
Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening

 , ,
, ,  ,
, 
Abstract
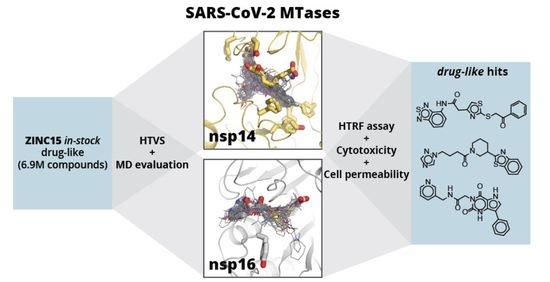
:
1. Introduction
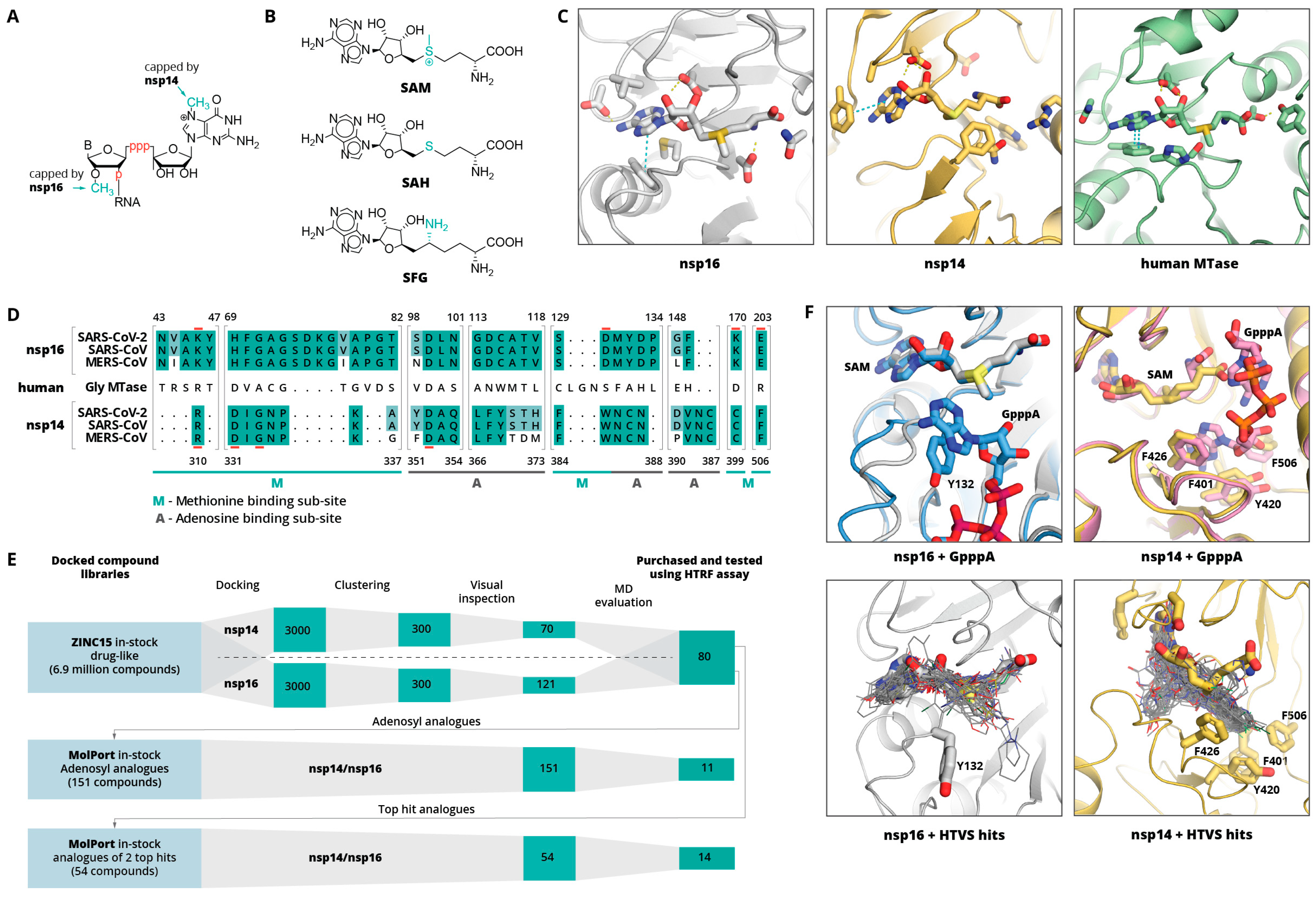
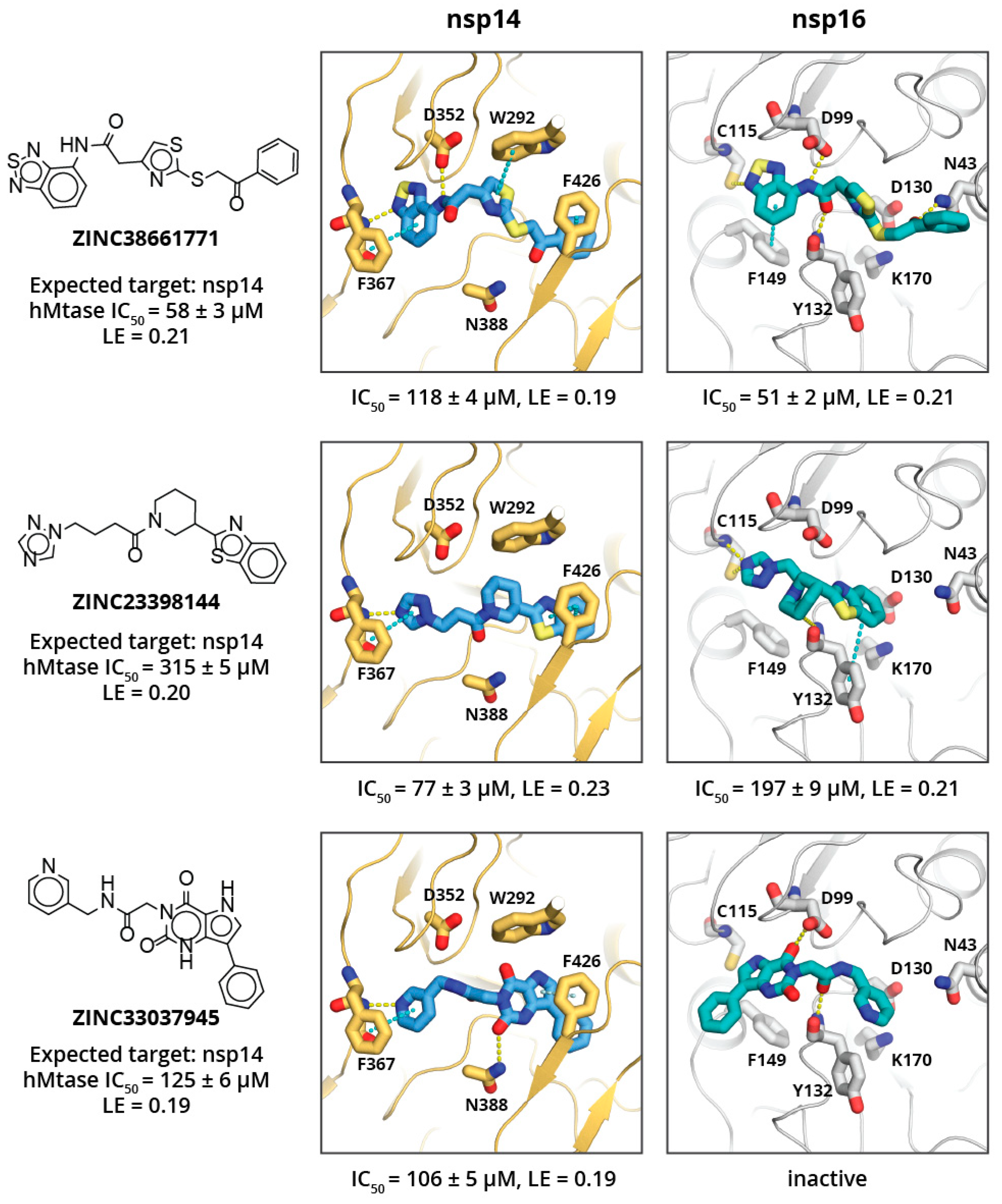
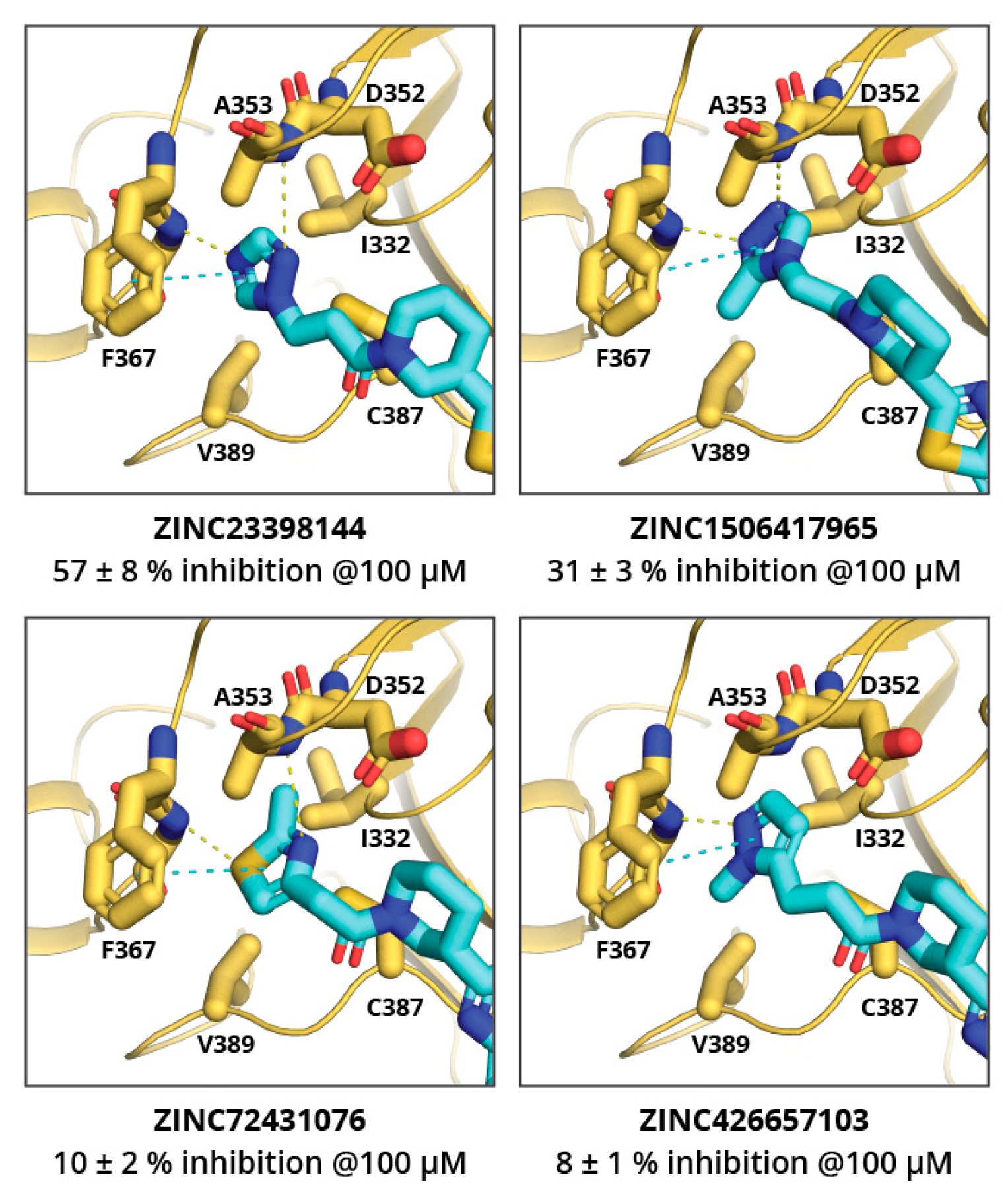
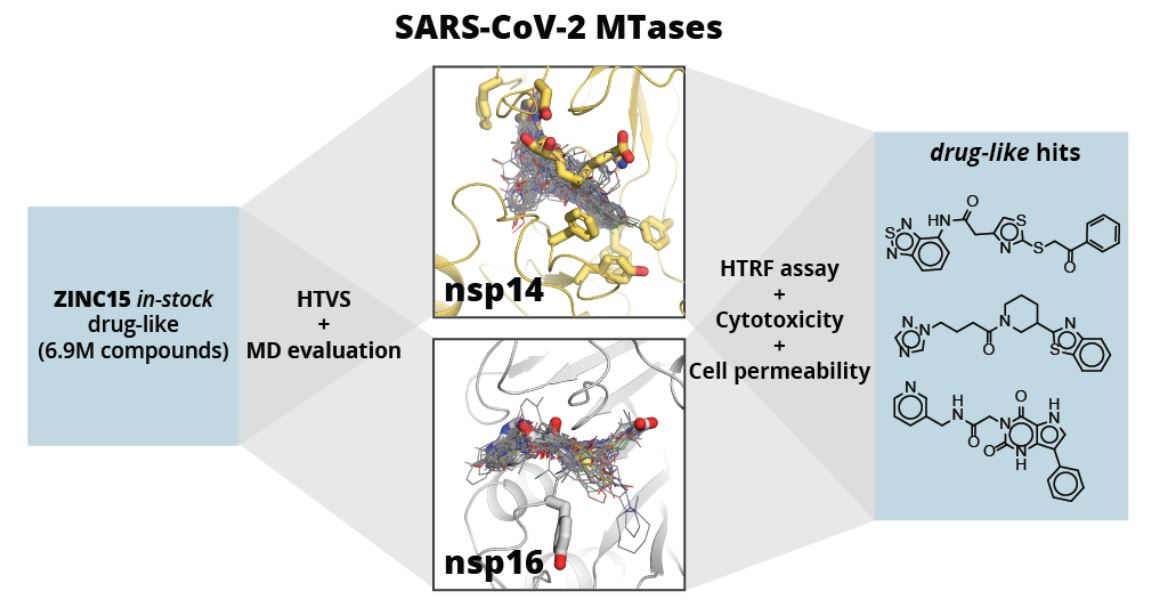
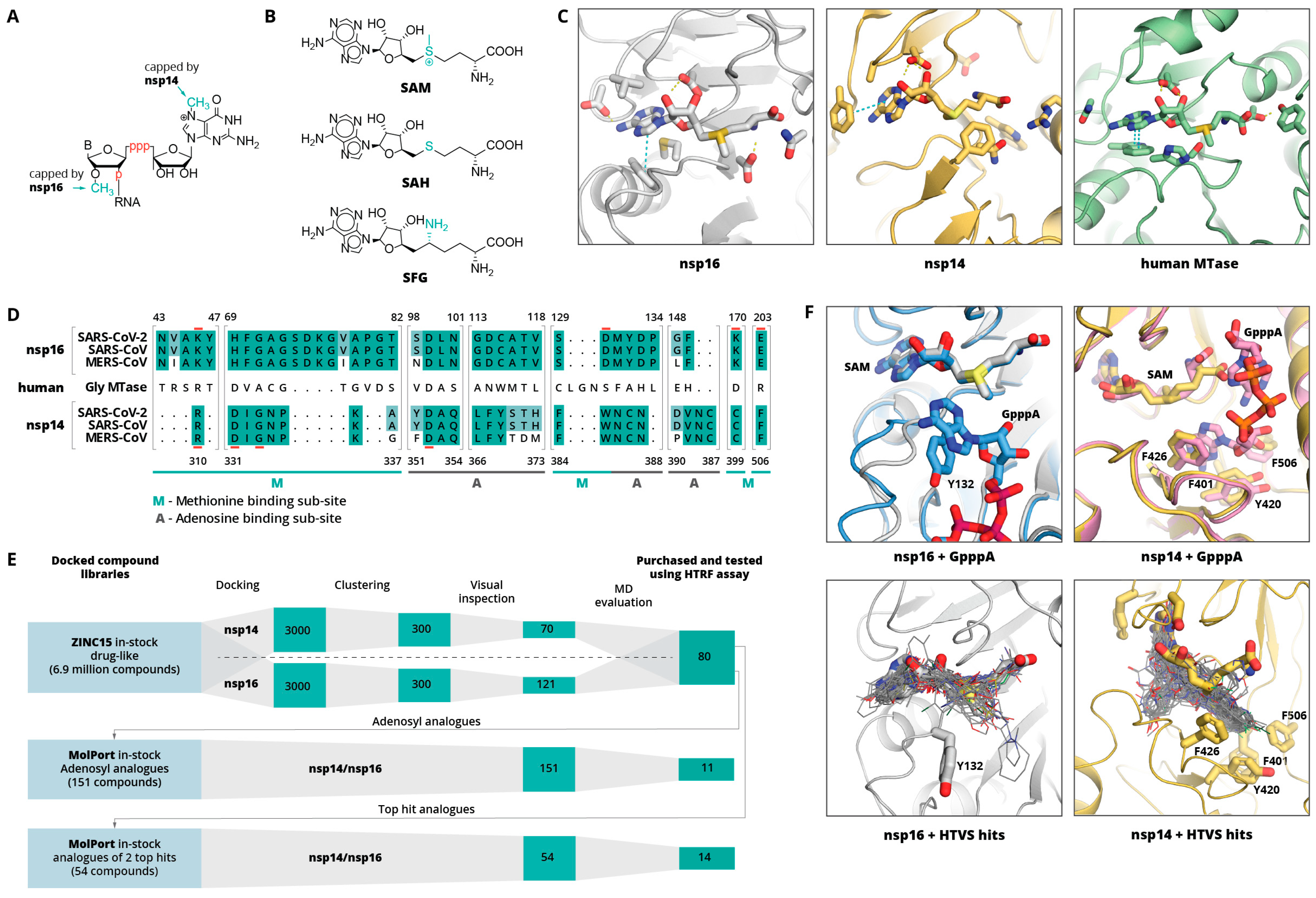
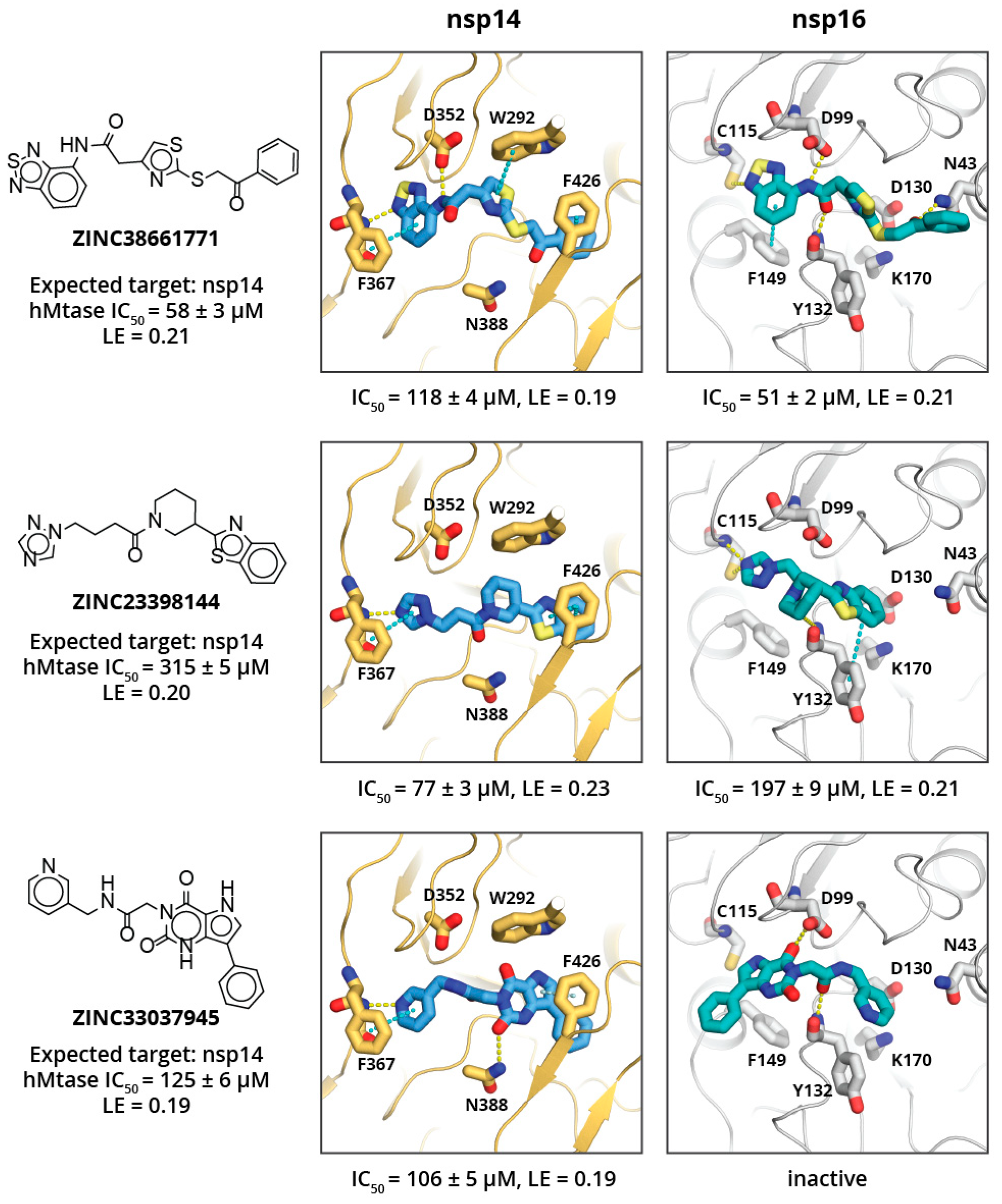
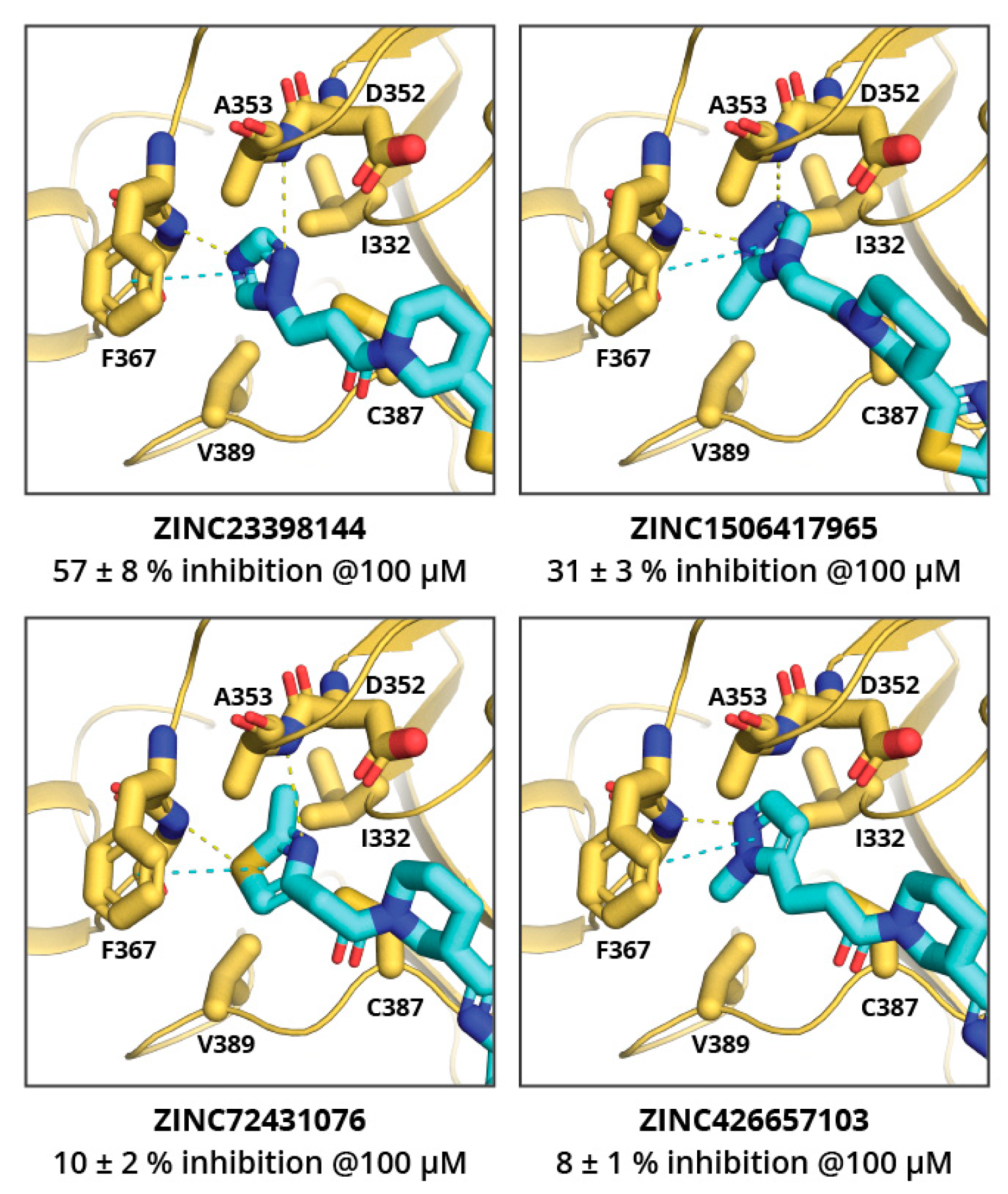
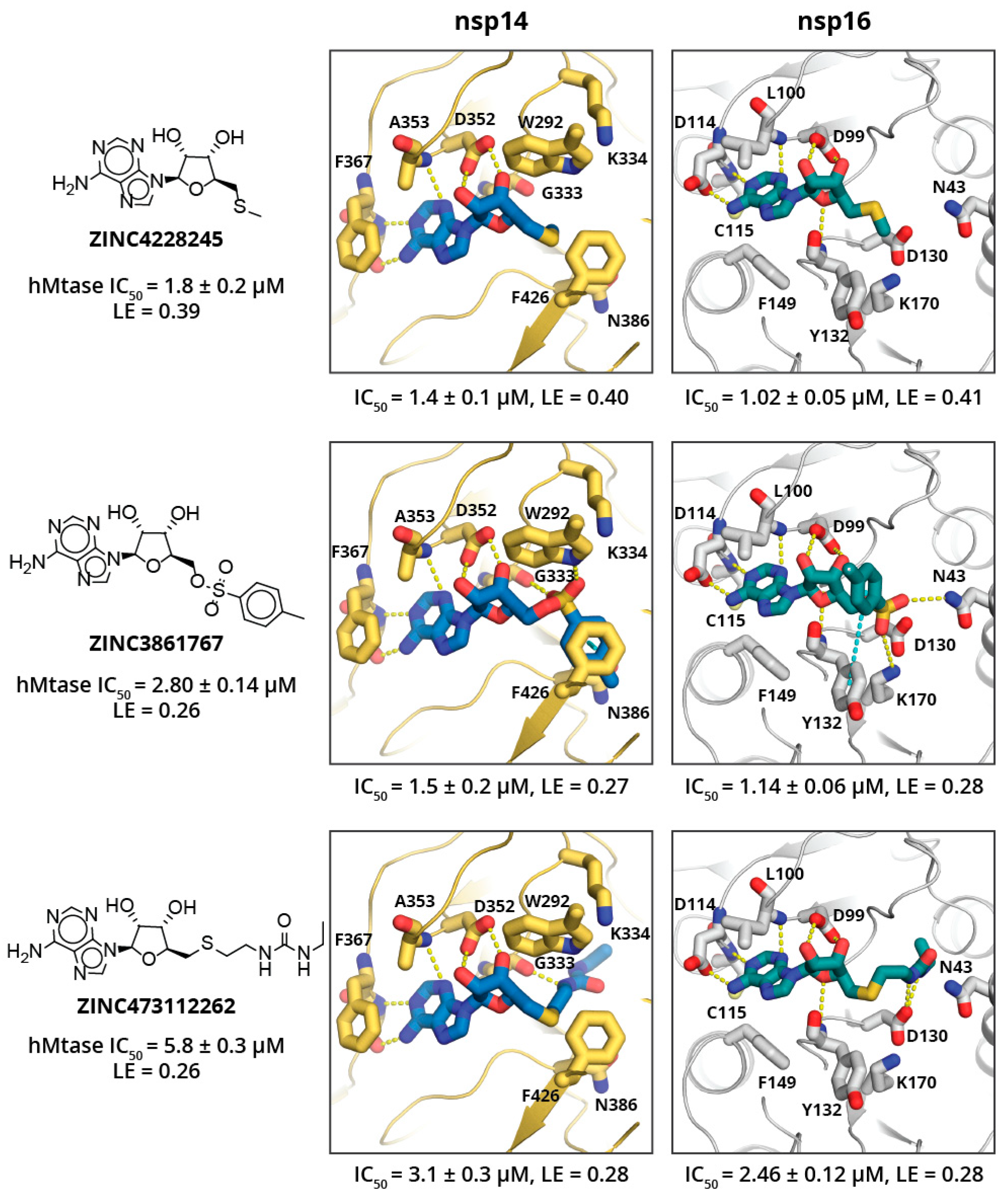
2. Results and Discussion
3. Materials and Methods
3.1. High-Throughput Virtual Screening (HTVS)
3.2. MD Calculations
3.3. Protein Expression and Purification
3.4. Homogeneous Time-Resolved Fluorescent Energy Transfer (Htrf) Assay
3.5. SARS-CoV-2 Nsp16/Nsp10 Methyltransferase Substrate RNA Production
| DEPC-treated water | 6 µL |
| 5x TranscriptAid reaction buffer | 4 µL |
| ATP, Tris buffered, 30 mM | 1 µL (1.5 mM final concentration) |
| CTP, Tris buffered, 100 mM | 1.5 µL (7.5 mM final concentration) |
| GTP, Tris buffered, 100 mM | 1.5 µL (7.5 mM final concentration) |
| UTP, Tris buffered, 100 mM | 1.5 µL (7.5 mM final concentration) |
| Cap analog G(5′)ppp(5′)A, 100 mM | 1.2 µL (6 mM final concentration) |
| Co25 template DNA, double stranded, preheated at 95 °C for 10 min, then slowly cooled down to 35 °C | 1 µg (0.5 µL) |
| TranscriptAid enzyme mix | 2 µL |
3.6. Cell Lines and Culture
3.7. Cytotoxicity Testing
3.8. Cell Permeability Testing
3.8.1. Compound Incubation in Cell Culture
3.8.2. LC/MS/MS Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef]
- Stadler, K.; Masignani, V.; Eickmann, M.; Becker, S.; Abrignani, S.; Klenk, H.D.; Rappuoli, R. SARS—Beginning to understand a new virus. Nat. Rev. Microbiol. 2003, 1, 209–218. [Google Scholar] [CrossRef]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. In Coronaviruses. Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2203, pp. 1–29. ISBN 9781071609002. [Google Scholar]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J.F. High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 13, eabe1202. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.; Lécine, P.; et al. Coronavirus Nsp10, a Critical Co-factor for Activation of Multiple Replicative Enzymes. J. Biol. Chem. 2014, 289, 25783–25796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.S.; Saikatendu, K.S.; Subramanian, V.; Neuman, B.W.; Brooun, A.; Griffith, M.; Moy, K.; Yadav, M.K.; Velasquez, J.; Buchmeier, M.J.; et al. Crystal Structure of Nonstructural Protein 10 from the Severe Acute Respiratory Syndrome Coronavirus Reveals a Novel Fold with Two Zinc-Binding Motifs. J. Virol. 2006, 80, 7894–7901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Lou, Z.; Sun, F.; Zhai, Y.; Yang, H.; Zhang, R.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Rao, Z. Dodecamer Structure of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein nsp10. J. Virol. 2006, 80, 7902–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menachery, V.D.; Debbink, K.; Baric, R.S. Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Res. 2014, 194, 191–199. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, J.; Sun, Y.; Wu, A.; Su, C.; Gao, G.; Cai, H.; Qiu, S.; Wu, Y.; Ahola, T.; et al. Structure-Function Analysis of Severe Acute Respiratory Syndrome Coronavirus RNA Cap Guanine-N7-Methyltransferase. J. Virol. 2013, 87, 6296–6305. [Google Scholar] [CrossRef] [Green Version]
- de Freitas, R.F.; Ivanochko, D.; Schapira, M. Methyltransferase inhibitors: Competing with, or exploiting the bound cofactor. Molecules 2019, 24, 4492. [Google Scholar] [CrossRef] [Green Version]
- Scheer, S.; Ackloo, S.; Medina, T.S.; Schapira, M.; Li, F.; Ward, J.A.; Lewis, A.M.; Northrop, J.P.; Richardson, P.L.; Kaniskan, H.Ü.; et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouadi, W.; Eydoux, C.; Coutard, B.; Martin, B.; Debart, F.; Vasseur, J.J.; Contreras, J.M.; Morice, C.; Quérat, G.; Jung, M.L.; et al. Toward the identification of viral cap-methyltransferase inhibitors by fluorescence screening assay. Antiviral Res. 2017, 144, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhu, X.; Lu, Y.; Zhang, X.; Jia, X.; Yang, T. Potential treatment with Chinese and Western medicine targeting NSP14 of SARS-CoV-2. J. Pharm. Anal. 2021, 11, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2662–2669. [Google Scholar] [CrossRef]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, R.; Sutto-Ortiz, P.; Guiraud, M.; Canard, B.; Vasseur, J.J.; Decroly, E.; Debart, F. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur. J. Med. Chem. 2020, 201, 112557. [Google Scholar] [CrossRef]
- Otava, T.; Šála, M.; Li, F.; Fanfrlík, J.; Devkota, K.; Perveen, S.; Chau, I.; Pakarian, P.; Hobza, P.; Vedadi, M.; et al. The Structure-Based Design of SARS-CoV-2 nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors. ACS Infect. Dis. 2021, 7, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Devkota, K.; Schapira, M.; Perveen, S.; Khalili Yazdi, A.; Li, F.; Chau, I.; Ghiabi, P.; Hajian, T.; Loppnau, P.; Bolotokova, A.; et al. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 In Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bisubstrate Inhibitors. SLAS Discov. Adv. Sci. Drug Discov. 2021, 247255522110262. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Pant, P.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S.; Sharma, P. Identification of promising drug candidates against NSP16 of SARS-CoV-2 through computational drug repurposing study. J. Biomol. Struct. Dyn. 2021, 39, 6713–6727. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.S.G.; Borchardt, R.T. Effects of S-adenosylhomocysteine analogs on vaccinia viral mRNA synthesis and methylation. Biochemistry 1982, 21, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.S.G.; Borchardt, R.T.; Stone, H.O. Inhibition of Newcastle disease virion messenger RNA (guanine-7-)-methyltransferase by analogs of S-adenosylhomocysteine. Biochemistry 1977, 16, 3928–3932. [Google Scholar] [CrossRef]
- Lin, Q.; Jiang, F.; Schultz, P.G.; Gray, N.S. Design of Allele-Specific Protein Methyltransferase Inhibitors. J. Am. Chem. Soc. 2001, 123, 11608–11613. [Google Scholar] [CrossRef]
- Bobiļeva, O.; Bobrovs, R.; Kaņepe, I.; Patetko, L.; Kalniņš, G.; Šišovs, M.; Bula, A.L.; Gri̅nberga, S.; Borodušķis, M.; Ramata-Stunda, A.; et al. Potent SARS-CoV-2 mRNA Cap Methyltransferase Inhibitors by Bioisosteric Replacement of Methionine in SAM Cosubstrate. ACS Med. Chem. Lett. 2021, 12, 1102–1107. [Google Scholar] [CrossRef]
- Canal, B.; McClure, A.W.; Curran, J.F.; Wu, M.; Ulferts, R.; Weissmann, F.; Zeng, J.; Bertolin, A.P.; Milligan, J.C.; Basu, S.; et al. Identifying SARS-CoV-2 Antiviral Compounds by Screening for Small Molecule Inhibitors of Nsp14/nsp10 Exoribonuclease. BioRxiv 2021. [Google Scholar] [CrossRef]
- Basu, S.; Mak, T.; Ulferts, R.; Wu, M.; Deegan, T.; Fujisawa, R.; Tan, K.W.; Lim, C.T.; Basier, C.; Canal, B.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp14 RNA cap methyltransferase. Biochem. J. 2021, 478, 2481–2497. [Google Scholar] [CrossRef]
- Perveen, S.; Khalili Yazdi, A.; Devkota, K.; Li, F.; Ghiabi, P.; Hajian, T.; Loppnau, P.; Bolotokova, A.; Vedadi, M. A High-Throughput RNA Displacement Assay for Screening SARS-CoV-2 nsp10-nsp16 Complex toward Developing Therapeutics for COVID-19. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 620–627. [Google Scholar] [CrossRef]
- Martin, W.R.; Cheng, F. Repurposing of FDA-Approved Toremifene to Treat COVID-19 by Blocking the Spike Glycoprotein and NSP14 of SARS-CoV-2. J. Proteome Res. 2020, 19, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Abhirami, R.; Boura, E.; Singh, S.K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 4582–4593. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Maurya, A.K.; Mishra, N.; Siddique, H.R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J. Recept. Signal Transduct. 2020, 40, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Morla, S.; Goyal, A.; Kumar, S. Computational guided drug repurposing for targeting 2′-O-ribose methyltransferase of SARS-CoV-2. Life Sci. 2020, 259, 118169. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Parihar, R.D.; Dhiman, U.; Dhamija, P.; Upadhyay, S.K.; Imran, M.; Behera, S.K.; Keshava Prasad, T.S. Docking of fda approved drugs targeting nsp-16, n-protein and main protease of SARS-CoV-2 as dual inhibitors. Biointerface Res. Appl. Chem. 2021, 11, 9848–9861. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Pakhomova, S.; Luka, Z.; Grohmann, S.; Wagner, C.; Newcomer, M.E. Glycine N-methyltransferases: A comparison of the crystal structures and kinetic properties of recombinant human, mouse and rat enzymes. Proteins Struct. Funct. Genet. 2004, 57, 331–337. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. Correction: In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Kerr, S.J. Competing methyltransferase systems. J. Biol. Chem. 1972, 247, 4248–4252. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Schrödinger Release 2020-4; Schrödinger, LLC: New York, NY, USA, 2020.
- Prime, Schrödinger Release 2020-4; Schrödinger, LLC: New York, NY, USA, 2020.
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2020.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.C.; Postma, J.P.M.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Wiersum, G.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Endres, M.; et al. The crystal structure of nsp10-nsp16 heterodimer from SARS-CoV-2 in complex with S-adenosylmethionine. BioRxiv 2020. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cytotoxicity (CC50), μM | Cell Permeability, % | |||
|---|---|---|---|---|---|
| 3T3 | HepG2 | A549 | 2 × 104 Cells/L | 4 × 104 Cells/L | |
| HTVS drug-like hits | |||||
| ZINC38661771 | 115.6 | 93.84 | 96.96 | <LOQ | <LOQ |
| ZINC23398144 | >100 | >100 | >100 | 0.7 | 0.6 |
| ZINC33037945 | >100 | >100 | >100 | 17.9 | 30.8 |
| SAM analogues | |||||
| ZINC4228245 | >200 | >200 | >200 | 0.7 | 0.8 |
| ZINC3861767 | >200 | >200 | >200 | 2.2 | 0.5 |
| ZINC473112262 | >100 | >100 | >100 | 0.3 | 0.3 |
| Sinefungin | 99.21 | >100 | 72.93 | <LOD | <LOD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobrovs, R.; Kanepe, I.; Narvaiss, N.; Patetko, L.; Kalnins, G.; Sisovs, M.; Bula, A.L.; Grinberga, S.; Boroduskis, M.; Ramata-Stunda, A.; et al. Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals 2021, 14, 1243. https://doi.org/10.3390/ph14121243
Bobrovs R, Kanepe I, Narvaiss N, Patetko L, Kalnins G, Sisovs M, Bula AL, Grinberga S, Boroduskis M, Ramata-Stunda A, et al. Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals. 2021; 14(12):1243. https://doi.org/10.3390/ph14121243
Chicago/Turabian StyleBobrovs, Raitis, Iveta Kanepe, Nauris Narvaiss, Liene Patetko, Gints Kalnins, Mihails Sisovs, Anna L. Bula, Solveiga Grinberga, Martins Boroduskis, Anna Ramata-Stunda, and et al. 2021. "Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening" Pharmaceuticals 14, no. 12: 1243. https://doi.org/10.3390/ph14121243
APA StyleBobrovs, R., Kanepe, I., Narvaiss, N., Patetko, L., Kalnins, G., Sisovs, M., Bula, A. L., Grinberga, S., Boroduskis, M., Ramata-Stunda, A., Rostoks, N., Jirgensons, A., Tars, K., & Jaudzems, K. (2021). Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals, 14(12), 1243. https://doi.org/10.3390/ph14121243