
Implications of Antigen Selection on T Cell-Based Immunotherapy


Abstract
:1. Introduction
2. T Cell-Mediated Immunity
2.1. Cancer Antigens
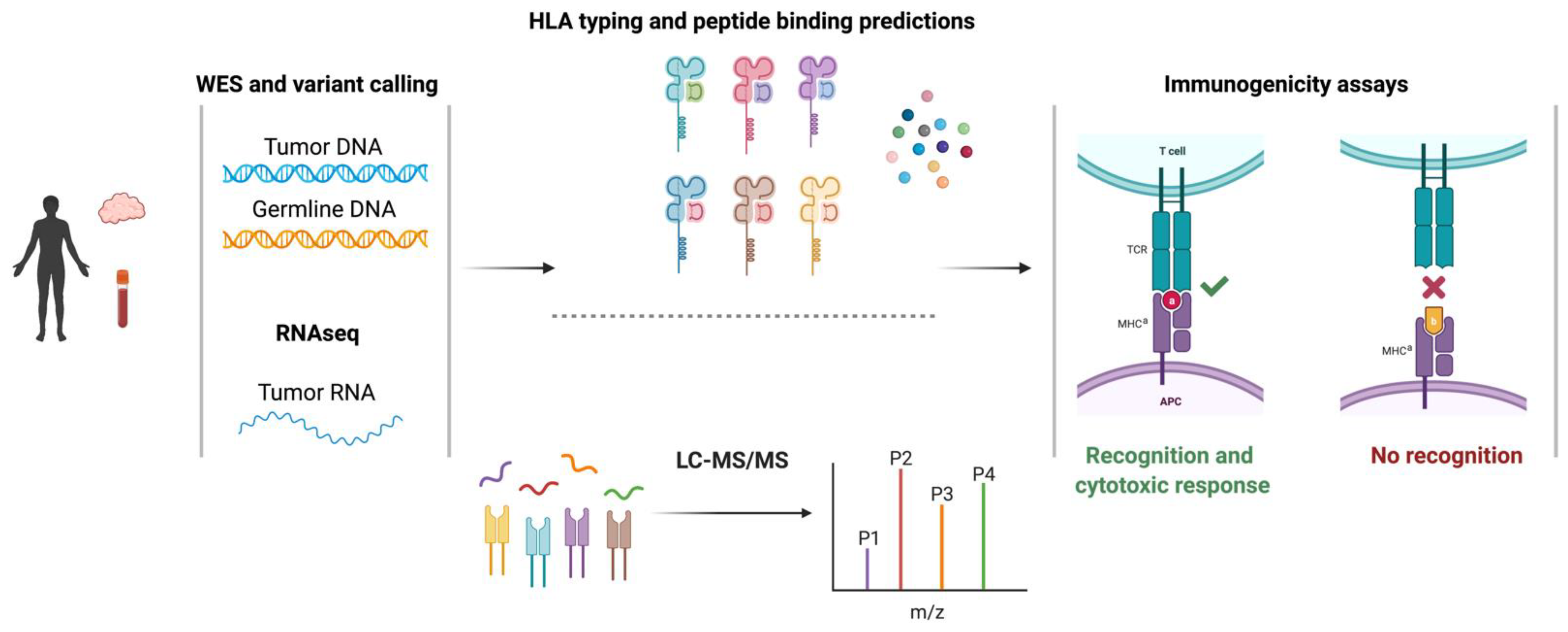
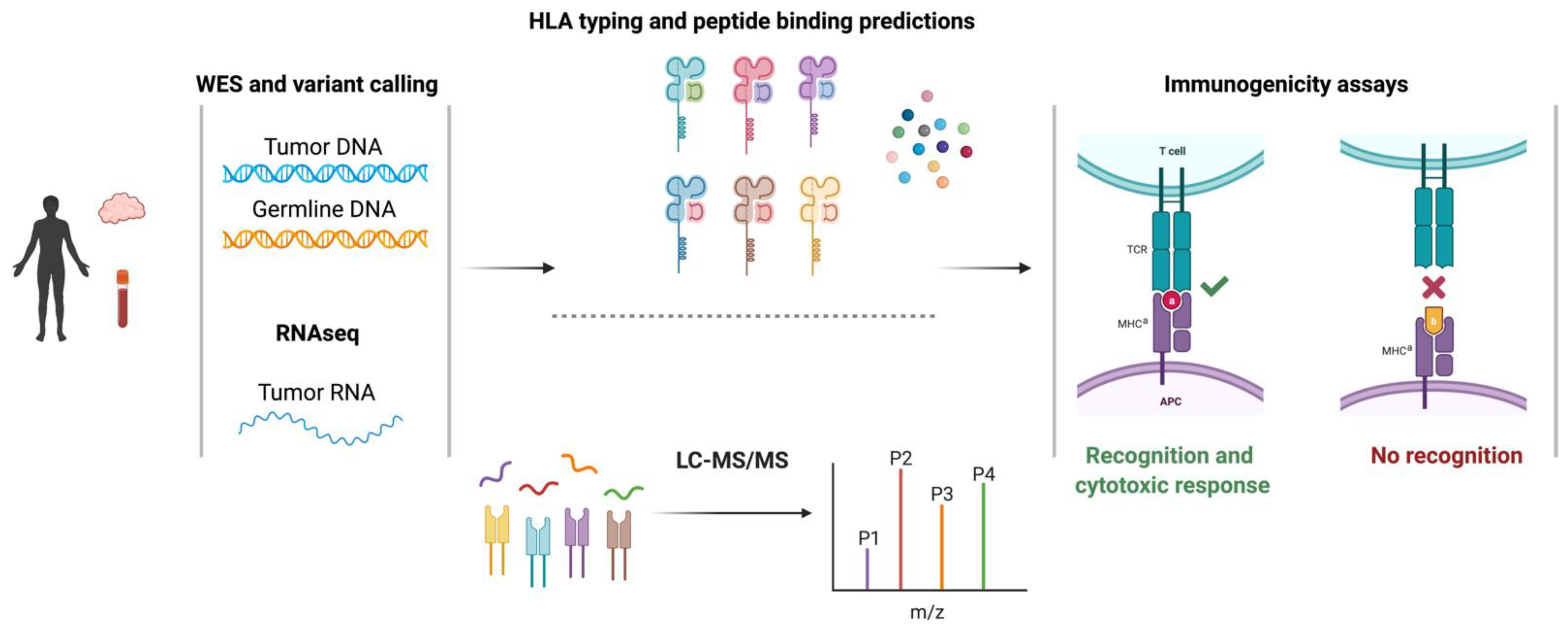
2.2. Identification of Single-Nucleotide Variant-Derived Neoantigens
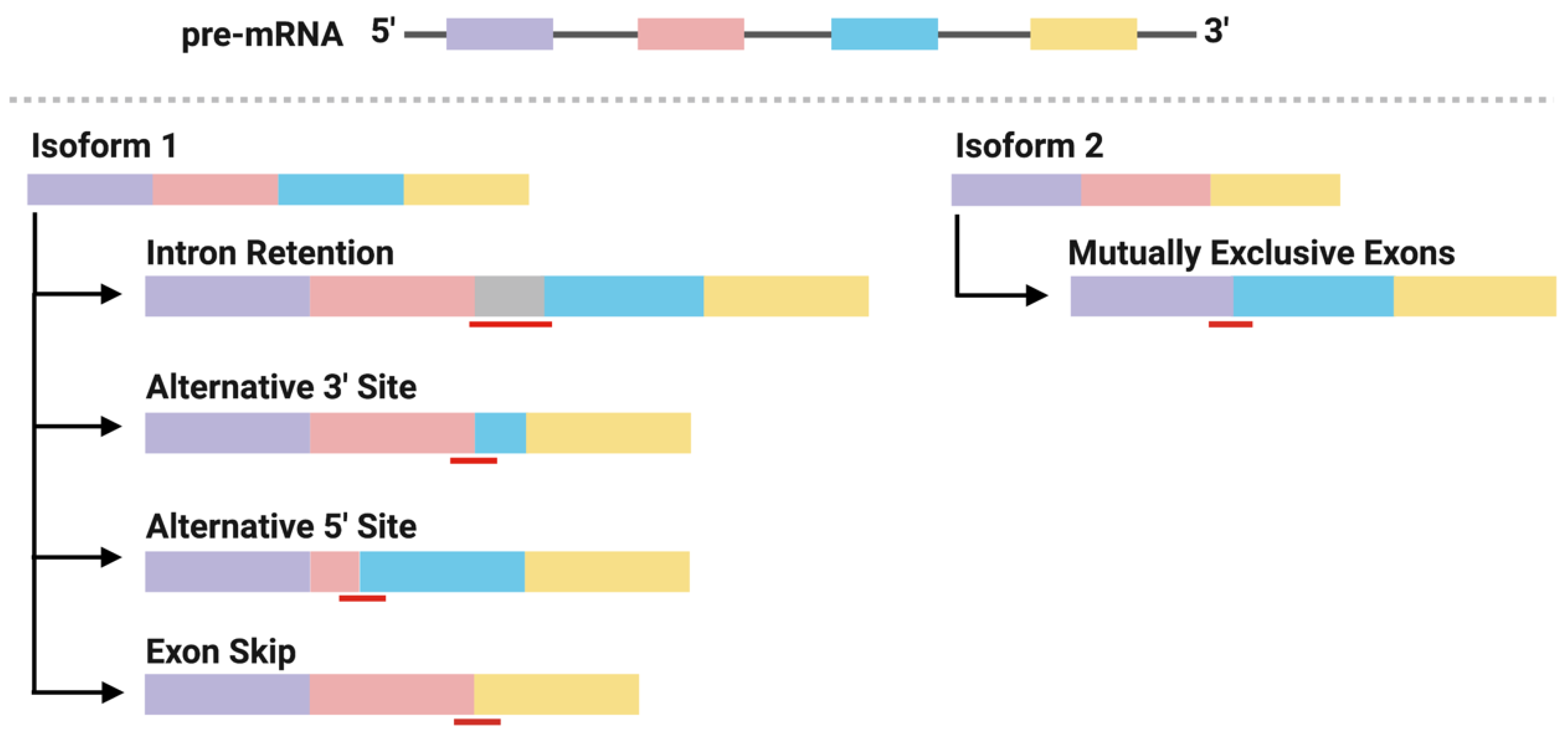
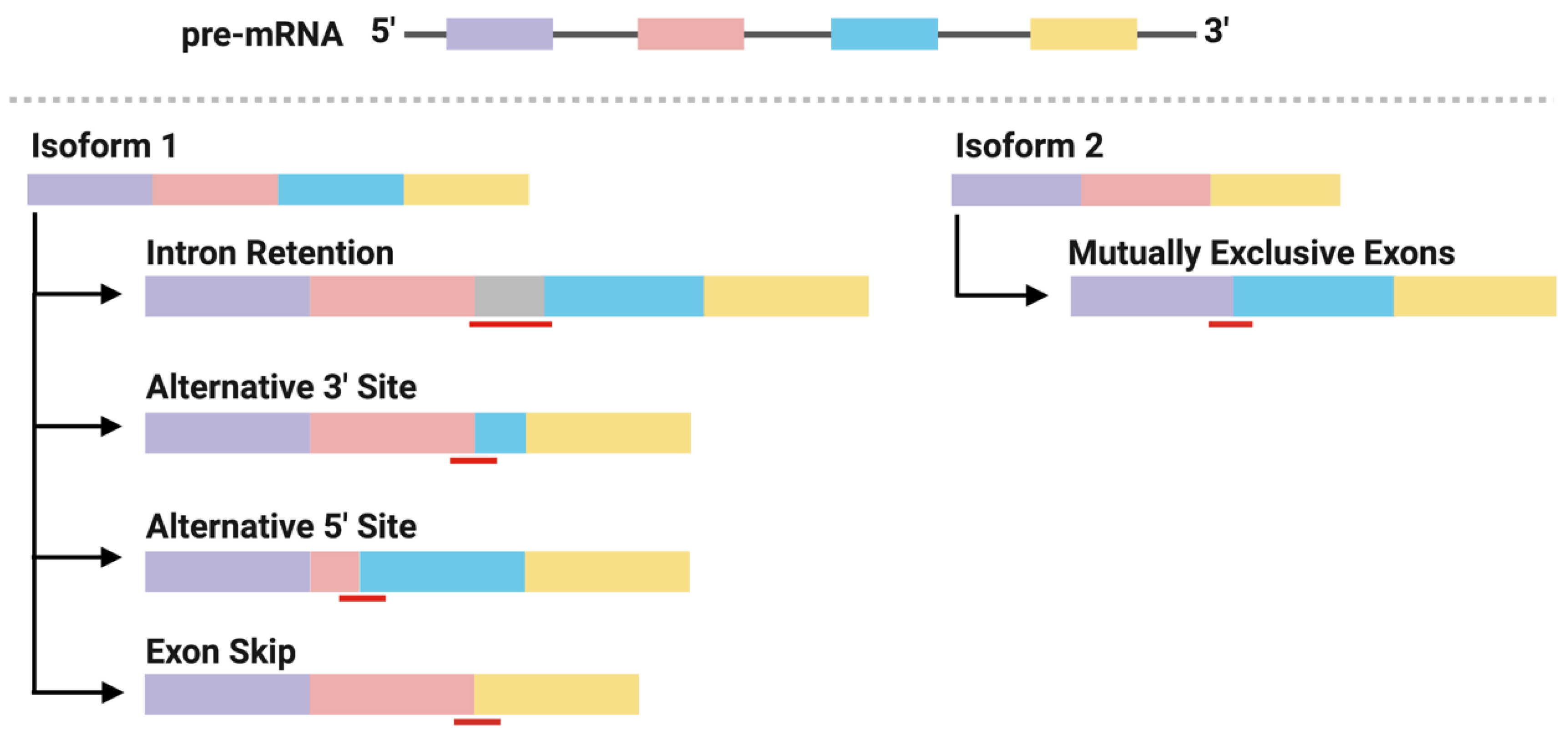
2.3. Neoantigens Derived from Alternative Splicing
2.4. Identification of Alternative Splicing-Derived Neoantigens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Server | Access (as of 27 September 2021) | Refs |
|---|---|---|
| MISO | http://hollywood.mit.edu/burgelab/miso/ | [56] |
| rMATS * | http://rnaseq-mats.sourceforge.net/rmats3.2.4/ | [57] |
| MAJIQ * | https://majiq.biociphers.org | [58] |
| LeafCutter | https://github.com/davidaknowles/leafcutter/ | [59] |
| SplAdder * | https://github.com/ratschlab/spladder | [60] |
| JUM | https://github.com/qqwang-berkeley/JUM | [61] |
| Whippet | https://github.com/timbitz/Whippet.jl | [62] |
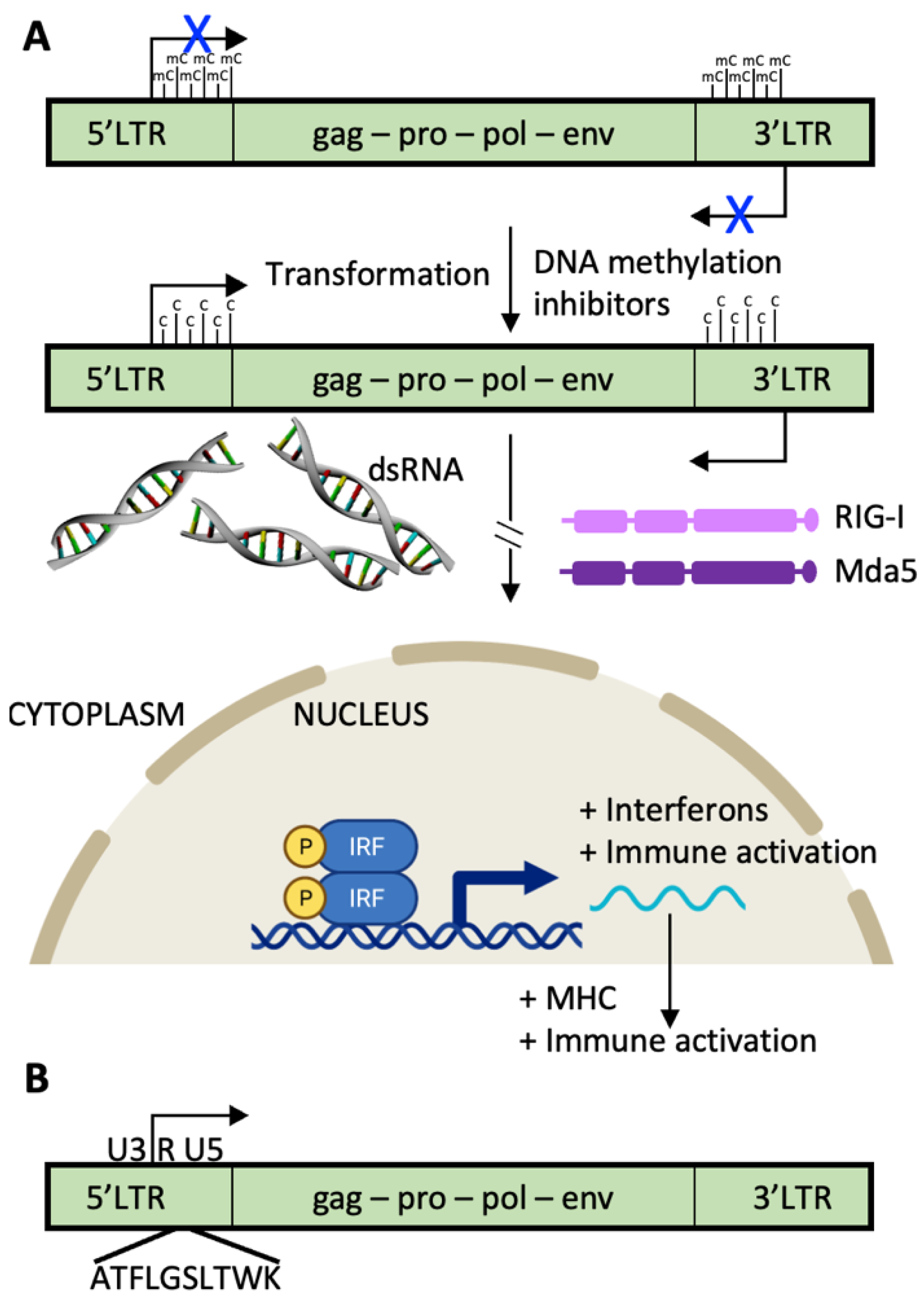
2.5. Endogenous Retroviruses
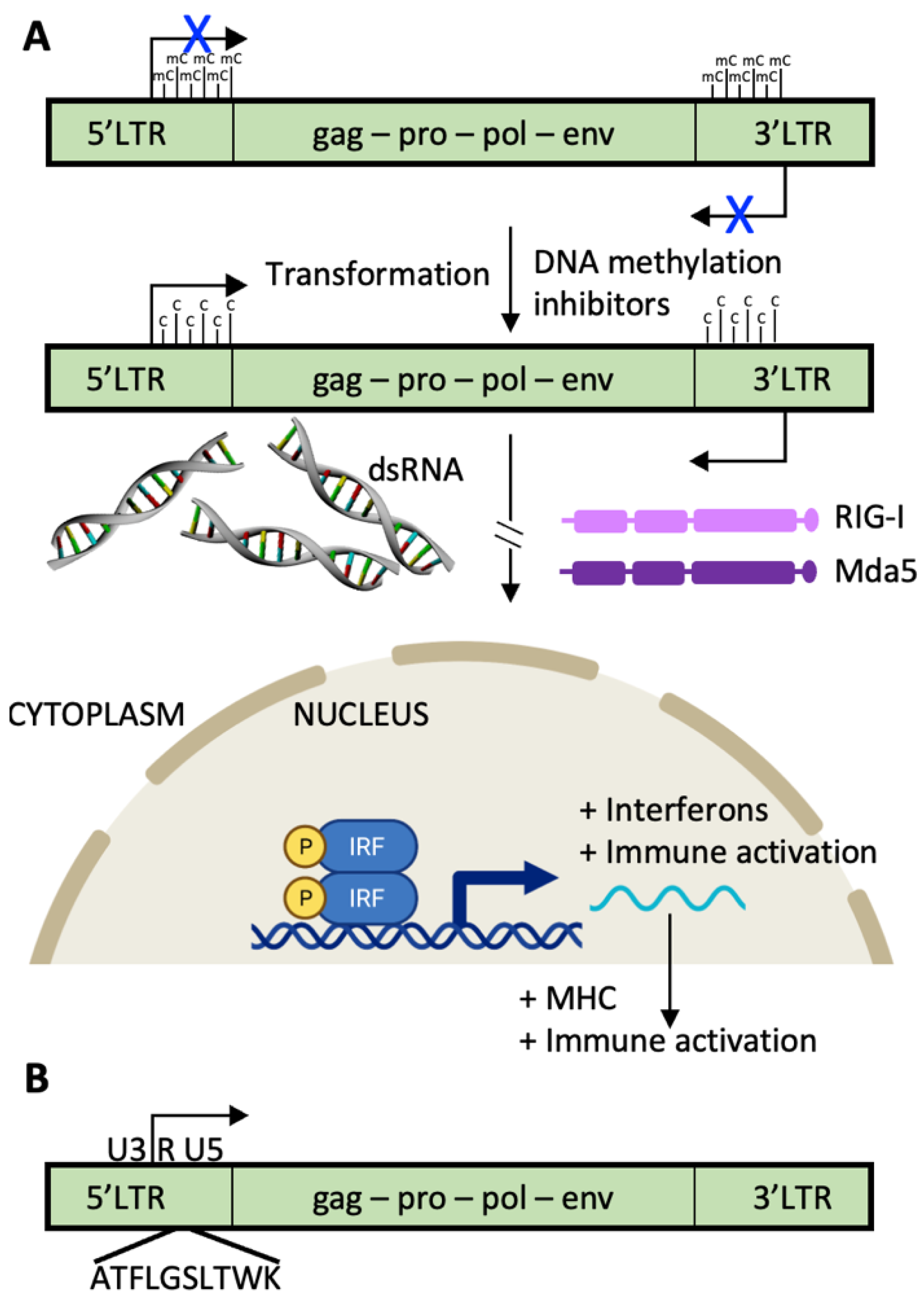
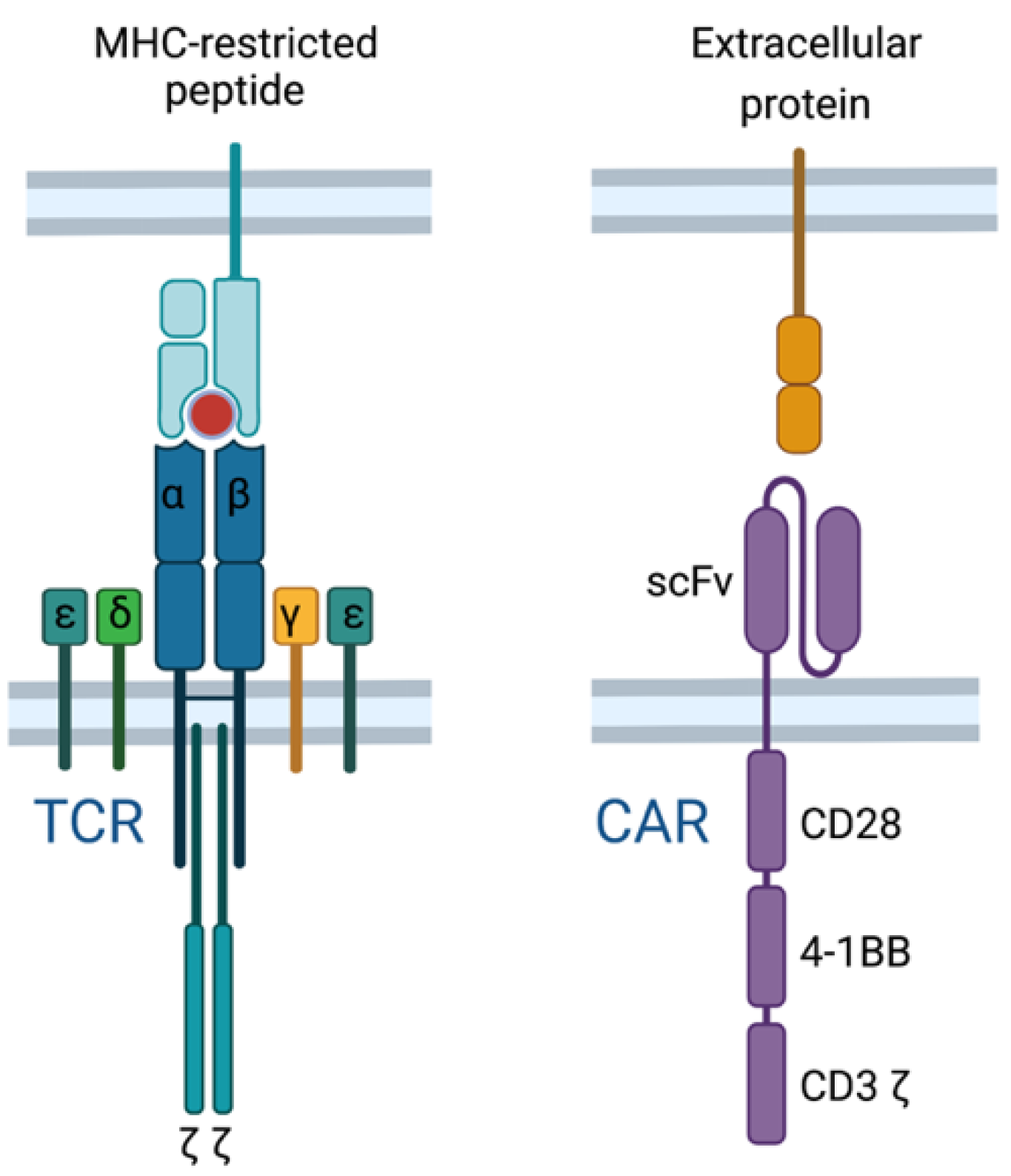
2.6. TCR T Cell vs. CAR T Cell Response
3. Summary, Conclusions, and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F. Tumor antigens discovery: Perspectives for cancer therapy. Mol. Med. 1997, 3, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Grigoriadis, A.; Caballero, O.L.; Hoek, K.S.; da Silva, L.; Chen, Y.T.; Shin, S.J.; Jungbluth, A.A.; Miller, L.D.; Clouston, D.; Cebon, J.; et al. CT-X antigen expression in human breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 13493–13498. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Gervois, N.; Schneider, J.; Escobar, P.; Valmori, D.; Pannetier, C.; Steinle, A.; Wolfel, T.; Lienard, D.; Brichard, V.; et al. Cytolytic T lymphocyte recognition of the immunodominant HLA-A*0201-restricted Melan-A/MART-1 antigenic peptide in melanoma. J. Immunol. 1997, 159, 2366–2374. [Google Scholar]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Bose, M.; Mukherjee, P. Potential of Anti-MUC1 Antibodies as a Targeted Therapy for Gastrointestinal Cancers. Vaccines 2020, 8, 659. [Google Scholar] [CrossRef]
- Feng, D.; Shaikh, A.S.; Wang, F. Recent Advance in Tumor-associated Carbohydrate Antigens (TACAs)-based Antitumor Vaccines. ACS Chem. Biol. 2016, 11, 850–863. [Google Scholar] [CrossRef]
- Hua, C.; Boussemart, L.; Mateus, C.; Routier, E.; Boutros, C.; Cazenave, H.; Viollet, R.; Thomas, M.; Roy, S.; Benannoune, N.; et al. Association of Vitiligo with Tumor Response in Patients with Metastatic Melanoma Treated with Pembrolizumab. JAMA Dermatol. 2016, 152, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Schultz, N.A.; Roslind, A.; Christensen, I.J.; Horn, T.; Hogdall, E.; Pedersen, L.N.; Kruhoffer, M.; Burcharth, F.; Wojdemann, M.; Johansen, J.S. Frequencies and prognostic role of KRAS and BRAF mutations in patients with localized pancreatic and ampullary adenocarcinomas. Pancreas 2012, 41, 759–766. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [Green Version]
- Gopanenko, A.V.; Kosobokova, E.N.; Kosorukov, V.S. Main Strategies for the Identification of Neoantigens. Cancers 2020, 12, 2879. [Google Scholar] [CrossRef]
- Park, J.; Chung, Y.J. Identification of neoantigens derived from alternative splicing and RNA modification. Genom. Inform. 2019, 17, e23. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Paul, S.; Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Sette, A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J. Immunol. 2013, 191, 5831–5839. [Google Scholar] [CrossRef] [Green Version]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Kesmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Venkatesh, G.; Grover, A.; Srinivasaraghavan, G.; Rao, S. MHCAttnNet: Predicting MHC-peptide bindings for MHC alleles classes I and II using an attention-based deep neural model. Bioinformatics 2020, 36, i399–i406. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Chong, C.; Guillaume, P.; Solleder, M.; Pak, H.; Gannon, P.O.; Kandalaft, L.E.; Coukos, G.; Gfeller, D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 2017, 13, e1005725. [Google Scholar] [CrossRef]
- Gfeller, D.; Guillaume, P.; Michaux, J.; Pak, H.S.; Daniel, R.T.; Racle, J.; Coukos, G.; Bassani-Sternberg, M. The Length Distribution and Multiple Specificity of Naturally Presented HLA-I Ligands. J. Immunol. 2018, 201, 3705–3716. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e7. [Google Scholar] [CrossRef]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Braunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Kubo, M.; Umebayashi, M.; Yew, P.Y.; Yoshimura, S.; Park, J.H.; Kiyotani, K.; Kai, M.; Yamada, M.; Oda, Y.; et al. Neoantigens elicit T cell responses in breast cancer. Sci. Rep. 2021, 11, 13590. [Google Scholar] [CrossRef]
- Broseus, L.; Ritchie, W. Challenges in detecting and quantifying intron retention from next generation sequencing data. Comput. Struct. Biotechnol. J. 2020, 18, 501–508. [Google Scholar] [CrossRef]
- Slansky, J.E.; Spellman, P.T. Alternative Splicing in Tumors—A Path to Immunogenicity? N. Engl. J. Med. 2019, 380, 877–880. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research, N.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Apcher, S.; Daskalogianni, C.; Lejeune, F.; Manoury, B.; Imhoos, G.; Heslop, L.; Fahraeus, R. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc. Natl. Acad. Sci. USA 2011, 108, 11572–11577. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Fitzgerald, E.B.; Kawakami, Y.; Rosenberg, S.A. The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J. Immunol. 1997, 159, 303–308. [Google Scholar]
- Coulie, P.G.; Lehmann, F.; Lethe, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Li, Y.F.; El-Gamil, M.; Ohnmacht, G.A.; Rosenberg, S.A.; Robbins, P.F. Melanoma-Reactive CD8+ T cells recognize a novel tumor antigen expressed in a wide variety of tumor types. J. Immunother. 2001, 24, 323–333. [Google Scholar] [CrossRef]
- Lupetti, R.; Pisarra, P.; Verrecchia, A.; Farina, C.; Nicolini, G.; Anichini, A.; Bordignon, C.; Sensi, M.; Parmiani, G.; Traversari, C. Translation of a retained intron in tyrosinase-related protein (TRP) 2 mRNA generates a new cytotoxic T lymphocyte (CTL)-defined and shared human melanoma antigen not expressed in normal cells of the melanocytic lineage. J. Exp. Med. 1998, 188, 1005–1016. [Google Scholar] [CrossRef]
- Guilloux, Y.; Lucas, S.; Brichard, V.G.; Van Pel, A.; Viret, C.; De Plaen, E.; Brasseur, F.; Lethe, B.; Jotereau, F.; Boon, T. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J. Exp. Med. 1996, 183, 1173–1183. [Google Scholar] [CrossRef]
- Aarnoudse, C.A.; van den Doel, P.B.; Heemskerk, B.; Schrier, P.I. Interleukin-2-induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int. J. Cancer 1999, 82, 442–448. [Google Scholar] [CrossRef]
- Wang, R.F.; Parkhurst, M.R.; Kawakami, Y.; Robbins, P.F.; Rosenberg, S.A. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J. Exp. Med. 1996, 183, 1131–1140. [Google Scholar] [CrossRef]
- Wang, R.F.; Johnston, S.L.; Zeng, G.; Topalian, S.L.; Schwartzentruber, D.J.; Rosenberg, S.A. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J. Immunol. 1998, 161, 3598–3606. [Google Scholar]
- Ronsin, C.; Chung-Scott, V.; Poullion, I.; Aknouche, N.; Gaudin, C.; Triebel, F. A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J. Immunol. 1999, 163, 483–490. [Google Scholar]
- Van Den Eynde, B.J.; Gaugler, B.; Probst-Kepper, M.; Michaux, L.; Devuyst, O.; Lorge, F.; Weynants, P.; Boon, T. A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J. Exp. Med. 1999, 190, 1793–1800. [Google Scholar] [CrossRef]
- Vauchy, C.; Gamonet, C.; Ferrand, C.; Daguindau, E.; Galaine, J.; Beziaud, L.; Chauchet, A.; Henry Dunand, C.J.; Deschamps, M.; Rohrlich, P.S.; et al. CD20 alternative splicing isoform generates immunogenic CD4 helper T epitopes. Int. J. Cancer 2015, 137, 116–126. [Google Scholar] [CrossRef]
- Kobayashi, J.; Torigoe, T.; Hirohashi, Y.; Idenoue, S.; Miyazaki, A.; Yamaguchi, A.; Hiratsuka, H.; Sato, N. Comparative study on the immunogenicity between an HLA-A24-restricted cytotoxic T-cell epitope derived from survivin and that from its splice variant survivin-2B in oral cancer patients. J. Transl. Med. 2009, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- David, J.K.; Maden, S.K.; Weeder, B.R.; Thompson, R.F.; Nellore, A. Putatively cancer-specific exon-exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer 2020, 2, zcaa001. [Google Scholar] [CrossRef] [Green Version]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Green, C.J.; Gazzara, M.R.; Barash, Y. MAJIQ-SPEL: Web-tool to interrogate classical and complex splicing variations from RNA-Seq data. Bioinformatics 2018, 34, 300–302. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.I.; Knowles, D.A.; Humphrey, J.; Barbeira, A.N.; Dickinson, S.P.; Im, H.K.; Pritchard, J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018, 50, 151–158. [Google Scholar] [CrossRef]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef]
- Wang, Q.; Rio, D.C. JUM is a computational method for comprehensive annotation-free analysis of alternative pre-mRNA splicing patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E8181–E8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.Y.; Gulden, P.H.; Woods, A.S.; Thomas, M.C.; Tong, C.D.; Wang, W.; Engelhard, V.H.; Pasternack, G.; Cotter, R.; Hunt, D.; et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc. Natl. Acad. Sci. USA 1996, 93, 9730–9735. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, J.A.; Sullivan, R.T.; Jordan, K.R.; McMahan, R.H.; Kemmler, C.B.; McDuffie, M.; Slansky, J.E. Age-dependent tolerance to an endogenous tumor-associated antigen. Vaccine 2008, 26, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Cherkasova, E.; Scrivani, C.; Doh, S.; Weisman, Q.; Takahashi, Y.; Harashima, N.; Yokoyama, H.; Srinivasan, R.; Linehan, W.M.; Lerman, M.I.; et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 2016, 76, 2177–2185. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Harashima, N.; Kajigaya, S.; Yokoyama, H.; Cherkasova, E.; McCoy, J.P.; Hanada, K.; Mena, O.; Kurlander, R.; Tawab, A.; et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J. Clin. Investig. 2008, 118, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.K.; Orskov, A.D.; Bjerregaard, A.M.; Unnikrishnan, A.; Holmberg-Thyden, S.; Borch, A.; Jensen, K.V.; Anande, G.; Bentzen, A.K.; Marquard, A.M.; et al. Human endogenous retroviruses form a reservoir of T cell targets in hematological cancers. Nat. Commun. 2020, 11, 5660. [Google Scholar] [CrossRef]
- Kolbe, A.R.; Bendall, M.L.; Pearson, A.T.; Paul, D.; Nixon, D.F.; Perez-Losada, M.; Crandall, K.A. Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses 2020, 12, 956. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Thomas, A.; Routh, E.D.; Pullikuth, A.; Jin, G.; Su, J.; Chou, J.W.; Hoadley, K.A.; Print, C.; Knowlton, N.; Black, M.A.; et al. Tumor mutational burden is a determinant of immune-mediated survival in breast cancer. Oncoimmunology 2018, 7, e1490854. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Adams, S.; Loibl, S.; Budczies, J.; Denkert, C.; Salgado, R. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: Clinical utility in an era of checkpoint inhibition. Ann. Oncol. 2021, 32, 1236–1244. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Li, M.; Esteva, F.J.; Hess, K.R.; Yin, B.; Rycaj, K.; Plummer, J.B.; Garza, J.G.; Ambs, S.; Johanning, G.L. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int. J. Cancer 2014, 134, 587–595. [Google Scholar] [CrossRef]
- Johanning, G.L.; Malouf, G.G.; Zheng, X.; Esteva, F.J.; Weinstein, J.N.; Wang-Johanning, F.; Su, X. Expression of human endogenous retrovirus-K is strongly associated with the basal-like breast cancer phenotype. Sci. Rep. 2017, 7, 41960. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, M.; Kong, Y.; Song, E.; Jayewickreme, T.; Kang, I.; Iwasaki, A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2018, 115, 12565–12572. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, M.; Kong, Y.; Iwasaki, A. Reply to Iniguez et al.: ERVmap is a validated approach to mapping proviral endogenous retroviruses in the human genome. Proc. Natl. Acad. Sci. USA 2019, 116, 21352–21353. [Google Scholar] [CrossRef]
- Bendall, M.L.; de Mulder, M.; Iniguez, L.P.; Lecanda-Sanchez, A.; Perez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Teran, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Jiang, Y.; Xiang, S.; Kaboli, P.J.; Shen, J.; Zhao, Y.; Wu, X.; Du, F.; Li, M.; Cho, C.H.; et al. Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front. Immunol. 2021, 12, 658753. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Fittall, M.W.; Van Loo, P. Translating insights into tumor evolution to clinical practice: Promises and challenges. Genome Med. 2019, 11, 20. [Google Scholar] [CrossRef]
- Zaidi, S.H.; Harrison, T.A.; Phipps, A.I.; Steinfelder, R.; Trinh, Q.M.; Qu, C.; Banbury, B.L.; Georgeson, P.; Grasso, C.S.; Giannakis, M.; et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat. Commun. 2020, 11, 3644. [Google Scholar] [CrossRef]

| Server | Training | Allele | Access (as of 27 September 2021) | Refs |
|---|---|---|---|---|
| NetMHCpan4.1 | BA and EL | MHC I | http://www.cbs.dtu.dk/services/NetMHCpan/ | [24] |
| MHCAttnNet | BA and EL | MHC I & II | https://github.com/gopuvenkat/MHCAttnNet | [25] |
| MixMHCpred2.1 | EL | MHC I | https://github.com/GfellerLab/MixMHCpred | [26,27] |
| MHCflurry | BA MHCI | MHC I | https://github.com/openvax/mhcflurry | [28] |
| NetMHCIIpan4.0 | BA and EL | MHC II | http://www.cbs.dtu.dk/services/NetMHCIIpan/ | [29] |
| MARIA | BA and EL | MHC II | https://maria.stanford.edu | [30] |
| Gene | Peptide | Tumor | Peptide Origin | Refs |
|---|---|---|---|---|
| GP100 | VYFFLPDHL | Melanoma | Intronic | [43] |
| MUM1 | EEKLIVVLF | Melanoma | Intronic | [44] |
| AIM2 | RSDSGQQARY | Melanoma | Intronic | [45] |
| TRP-2 | EVISCKLIKR | Melanoma | Intronic | [46] |
| MGAT5 | VLPDVFIRC/VLPDVFIRCV | Melanoma | Intronic | [47] |
| LAGE1 | MLMAQEALAFL | Melanoma | aORF | [48] |
| TRP1 | MSLQRQFLR | Melanoma | aORF | [49] |
| NYESO1 | LAAQERRVPR | Breast and melanoma | aORF | [50] |
| iCE | SPRWWPTCL | Renal cell carcinoma | aORF | [51] |
| RU2 | LPRWPPPQL | Renal cell carcinoma | Intronic | [52] |
| CD20 | RMSSLELVI | Lymphoma | aORF | [53] |
| Survivin-2B | AYACNTSTL | Oral cancer | aORF | [54] |
| Server | Access (as of 27 September 2021) | Refs |
|---|---|---|
| ERVmap | http://mtokuyama.github.io/ERVmap/ | [74] |
| Telescope | http://github.com/mlbendall/telescope | [75] |
| REdiscoverTE | https://research-pub.gene.com/REdiscoverTEpaper | [77] |
| Transgenic TCR T Cells | CAR T Cells | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camp, F.A.; Slansky, J.E. Implications of Antigen Selection on T Cell-Based Immunotherapy. Pharmaceuticals 2021, 14, 993. https://doi.org/10.3390/ph14100993
Camp FA, Slansky JE. Implications of Antigen Selection on T Cell-Based Immunotherapy. Pharmaceuticals. 2021; 14(10):993. https://doi.org/10.3390/ph14100993
Chicago/Turabian StyleCamp, Faye A., and Jill E. Slansky. 2021. "Implications of Antigen Selection on T Cell-Based Immunotherapy" Pharmaceuticals 14, no. 10: 993. https://doi.org/10.3390/ph14100993
APA StyleCamp, F. A., & Slansky, J. E. (2021). Implications of Antigen Selection on T Cell-Based Immunotherapy. Pharmaceuticals, 14(10), 993. https://doi.org/10.3390/ph14100993