
Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
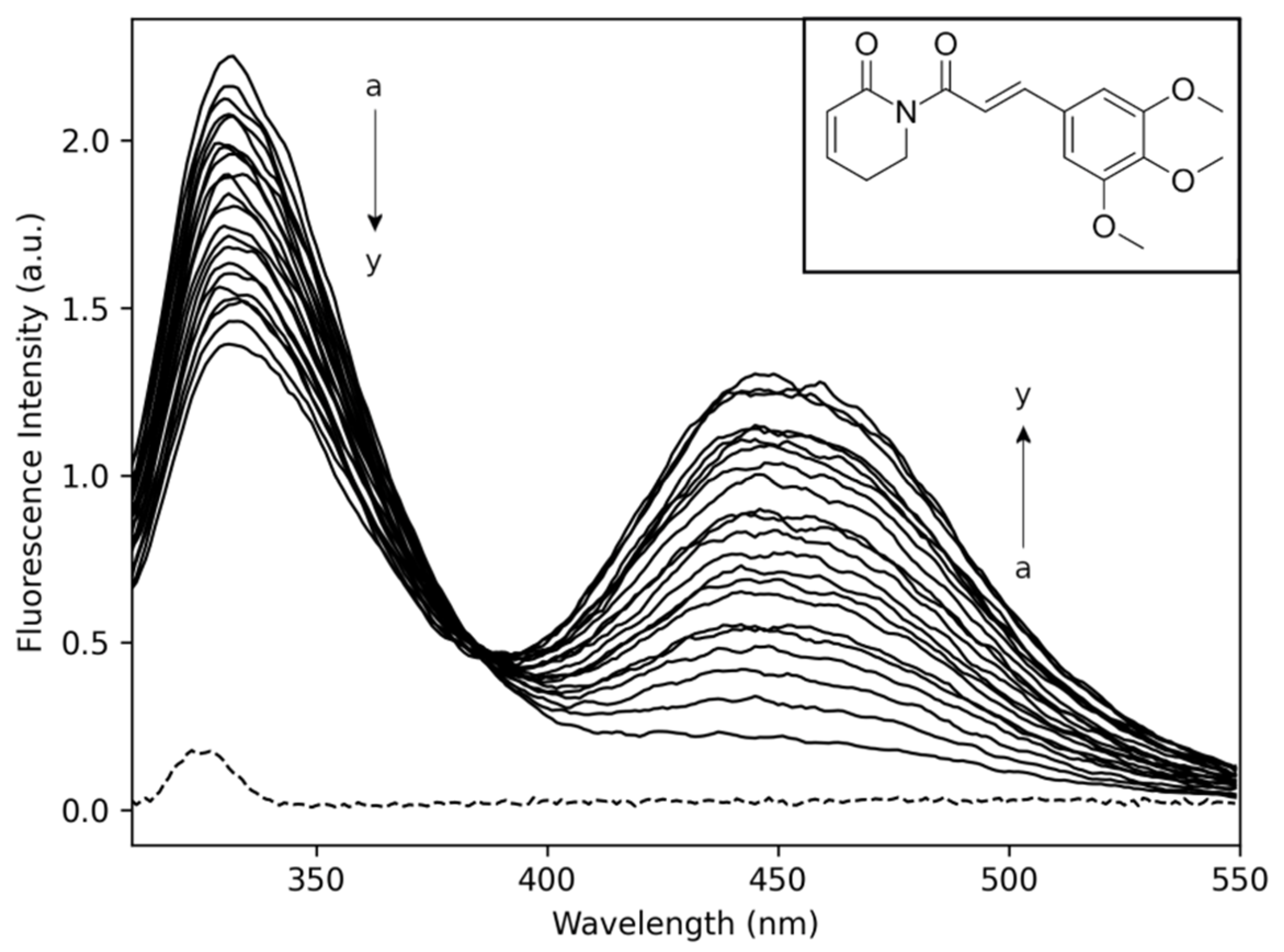
2.1. Fluorescence Spectroscopy
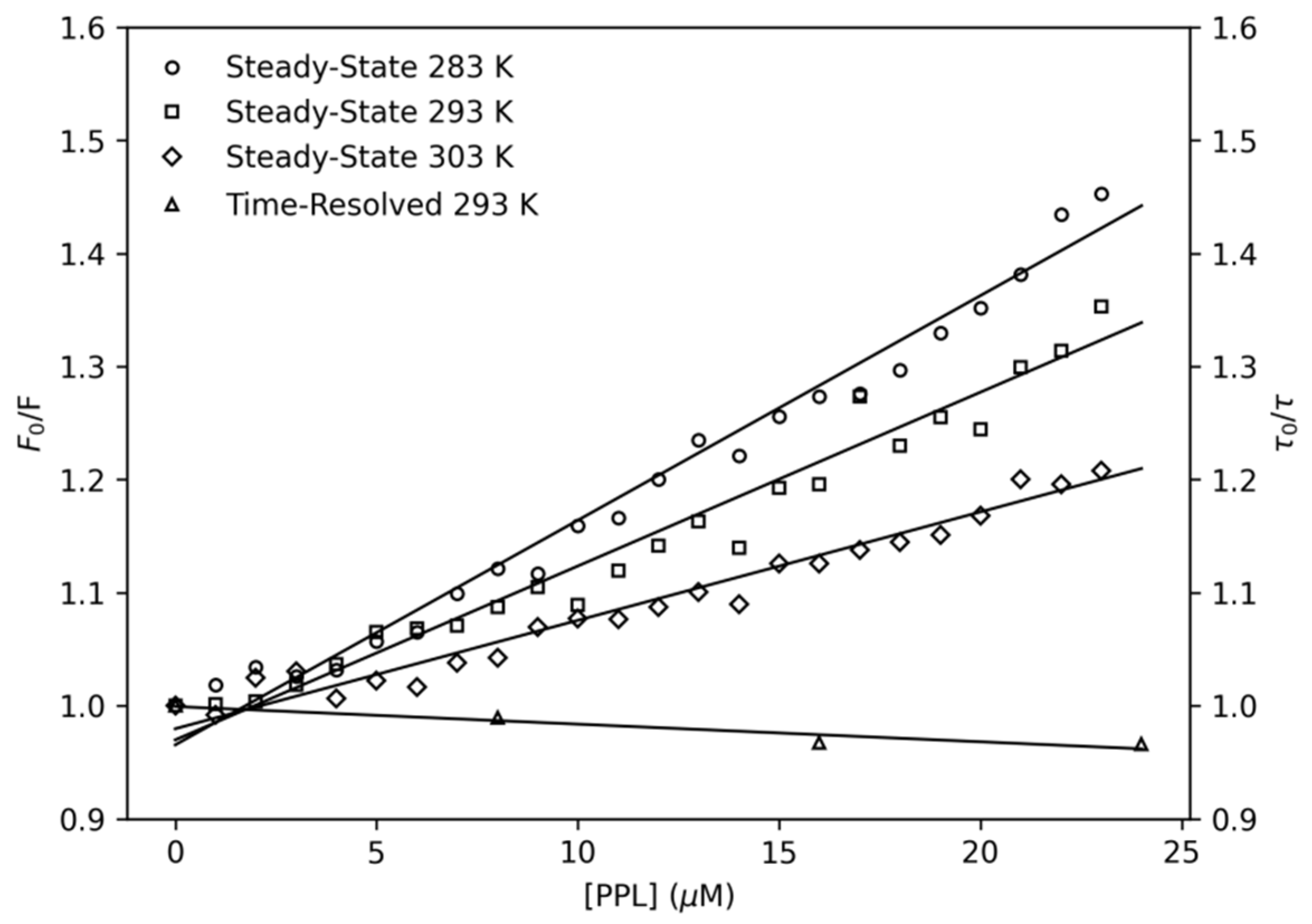
2.1.1. Thermodynamic Parameters
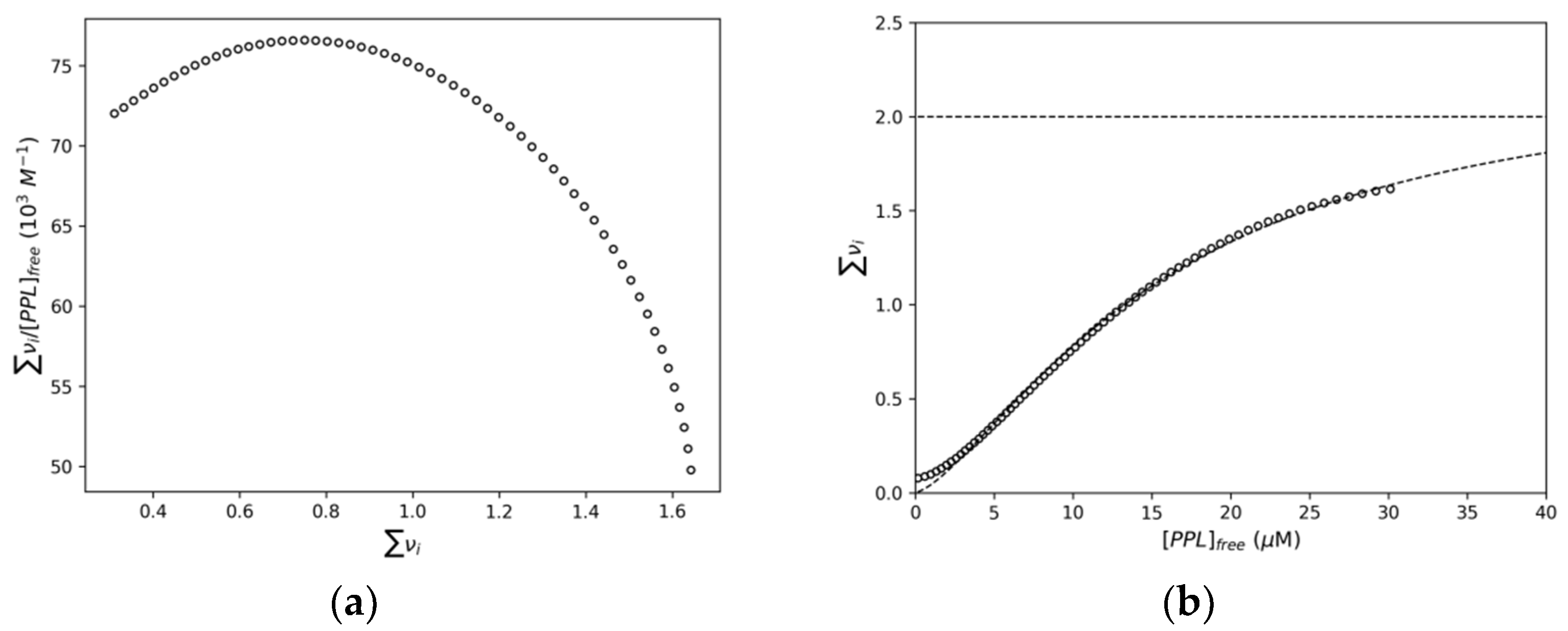
2.1.2. Interaction Density Function (IDF)
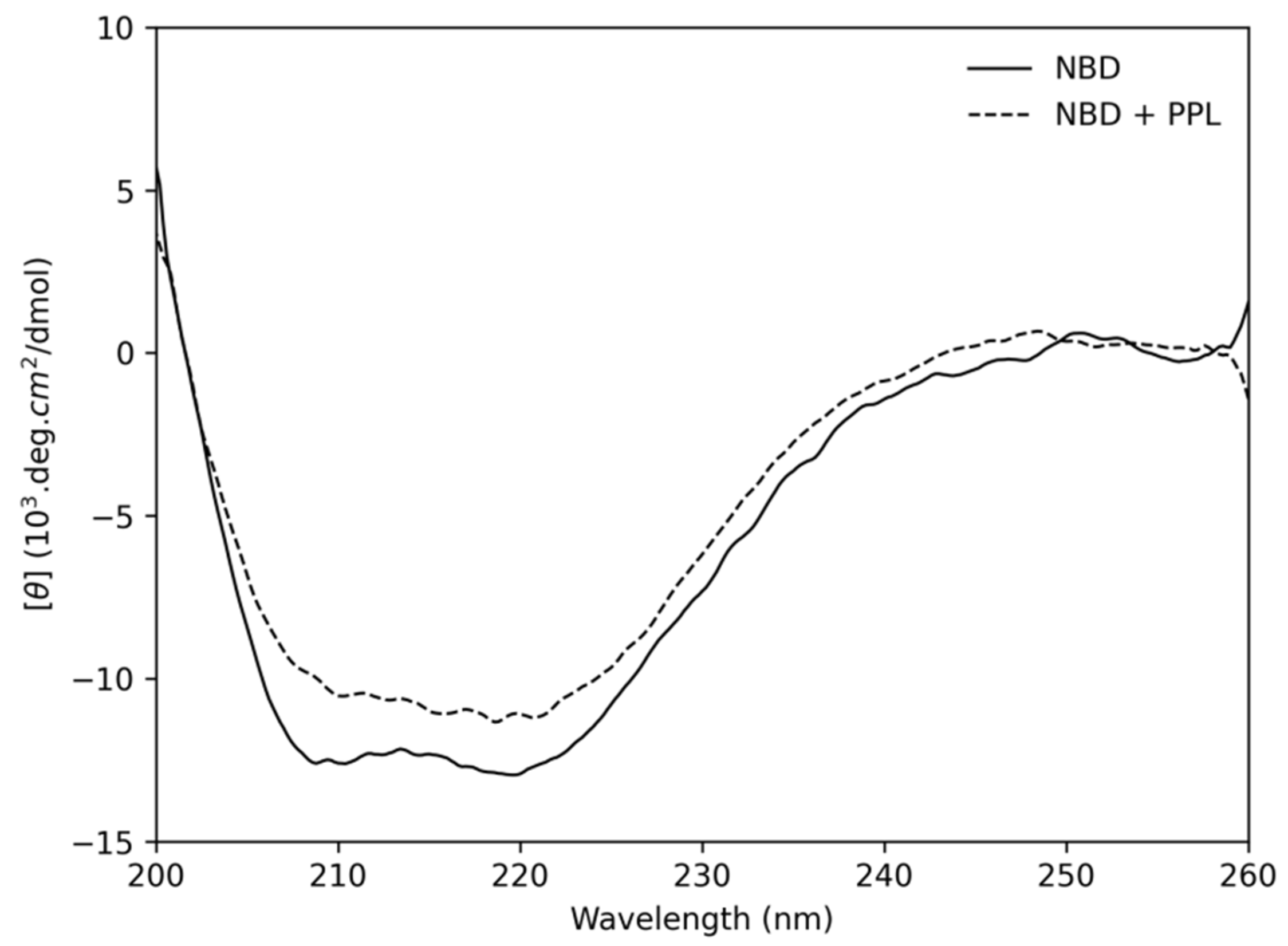
2.2. Circular Dichroism
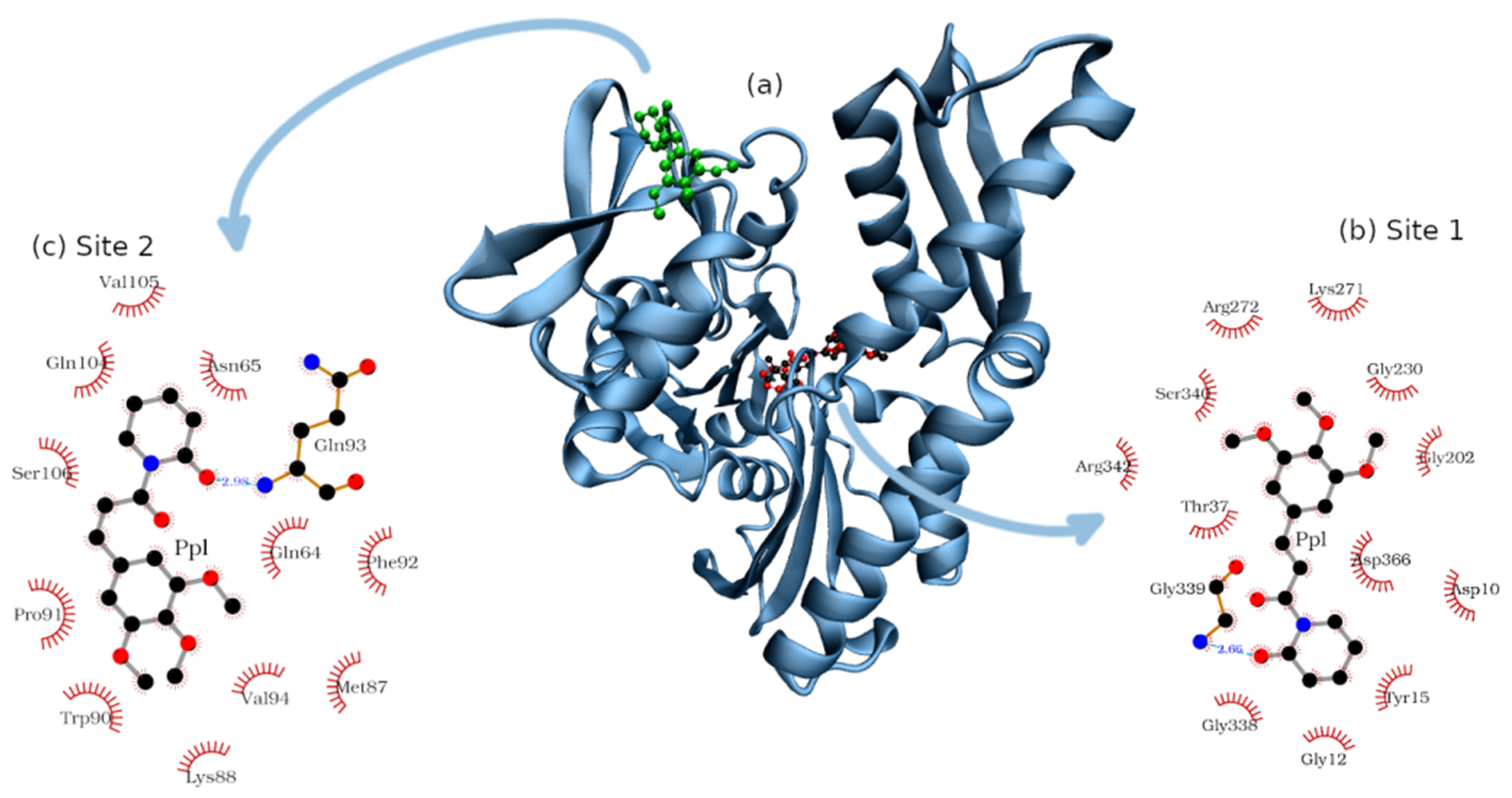
2.3. Molecular Docking
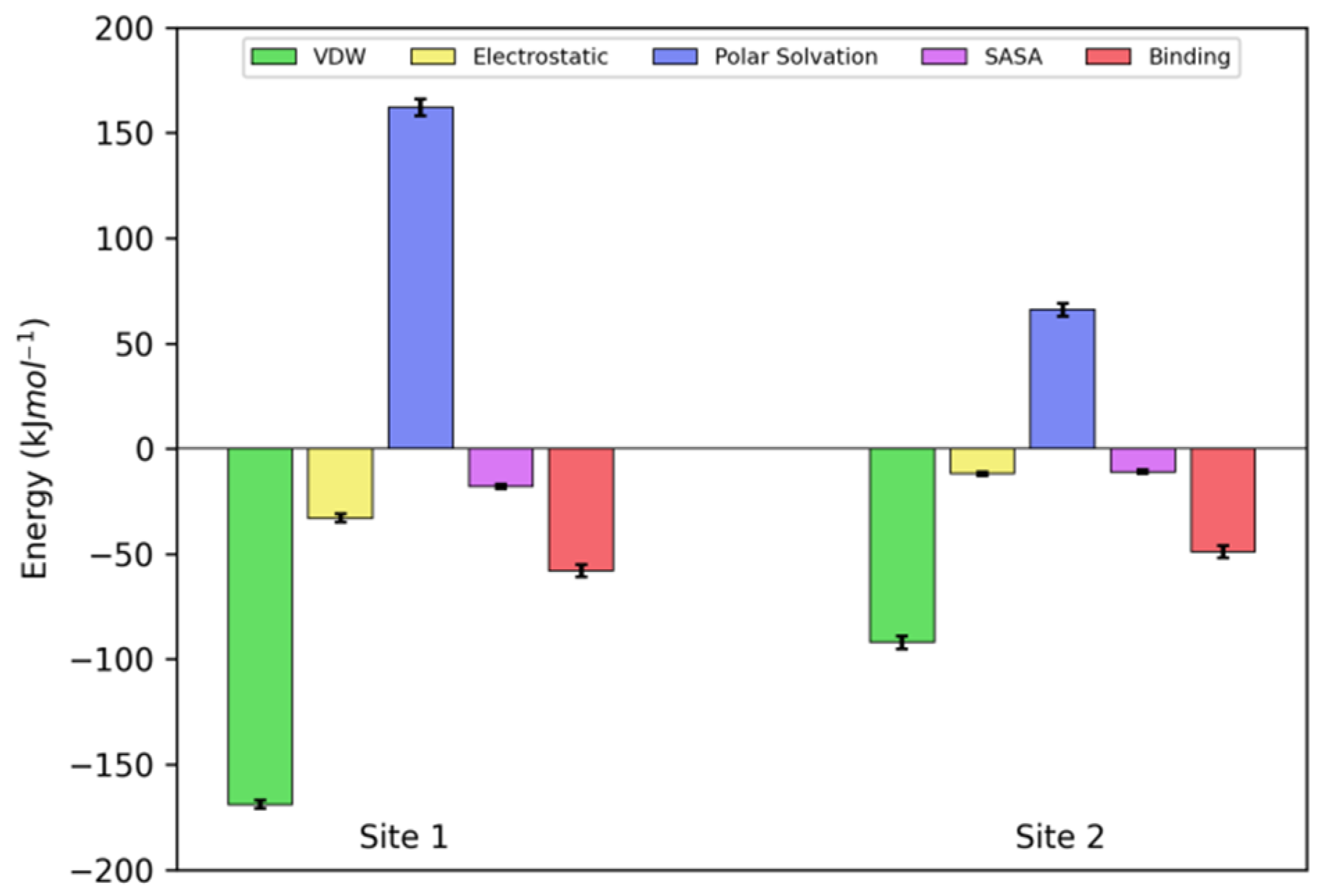
2.4. Molecular Dynamics
3. Materials and Methods
3.1. Reagents
3.2. Experimental Methods
3.2.1. Steady-State Fluorescence Spectroscopy
3.2.2. Time-Resolved Fluorescence
3.2.3. Circular Dichroism
3.3. Computational Methods
3.3.1. Molecular Docking
3.3.2. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyer, T.N.; Da Silva, A.L. Resposta celular ao estresse. Rev. Assoc. Med. Bras. 1999, 45, 181–188. (In Portuguese) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Cell. Mol. Life Sci. 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Karp, G. Biologia Celular e Molecular: Conceitos e Experimentos, 1st ed.; Manole: Barueri, Brazil, 2005; pp. 53–54. (In Portuguese) [Google Scholar]
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.D.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 2016, 6, 34701. [Google Scholar] [CrossRef]
- Somensi, N.; Brum, P.O.; de Miranda Ramos, V.; Gasparotto, J.; Zanotto-Filho, A.; Rostirolla, D.C.; da Silva Morrone, M.; Moreira, J.C.F.; Gelain, D.P. Extracellular HSP70 activates ERK1/2, NF-kB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell. Physiol. Biochem. 2017, 42, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Hulina, A.; Rajković, M.G.; Despot, D.J.; Jelić, D.; Dojder, A.; Čepelak, I.; Rumora, L. Extracellular Hsp70 induces inflammation and modulates LPS/LTA-stimulated inflammatory response in THP-1 cells. Cell Stress Chaperones 2018, 23, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, S. Heat shock protein 70 as a double agent acting inside and outside the cell: Insights into autoimmunity. Int. J. Mol. Sci. 2020, 21, 5298. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, H.; Cao, L. Recent advances in heat shock proteins in cancer diagnosis, prognosis, metabolism and treatment. Biomed. Pharmacother. 2021, 142, 112074. [Google Scholar] [CrossRef]
- Cheeseman, M.D.; Westwood, I.M.; Barbeau, O.; Rowlands, M.; Dobson, S.; Jones, A.M.; Jeganathan, F.; Burke, R.; Kadi, N.; Workman, P.; et al. Exploiting protein conformational change to optimize adenosine-derived inhibitors of HSP70. J. Med. Chem. 2016, 59, 4625–4636. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Adams, D.J.; Dai, M.; Pellegrino, G.; Wagner, B.K.; Stern, A.M.; Shamji, A.F.; Schreiber, S.L. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci. USA 2012, 109, 15115–15120. [Google Scholar] [CrossRef] [Green Version]
- Piska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Pękala, E. Piperlongumine (piplartine) as a lead compound for anticancer agents—Synthesis and properties of analogues: A mini-review. Eur. J. Med. Chem. 2018, 156, 13–20. [Google Scholar] [CrossRef]
- Wu, Y.; Min, X.; Zhuang, C.; Li, J.; Yu, Z.; Dong, G.; Yao, J.; Wang, S.; Liu, Y.; Wu, S.; et al. Design, synthesis and biological activity of piperlongumine derivatives as selective anticancer agents. Eur. J. Med. Chem. 2014, 82, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Tyagi, A.K. Historical spice as a future drug: Therapeutic potential of piperlongumine. Curr. Pharm. Des. 2016, 22, 4151–4159. [Google Scholar] [CrossRef] [PubMed]
- Zazeri, G.; Povinelli, A.P.R.; Le Duff, C.S.; Tang, B.; Cornelio, M.L.; Jones, A.M. Synthesis and spectroscopic analysis of piperine-and piperlongumine-inspired natural product scaffolds and their molecular docking with IL-1β and NF-κB proteins. Molecules 2020, 25, 2841. [Google Scholar] [CrossRef]
- Sun, L.-D.; Wang, F.; Dai, F.; Wang, Y.-H.; Lin, D.; Zhou, B. Development and mechanism investigation of a new piperlongumine derivative as a potent anti-inflammatory agent. Biochem. Pharmacol. 2015, 95, 156–169. [Google Scholar] [CrossRef]
- Zheng, J.; Son, D.J.; Gu, S.M.; Woo, J.R.; Ham, Y.W.; Lee, H.P.; Kim, W.J.; Jung, J.K.; Hong, J.T. Piperlongumine inhibits lung tumor growth via inhibition of nuclear factor kappa B signaling pathway. Sci. Rep. 2016, 6, 26357. [Google Scholar] [CrossRef]
- Ginzburg, S.; Golovine, K.V.; Makhov, P.B.; Uzzo, R.G.; Kutikov, A.; Kolenko, V.M. Piperlongumine inhibits NF-κB activity and attenuates aggressive growth characteristics of prostate cancer cells. Prostate 2014, 74, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Si, D.; Wang, Y.; Zhou, Y.-H.; Guo, Y.; Wang, J.; Zhou, H.; Li, Z.-S.; Fawcett, J.P. Mechanism of CYP2C9 inhibition by flavones and flavonols. Drug Metab. Dispos. 2009, 37, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.M.; Lee, H.P.; Ham, Y.W.; Son, D.J.; Kim, H.Y.; Oh, K.W.; Han, S.-B.; Yun, J.; Hong, J.T. Piperlongumine improves lipopolysaccharide-induced amyloidogenesis by suppressing NF-KappaB pathway. Neuromol. Med. 2018, 20, 312–327. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2004; Available online: https://www.academia.edu/34533342/Principles_of_Fluorescence_Spectroscopy_Third_Edition (accessed on 6 December 2021).
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Povinelli, A.P.R.; Zazeri, G.; Cornélio, M.L. Molecular Mechanism of Flavonoids Using Fluorescence Spectroscopy and Computational Tools. In Flavonoids A Coloring Model for Cheering Up Life; IntechOpen: London, UK, 2019. [Google Scholar]
- Caruso, Í.P.; Vilegas, W.; de Oliveira, L.C.; Cornélio, M.L. Fluorescence spectroscopic and dynamics simulation studies on isoorientin binding with human serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 228, 117738. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Lohman, T.M.; Bujalowski, W. Thermodynamic methods for model-independent determination of equilibrium binding isotherms for protein-DNA interactions: Spectroscopic approaches to monitor binding. Methods Enzymol. 1991, 208, 258–290. [Google Scholar]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; de Freitas Lima, M.; Cornélio, M.L. Detailed Characterization of the Cooperative Binding of Piperine with Heat Shock Protein 70 by Molecular Biophysical Approaches. Biomedicines 2020, 8, 629. [Google Scholar] [CrossRef]
- Bordbar, A.K.; Saboury, A.A.; Moosavi-Movahedi, A.A. The shapes of Scatchard plots for systems with two sets of binding sites. Biochem. Educ. 1996, 24, 172–175. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, P.A.; de Freitas Lima, M.; Cornélio, L.M. Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine. Int. J. Mol. Sci. 2019, 20, 2856. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, D.; Jordan, R.; McMacken, R.; Freire, E. Thermodynamic and structural analysis of the folding/unfolding transitions of the Escherichia coli molecular chaperone DnaK. J. Mol. Biol. 1993, 232, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Palleros, D.R.; Reid, K.L.; McCarty, J.S.; Walker, G.C.; Fink, A.L. DnaK, hsp73, and their molten globules. Two different ways heat shock proteins respond to heat. J. Biol. Chem. 1992, 267, 5279–5285. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; de Freitas Lima, M.; Cornélio, M.L. The Cytokine IL-1β and Piperine Complex Surveyed by Experimental and Computational Molecular Biophysics. Biomolecules 2020, 10, 1337. [Google Scholar] [CrossRef] [PubMed]
- Povinelli, A.P.R.; Zazeri, G.; de Freitas Lima, M.; Cornélio, M.L. Details of the cooperative binding of piperlongumine with rat serum albumin obtained by spectroscopic and computational analyses. Sci. Rep. 2019, 9, 15667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Lindstrom, W.; Huey, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. User Guide AutoDock Version 4.2. Automated Docking of Flexible Ligands to Flexible Receptors. 2010. Available online: https://autodock.scripps.edu/wp-content/uploads/sites/56/2021/10/AutoDock4.2.6_UserGuide.pdf (accessed on 6 December 2021).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 24503. [Google Scholar] [CrossRef]
- Batcho, P.F.; Case, D.A.; Schlick, T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001, 115, 4003–4018. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | KSV (×104 M−1) | Kq (×1012 M−1·s−1) | Ka (×104 M−1) |
|---|---|---|---|
| 283 | 1.9 ± 0.1 | 8.9 ± 0.1 | 2.5 ± 0.8 |
| 293 | 1.5 ± 0.1 | 6.9 ± 0.1 | 2.2 ± 0.5 |
| 303 | 1.0 ± 0.1 | 4.3 ± 0.1 | 1.8 ± 0.2 |
| T (K) | ∆G (kJ·mol−1) | ∆H (kJ·mol−1) | ∆S (J·mol−1·K-1) | T·∆S (kJ·mol−1) |
|---|---|---|---|---|
| 283 | −23.7 ± 1.3 | −11.9 ± 0.9 | 42.4 ± 3.4 | 11.9 ± 0.9 |
| 293 | −24.2 ± 1.3 | 12.4 ± 0.9 | ||
| 303 | −24.6 ± 1.3 | 12.8 ± 1.0 |
| Binding Sites | Amino Acids | |||
|---|---|---|---|---|
| Non-Polar | Positively Charged | Negatively Charged | Polar | |
| 1 | Gly12, 202, 230, 338, 339 | Lys271; Arg272, 342 | Asp10, 366 | Tyr15; Thr37; Ser340 |
| 2 | Val94, 105; Met87; Pro91; Trp90; Phe92 | Lys88 | - | Asn65; Gln64; 93, 104; Ser106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Povinelli, A.P.R.; Zazeri, G.; Jones, A.M.; Cornélio, M.L. Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study. Pharmaceuticals 2021, 14, 1298. https://doi.org/10.3390/ph14121298
Povinelli APR, Zazeri G, Jones AM, Cornélio ML. Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study. Pharmaceuticals. 2021; 14(12):1298. https://doi.org/10.3390/ph14121298
Chicago/Turabian StylePovinelli, Ana Paula Ribeiro, Gabriel Zazeri, Alan M. Jones, and Marinnio Lopes Cornélio. 2021. "Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study" Pharmaceuticals 14, no. 12: 1298. https://doi.org/10.3390/ph14121298
APA StylePovinelli, A. P. R., Zazeri, G., Jones, A. M., & Cornélio, M. L. (2021). Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study. Pharmaceuticals, 14(12), 1298. https://doi.org/10.3390/ph14121298