
Effect of Selected Silyl Groups on the Anticancer Activity of 3,4-Dibromo-5-Hydroxy-Furan-2(5H)-One Derivatives

 , and
, and
Abstract
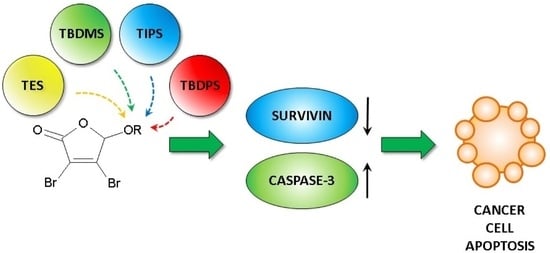
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Antiproliferative Activity
2.3. Cell Cycle Analysis and Apoptosis Induction
2.4. Colony-Formation Assay
2.5. Effect of Silyl Group on Compounds Selectivity
3. Discussion
4. Materials and Methods
4.1. Chemistry
Silyl Ethers of 3,4-Dibromo-5-hydroxy-furan-2(5H)-one 3a–d
3,4-Dibromo-5-(tert-butyldimethylsilyloxy)-furan-2(5H)-one (3a)
3,4-Dibromo-5-(triisopropylsilyloxy)-furan-2(5H)-one (3b)
3,4-Dibromo-5-(tert-butyldiphenylsilyloxy)-furan-2(5H)-one (3c)
3,4-Dibromo-5-(trietylsilyloxy)-furan-2(5H)-one (3d)
4.2. Cell Lines
4.3. MTT Assay
4.4. Cell Cycle Analysis
4.5. Annexin V-FITC Apoptosis Assay
4.6. Analysis of the Expression of Apoptosis-Related Proteins
4.7. Clonogenic Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Bernards, R. Drug Resistance to Targeted Therapies: Déjà vu All over Again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracegirdle, J.; Stevenson, L.J.; Page, M.J.; Owen, J.G.; Keyzers, R.A. Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum Kuranui. Mar. Drugs 2020, 18, 337. [Google Scholar] [CrossRef] [PubMed]
- Klapper, M.; Schlabach, K.; Paschold, A.; Zhang, S.; Chowdhury, S.; Menzel, K.; Rosenbaum, M.A.; Stallforth, P. Biosynthesis of Pseudomonas-Derived Butenolides. Angew. Chem. Int. Ed. 2020, 59, 5607–5610. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Nam, N.-H.; You, Y.-J.; Ahn, B.-Z. Synthesis and Cytotoxicity of 3,4-Diaryl-2(5H)-Furanones. Bioorg. Med. Chem. Lett. 2002, 12, 719–722. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Cao, L.; Mei, W.-J.; Wu, H.-Q.; Luo, S.-H.; Zhan, H.-Y.; Wang, Z.-Y. Bis-2(5H)-Furanone Derivatives as New Anticancer Agents: Design, Synthesis, Biological Evaluation, and Mechanism Studies. Chem. Biol. Drug Des. 2018, 92, 1232–1240. [Google Scholar] [CrossRef]
- Wei, M.-X.; Yu, J.-Y.; Liu, X.-X.; Li, X.-Q.; Zhang, M.-W.; Yang, P.-W.; Yang, J.-H. Synthesis of Artemisinin-Piperazine-Furan Ether Hybrids and Evaluation of in Vitro Cytotoxic Activity. Eur. J. Med. Chem. 2021, 215, 113295. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Luo, S.-H.; Mei, W.-J.; Cao, L.; Wu, H.-Q.; Wang, Z.-Y. Synthesis and Biological Evaluation of 4-Biphenylamino-5-Halo-2(5H)-Furanones as Potential Anticancer Agents. Eur. J. Med. Chem. 2017, 139, 84–94. [Google Scholar] [CrossRef]
- Pan, J.; Ren, D. Structural Effects on Persister Control by Brominated Furanones. Bioorg. Med. Chem. Lett. 2013, 23, 6559–6562. [Google Scholar] [CrossRef]
- Steenackers, H.P.; Levin, J.; Janssens, J.C.; Weerdt, A.D.; Balzarini, J.; Vanderleyden, J.; De Vos, D.E.; De Keersmaecker, S.C. Structure–Activity Relationship of Brominated 3-Alkyl-5-Methylene-2(5H)-Furanones and Alkylmaleic Anhydrides as Inhibitors of Salmonella Biofilm Formation and Quorum Sensing Regulated Bioluminescence in Vibrio Harveyi. Bioorg. Med. Chem. 2010, 18, 5224–5233. [Google Scholar] [CrossRef]
- Pour, M.; Špulák, M.; Balšánek, V.; Kuneš, J.; Buchta, V.; Waisser, K. 3-Phenyl-5-Methyl-2H,5H-Furan-2-Ones: Tuning Antifungal Activity by Varying Substituents on the Phenyl Ring. Bioorg. Med. Chem. Lett. 2000, 10, 1893–1895. [Google Scholar] [CrossRef]
- Pour, M.; Špulák, M.; Buchta, V.; Kubanová, P.; Vopršalová, M.; Wsól, V.; Fáková, H.; Koudelka, P.; Pourová, H.; Schiller, R. 3-Phenyl-5-Acyloxymethyl-2H,5H-Furan-2-ones: Synthesis and Biological Activity of a Novel Group of Potential Antifungal Drugs. J. Med. Chem. 2001, 44, 2701–2706. [Google Scholar] [CrossRef] [PubMed]
- Surmiak, E.; Twarda-Clapa, A.; Zak, K.M.; Musielak, B.; Tomala, M.D.; Kubica, K.; Grudnik, P.; Madej, M.; Jablonski, M.; Potempa, J.; et al. A Unique Mdm2-Binding Mode of the 3-Pyrrolin-2-One- and 2-Furanone-Based Antagonists of the P53-Mdm2 Interaction. ACS Chem. Biol. 2016, 11, 3310–3318. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Elleman, A.V.; Ghebreselasie, K.; Daniel, C.K.; Crews, B.C.; Nance, K.D.; Huda, T.; Marnett, L.J. Design of Fluorine-Containing 3,4-Diarylfuran-2(5H)-ones as Selective COX-1 Inhibitors. ACS Med. Chem. Lett. 2014, 5, 1254–1258. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Orta, M.L.; Pastor, N.; Austin, C.A.; Mateos, S.; López-Lázaro, M. Alpha, Beta-Unsaturated Lactones 2-Furanone and 2-Pyrone Induce Cellular DNA Damage, Formation of Topoisomerase I- and II-DNA Complexes and Cancer Cell Death. Toxicol. Lett. 2013, 222, 64–71. [Google Scholar] [CrossRef]
- Husain, A.; Khan, S.A.; Iram, F.; Iqbal, A.; Asif, M. Insights into the Chemistry and Therapeutic Potential of Furanones: A Versatile Pharmacophore. Eur. J. Med. Chem. 2019, 171, 66–92. [Google Scholar] [CrossRef]
- Zhang, J.; Blazecka, P.G.; Belmont, D.; Davidson, J.G. Reinvestigation of Mucohalic Acids, Versatile and Useful Building Blocks for Highly Functionalized α,β-Unsaturated γ-Butyrolactones. Org. Lett. 2002, 4, 4559–4561. [Google Scholar] [CrossRef]
- Li, A.P. Screening for Human ADME/Tox Drug Properties in Drug Discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon Molecules with Medicinal Applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef]
- Fujii, S.; Hashimoto, Y. Progress in the Medicinal Chemistry of Silicon: C/Si Exchange and Beyond. Future Med. Chem. 2017, 9, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Byczek-Wyrostek, A.; Kitel, R.; Rumak, K.; Skonieczna, M.; Kasprzycka, A.; Walczak, K. Simple 2(5H)-Furanone Derivatives with Selective Cytotoxicity towards Non-Small Cell Lung Cancer Cell Line A549—Synthesis, Structure-Activity Relationship and Biological Evaluation. Eur. J. Med. Chem. 2018, 150, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Hattum, A.H.V.; Pinedo, H.M. New Highly Lipophilic Camptothecin BNP1350 Is an Effective Drug in Experimental Human Cancer. Int. J. Cancer 2000, 88, 260–266. [Google Scholar] [CrossRef]
- Goj, K.; Rusin, A.; Szeja, W.; Kitel, R.; Komor, R.; Grynkiewicz, G. Synthesis of Genistein 2,3-anhydroglycoconjugates—Potential antiproliferative agents. Acta Pol. Pharm. Drug Res. 2012, 69, 1239–1247. [Google Scholar]
- Rodríguez-Barrios, F.; Pérez, C.; Lobatón, E.; Velázquez, S.; Chamorro, C.; San-Félix, A.; Pérez-Pérez, M.-J.; Camarasa, M.-J.; Pelemans, H.; Balzarini, J.; et al. Identification of a Putative Binding Site for [2′,5′-Bis-O-(Tert-Butyldimethylsilyl)-β-D-Ribofuranosyl]-3′-Spiro-5″-(4″-Amino-1″,2″-Oxathiole-2″,2″-Dioxide) Thymine (TSAO) Derivatives at the P51−p66 Interface of HIV-1 Reverse Transcriptase. J. Med. Chem. 2001, 44, 1853–1865. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Rim, A.S.; Dharia, C.; Clark, A.D.; Camarasa, M.-J.; Balzarini, J.; Arnold, E. Crystal Structure of Tert -Butyldimethylsilyl-Spiroaminooxathioledioxide-Thymine (TSAO-T) in Complex with HIV-1 Reverse Transcriptase (RT) Redefines the Elastic Limits of the Non-Nucleoside Inhibitor-Binding Pocket. J. Med. Chem. 2011, 54, 2727–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
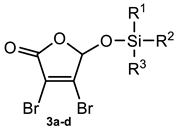 | |||
|---|---|---|---|
| Target Compound | R1 | R2 | R3 |
| 3a | Me | tert-Bu | Me |
| 3b | i-Pr | i-Pr | i-Pr |
| 3c | Ph | tert-Bu | Ph |
| 3d | Et | Et | Et |
| Compound | ||||||
|---|---|---|---|---|---|---|
| Cell Line | MBA | 3a | 3b | 3c | 3d | 5FU |
| HCT 116 wt | 20.5 ± 5.2 | 1.3 ± 0.12 | 15.1 ± 1.2 | 6.3 ± 0.3 | 1.6 ± 0.4 | 6.3 ± 1.0 |
| HCT 116 p53−/− | 25.4 ± 6.0 | 21.4 ± 3.4 | 10.8 ± 1.0 | 4.0 ± 0.3 | 4.4 ± 0.4 | 13.4 ± 1.5 |
| HT-29 | 97.1 ± 6.6 | 88.7 ± 8.2 | 7.3 ± 0.8 | 3.9 ± 0.2 | nt | 32.8 ± 4.7 |
| MCF-7 | 141.8 ± 22.6 | 186.4 ± 14.2 | 7.7 ± 0.9 | 10.9 ± 0.7 | 89.2 ± 6.0 | 29.7 ± 1.4 |
| SJSA-1 | 212.8 ± 8.8 | 196.3 ± 5.4 | 9.9 ± 1.1 | 14.9 ± 1.5 | nt | nt |
| U2OS | 97.6 ± 1.1 | nt | 9.3 ± 0.2 | 10.2 ± 0.8 | nt | 89.8 ± 2.1 |
| HepG2 | 91.7 ± 2.2 | 135.4 ± 6.0 | 15.6 ± 1.2 | nt | nt | nt |
| Hep3B | 125.4 ± 10.5 | 135.3 ± 6.1 | 21.3 ± 3.5 | 65.6 ± 2.2 | nt | nt |
| Compound | Cell Line | SI | cLogP a | |
|---|---|---|---|---|
| A549 | BEAS-2B | |||
| MBA | 203 ± 0.3 | 39.7 ± 9.7 | 0.2 | 1.02 |
| 3a | 23.4 ± 6.6 | 141.7 ± 23.1 | 6.05 | 3.08 |
| 3b | 30.4 ± 4.2 | 12.5 ± 1.4 | 0.41 | 4.06 |
| 3c | 29.1 ± 1.0 | 20.3 ± 0.6 | 0.70 | 4.68 |
| 3d | 4.7 ± 0.1 | 8.1 ± 1.2 | 1.72 | 3.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitel, R.; Byczek-Wyrostek, A.; Hopko, K.; Kasprzycka, A.; Walczak, K. Effect of Selected Silyl Groups on the Anticancer Activity of 3,4-Dibromo-5-Hydroxy-Furan-2(5H)-One Derivatives. Pharmaceuticals 2021, 14, 1079. https://doi.org/10.3390/ph14111079
Kitel R, Byczek-Wyrostek A, Hopko K, Kasprzycka A, Walczak K. Effect of Selected Silyl Groups on the Anticancer Activity of 3,4-Dibromo-5-Hydroxy-Furan-2(5H)-One Derivatives. Pharmaceuticals. 2021; 14(11):1079. https://doi.org/10.3390/ph14111079
Chicago/Turabian StyleKitel, Radoslaw, Anna Byczek-Wyrostek, Katarzyna Hopko, Anna Kasprzycka, and Krzysztof Walczak. 2021. "Effect of Selected Silyl Groups on the Anticancer Activity of 3,4-Dibromo-5-Hydroxy-Furan-2(5H)-One Derivatives" Pharmaceuticals 14, no. 11: 1079. https://doi.org/10.3390/ph14111079