Vayghan, H.; Varshosaz, P.; Hajizadeh-Sharafabad, F.; Razmi, H.R.; Amirpour, M.; Tavakoli-Rouzbehani, O.M.; Alizadeh, M.; Maleki, V. A Comprehensive insight into the effect of glutamine supplementation on metabolic variables in diabetes mellitus: A systematic review. Nutr. Metab. 2020, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Samocha-Bonet, D.; Chisholm, D.J.; Gribble, F.M.; Coster, A.C.F.; Carpenter, K.H.; Jones, G.R.D.; Holst, J.J.; Greenfield, J.R. Glycemic effects and safety of l-glutamine supplementation with or without sitagliptin in type 2 diabetes patients-a randomized study. PLoS ONE 2014, 9, e113366. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhang, Y.; Wu, D.; Wang, C.; Yang, Y.J.; Fan, S.J.; Xia, L.; Wei, Y.; Peng, X. 1H-NMR metabolomics identifies significant changes in hypermetabolism after glutamine administration in burned rats. Am. J. Transl. Res. 2019, 11, 7286–7299. [Google Scholar] [PubMed]
- Wei, S.; Liu, L.; Zhang, J.; Bowers, J.; Gowda, G.A.N.; Seeger, H.; Fehm, T.; Neubauer, H.J.; Vogel, U.; Clare, S.E.; et al. Metabolomics approach for predicting response to neoadjuvant chemotherapy for breast cancer. Mol. Oncol. 2013, 7, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappello, T.; Giannetto, A.; Parrino, V.; De Marco, G.; Mauceri, A.; Maisano, M. Food safety using NMR-based metabolomics: Assessment of the Atlantic bluefin tuna, Thunnus thynnus, from the Mediterranean sea. Food Chem. Toxicol. 2018, 115, 391–397. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef]
- Murugesu, S.; Ibrahim, Z.; Uddin, Q.; Uzir, B.F.; Nik, N.I.; Perumal, V.; Abas, F.; Shaari, K.; Khatib, A. Identification of α-glucosidase inhibitors from Clinacanthus nutans leaf extract using liquid chromatography-mass spectrometry-based metabolomics and protein-ligand interaction with molecular docking. J. Pharm. Anal. 2019, 9, 91–99. [Google Scholar] [CrossRef]
- Ali, M.Y.; Kim, D.H.; Seong, S.H.; Kim, H.-R.; Jung, H.A.; Choi, J.S. α-Glucosidase and protein tyrosine phosphatase 1b inhibitory activity of plastoquinones from marine brown alga Sargassum serratifolium. Mar. Drugs 2017, 15, 368. [Google Scholar] [CrossRef] [Green Version]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1b and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef]
- Nipun, T.S.; Khatib, A.; Ibrahim, Z.; Ahmed, Q.U.; Redzwan, I.E.; Saiman, M.Z.; Supandi, F.; Primaharinastiti, R.; El-Seedi, H.R. Characterization of α-glucosidase inhibitors from Psychotria malayana jack leaves extract using LC-MS-based multivariate data analysis and in-silico molecular docking. Molecules 2020, 25, 5885. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2014, 36, 78–95. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing autodock and vina in ligand/decoy discrimination for virtual screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Narkowicz, C.K.; Al-Aubaidy, H.A.; Jelinek, H.F.; Nichols, D.S.; Burgess, J.R.; Jacobson, G.A. Phytosterol supplements do not inhibit dipeptidyl peptidase-4. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Adnan, S.; Shah, A.; Afifi, M.; Imran, S.; Sultan, S.; Rahim, F.; Mohammed, K. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 2018, 77, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Alyar, S.; Sen, T.; Ozmen, Ü.O.; Alyar, H.; Adem, S.; Sen, C. Synthesis, spectroscopic characterizations, enzyme inhibition, molecular docking study and dft calculations of new schiff bases of sulfa drugs. J. Mol. Struct. 2019, 1185, 416–424. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Han, L.; Fang, C.; Zhu, R.; Peng, Q.; Li, D.; Wang, M. Inhibitory effect of phloretin on α-glucosidase: Kinetics, interaction mechanism and molecular docking. Int. J. Biol. Macromol. 2017, 95, 520–527. [Google Scholar] [CrossRef]
- Imran, S.; Taha, M.; Ismail, H.; Kashif, S.M.; Rahim, F.; Jamil, W.; Wahab, H.; Khan, K.M. Synthesis, In vitro and docking studies of new flavone ethers as α-glucosidase inhibitors. Chem. Biol. Drug Des. 2016, 87, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Lestari, W.; Dewi, R.T.; Broto, L.; Kardono, S.; Yanuar, A. Docking sulochrin and its derivative as α-glucosidase inhibitors of Saccharomyces cerevisiae. Indones. J. Chem. 2017, 17, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, G.; Pan, J.; Wang, Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.W.; Awin, T.; Munirah, S.; Faudzi, M.; Shaari, M.M.K.; Abas, F. Synthesis and biological evaluation of asymmetrical diarylpentanoids as antiin fl ammatory, anti-α-glucosidase, and antioxidant agents. Med. Chem. Res. 2019, 28, 2002–2009. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; Mccammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of poisson–boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Methanol in Water | (%) of Inhibition * |
|---|---|
| 0% | 39.6 ± 3.32 c |
| 25% | 49.6 ± 2.37 b |
| 50% | 42.0 ± 4.04 b,c |
| 75% | 36.1 ± 3.37 c |
| 100% | 71.7 ± 6.56 a |
| Quercetin | 74.7 ± 1.14 a |
| Compound No. | Putative Metabolites | RT (min) | % of Area | SI | MF |
|---|---|---|---|---|---|
| 1 | 1,3,5-benzenetriol | 37.213 | 0.61 | 98 | C6H6O3 |
| 2 | Palmitic acid | 58.142 | 0.04 | 98 | C16H32O2 |
| 3 | Cholesta-7,9(11)-diene-3-ol | 78.294 | 3.42 | 94 | C28H48O |
| 4 | 1-monopalmitin | 81.935 | 0.17 | 93 | C19H38O4 |
| 5 | β-tocopherol | 100.870 | 0.10 | 93 | C28H48O2 |
| 6 | α-tocopherol | 101.104 | 0.24 | 98 | C29H50O2 |
| 7 | 24-epicampesterol | 104.362 | 0.03 | 90 | C28H48O |
| 8 | Stigmast-5-ene | 107.083 | 0.31 | 99 | C29H50 |
| 9 | Myo-inositol | 60.234 | 0.15 | 94 | C6H12O6 |
| Sample Name | IC50 (μg/mL) * |
|---|---|
| Palmitic acid | 8.04 ± 0.12 a |
| Methanol extracts | 2.83 ± 0.32 b |
| Quercetin | 1.86 ± 0.04 c |
| Concentrations of 1-Monopalmitin (μg/mL) Added to 2 μg/mL of Methanol Extract | % of AGI * | Concentrations of 1-Monopalmitin (μg/mL) | % of AGI |
|---|---|---|---|
| 4 | 75.59 ± 1.95 a | 4 | 0 |
| 2 | 61.89 ± 0.77 b | 2 | 0 |
| 1 | 59.03 ± 1.23 c | 1 | 0 |
| 0.5 | 41.14 ± 1.18 d | 0.5 | 0 |
| 0.25 | 34.36 ± 0.49 e | 0.25 | 0 |
| 0 | 31.84 ± 0.87 e | - | - |
| Concentrations of α-Tocopherol (μg/mL) Added to 2 μg/mL of Methanol Extract | % of AGI * | Concentrations of α-Tocopherol (μg/mL) | % of AGI * |
|---|---|---|---|
| 4 | 97.08 ± 0.60 a | 4 | 10.93 ± 0.63 g |
| 2 | 90.94 ± 0.91 b | 2 | 7.84 ± 0.82 h |
| 1 | 85.22 ± 0.49 c | 1 | 6.07 ± 0.85 h |
| 0.5 | 74.55 ± 0.37 d | 0.5 | 3.61 ± 0.85 i |
| 0.25 | 56.99 ± 1.46 e | 0.25 | 0 |
| 0 | 31.18 ± 1.09 f | - | - |
| Compound | Putative Metabolites | Proton Number | Chemical Shift (ppm) | Multiplicity * | Coupling Constant (J) | References |
|---|---|---|---|---|---|---|
| 9 | Myo-inositol | H-1 H-5 | 3.51 3.20 | dd t | 10.0 Hz; 4.0 Hz 9.0 Hz | [25,26,27] |
| 10 | 4-hydroxyphenyl-pyruvic acid | H-3′; H-5′ H-2′; H-6′ H-3 | 6.78 7.11 4.03 | d d s | 8.0 Hz 8.0 Hz - | [28,29] |
| 11 | Glutamine | H-4 H-3 | 2.44 2.12 | m m | - - | [26,30,31,32,33,34] |
| 12 | Sucrose | H-2 (glucose moiety) | 5.40 | d | 4.0 Hz | [26,32,35,36] |
| 13 | β-glucose | H-2 | 4.58 | d | 8.0 Hz | [26,32,35,36] |
| 14 | α-glucose | H-2 | 5.18 | d | 4.0 Hz | [26,32,35,36] |
| Compound | Binding Affinity (kcal/mol) |
|---|---|
| Control ligand (ADG) | −6.0 |
| Quercetin | −8.4 |
| 1 | −5.5 |
| 2 | −6.1 |
| 3 | −9.1 |
| 4 | −6.1 |
| 5 | −8.6 |
| 6 | −7.9 |
| 7 | −7.7 |
| 8 | −9.4 |
| 10 | −6.5 |
| 11 | −5.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nipun, T.S.; Khatib, A.; Ibrahim, Z.; Ahmed, Q.U.; Redzwan, I.E.; Primaharinastiti, R.; Saiman, M.Z.; Fairuza, R.; Widyaningsih, T.D.; AlAjmi, M.F.; et al. GC-MS- and NMR-Based Metabolomics and Molecular Docking Reveal the Potential Alpha-Glucosidase Inhibitors from Psychotria malayana Jack Leaves. Pharmaceuticals 2021, 14, 978. https://doi.org/10.3390/ph14100978
Nipun TS, Khatib A, Ibrahim Z, Ahmed QU, Redzwan IE, Primaharinastiti R, Saiman MZ, Fairuza R, Widyaningsih TD, AlAjmi MF, et al. GC-MS- and NMR-Based Metabolomics and Molecular Docking Reveal the Potential Alpha-Glucosidase Inhibitors from Psychotria malayana Jack Leaves. Pharmaceuticals. 2021; 14(10):978. https://doi.org/10.3390/ph14100978
Chicago/Turabian StyleNipun, Tanzina Sharmin, Alfi Khatib, Zalikha Ibrahim, Qamar Uddin Ahmed, Irna Elina Redzwan, Riesta Primaharinastiti, Mohd Zuwairi Saiman, Raudah Fairuza, Tri Dewanti Widyaningsih, Mohamed F. AlAjmi, and et al. 2021. "GC-MS- and NMR-Based Metabolomics and Molecular Docking Reveal the Potential Alpha-Glucosidase Inhibitors from Psychotria malayana Jack Leaves" Pharmaceuticals 14, no. 10: 978. https://doi.org/10.3390/ph14100978
APA StyleNipun, T. S., Khatib, A., Ibrahim, Z., Ahmed, Q. U., Redzwan, I. E., Primaharinastiti, R., Saiman, M. Z., Fairuza, R., Widyaningsih, T. D., AlAjmi, M. F., Khalifa, S. A. M., & El-Seedi, H. R. (2021). GC-MS- and NMR-Based Metabolomics and Molecular Docking Reveal the Potential Alpha-Glucosidase Inhibitors from Psychotria malayana Jack Leaves. Pharmaceuticals, 14(10), 978. https://doi.org/10.3390/ph14100978