
Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes

 , ,
, ,
Abstract
1. Introduction
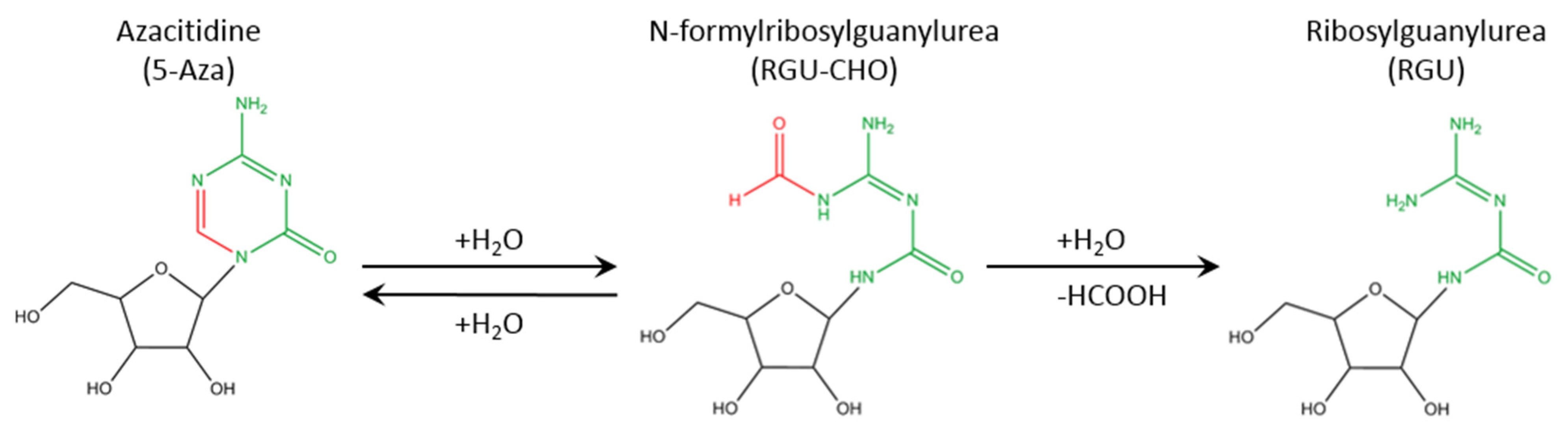
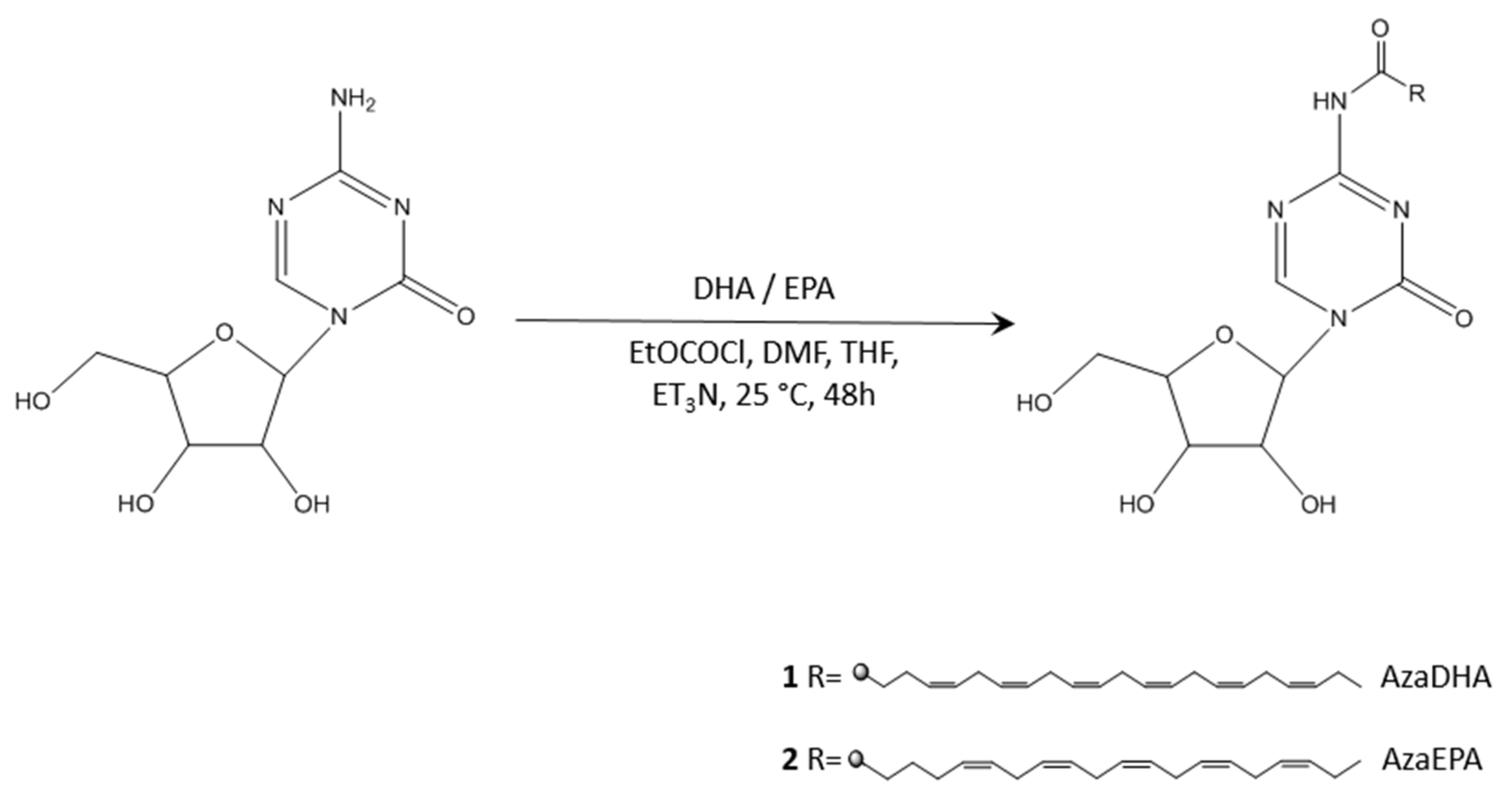
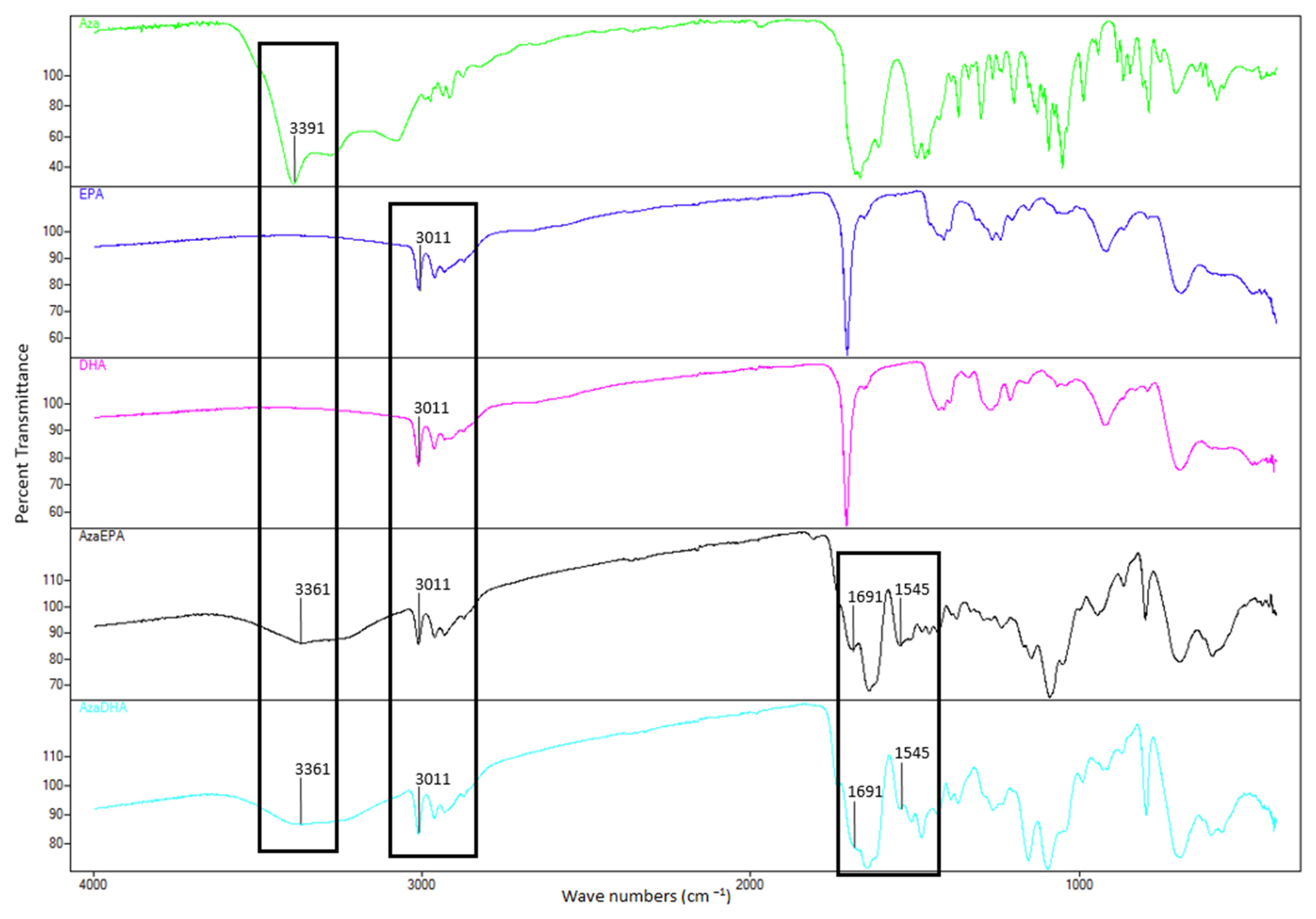
2. Results and Discussion
3. Materials and Methods
3.1. Materials
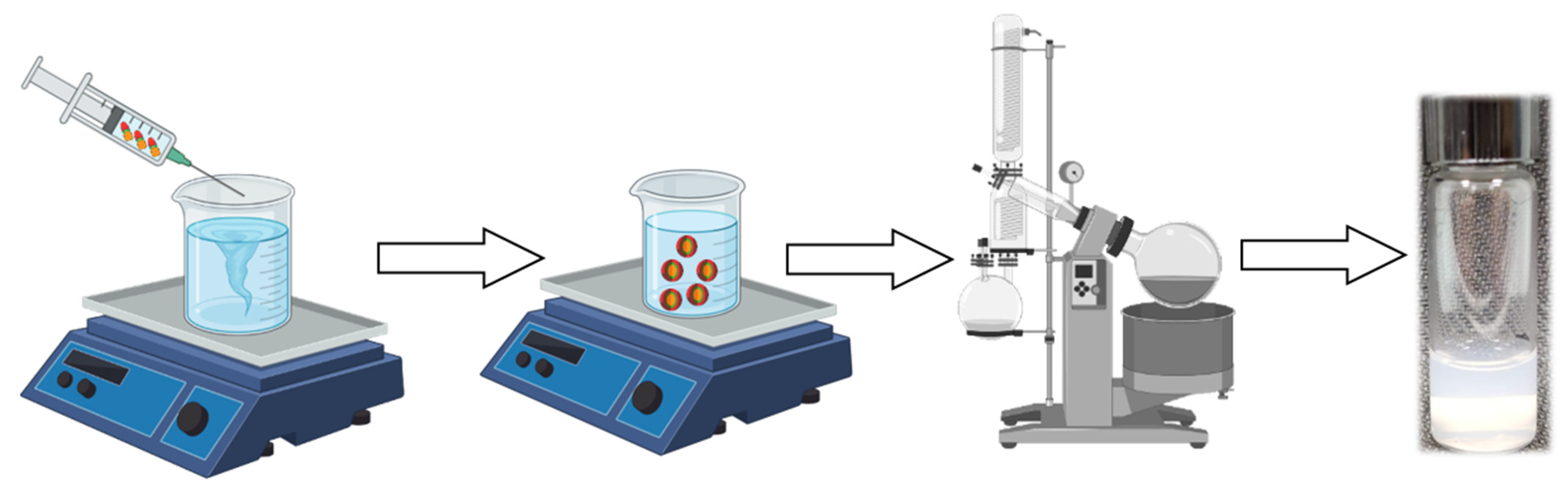
3.2. Synthesis and Purification
3.3. 1H and 13C NMR
3.4. Elemental Analysis
3.5. Fourier-Transform Infrared Spectroscopy
3.6. Mass Spectrometry
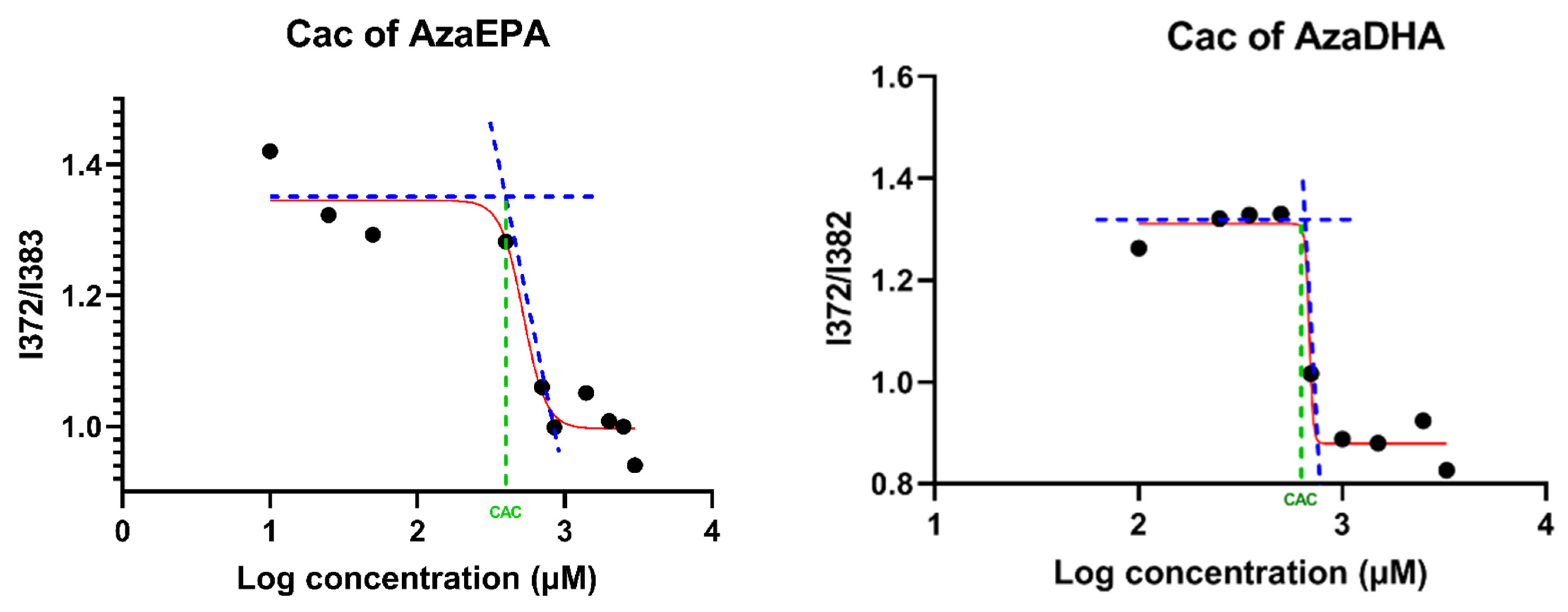
3.7. Critical Aggregation Concentration (CAC)
3.8. Self-Assembly Formulation
3.9. Dynamic Light Scattering (DLS)
3.10. Cryogenic Transmission Electron Microscopy (Cryo-TEM)
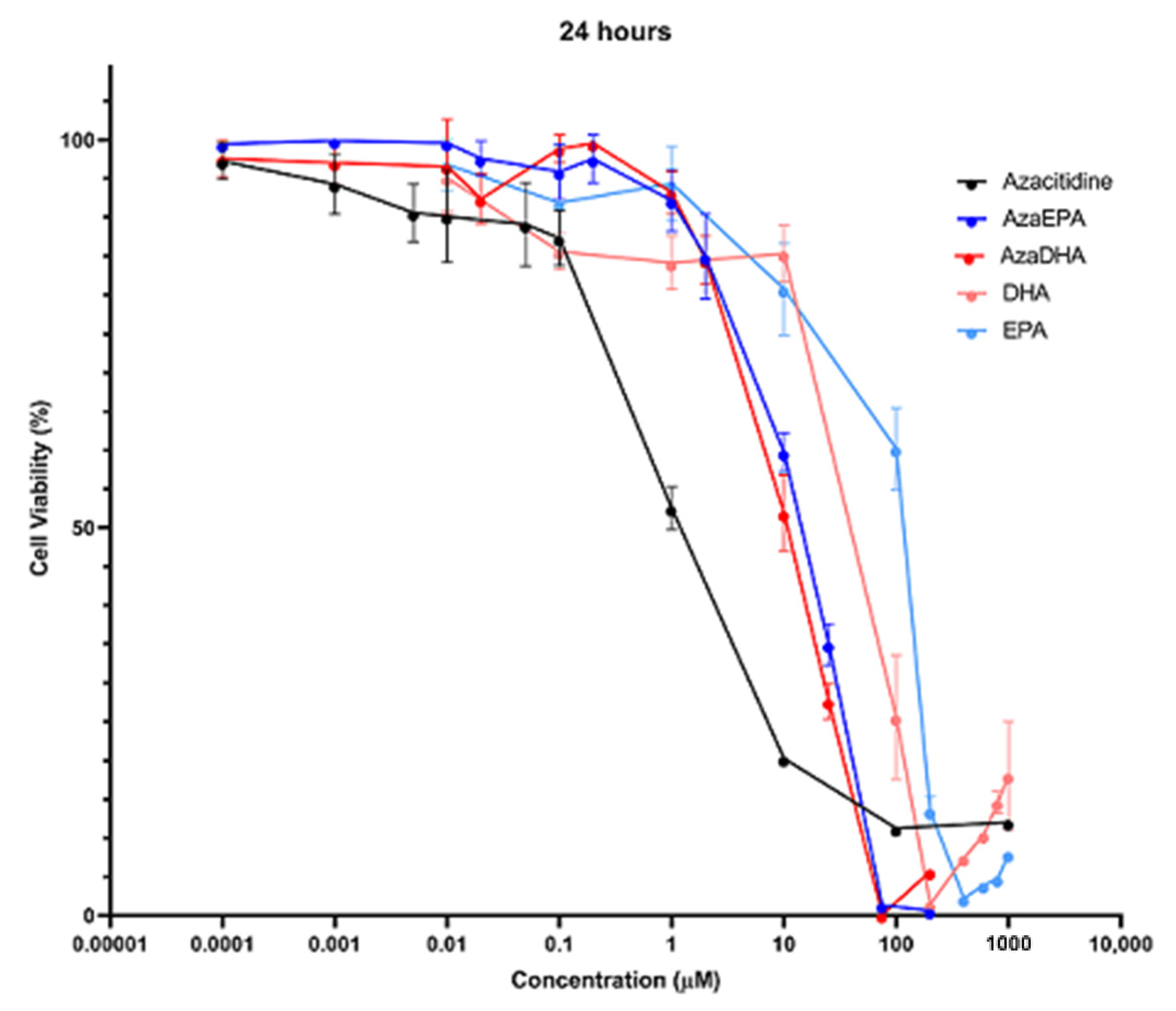
3.11. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hellström-Lindberg, E.; Tobiasson, M.; Greenberg, P. Myelodysplastic syndromes: Moving towards personalized management. Haematologica 2020, 105, 1765–1779. [Google Scholar] [CrossRef]
- Hong, M.; He, G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J. Transl. Intern. Med. 2017, 5, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Thépot, S.; Achour, B.; Quesnel, B.; Dreyfus, F.; Turlure, P.; Taksin, A.-L.; Vey, N.; Koka, A.M.; De Botton, S.; et al. Azacytidine (AZA) in MDS (including RAEB-t and CMML) in Patients (pts) ≥ 80 Years: Results of the French ATU Program. Blood 2009, 114, 1773. [Google Scholar] [CrossRef]
- Platzbecker, U.; Kubasch, A.S.; Homer-Bouthiette, C.; Prebet, T. Current challenges and unmet medical needs in myelodysplastic syndromes. Leukemia 2021, 35, 2182–2198. [Google Scholar] [CrossRef]
- Musto, P.; Maurillo, L.; Spagnoli, A.; Gozzini, A.; Rivellini, F.; Lunghi, M.; Villani, O.; Aloe-Spiriti, M.A.; Venditti, A.; Santini, V.; et al. Azacitidine for the treatment of lower risk myelodysplastic syndromes. Cancer 2010, 116, 1485–1494. [Google Scholar] [CrossRef]
- Waespe, N.; Akker, M.V.D.; Klaassen, R.J.; Lieberman, L.; Irwin, M.S.; Ali, S.S.; Abdelhaleem, M.; Zlateska, B.; Liebman, M.; Cada, M.; et al. Response to treatment with azacitidine in children with advanced myelodysplastic syndrome prior to hematopoietic stem cell transplantation. Haematologica 2016, 101, 1508–1515. [Google Scholar] [CrossRef]
- Keruakous, A.R.; Holter-Chakrabarty, J.; Schmidt, S.A.; Khawandanah, M.O.; Selby, G.; Yuen, C. Azacitidine maintenance therapy post-allogeneic stem cell transplantation in poor-risk acute myeloid leukemia. Hematol. Stem Cell Ther. 2021, in press. [Google Scholar] [CrossRef]
- Oshrine, B.R.; Shyr, D.; Hale, G.; Petrovic, A. Low-dose azacitidine for relapse prevention after allogeneic hematopoietic cell transplantation in children with myeloid malignancies. Pediatr. Transplant. 2019, 23, e13423. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 1–29. [Google Scholar] [CrossRef]
- Pleyer, L.; Döhner, H.; Dombret, H.; Seymour, J.F.; Schuh, A.C.; Beach, C.L.; Swern, A.S.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; et al. Azacitidine for Front-Line Therapy of Patients with AML: Reproducible Efficacy Established by Direct Comparison of International Phase 3 Trial Data with Registry Data from the Austrian Azacitidine Registry of the AGMT Study Group. Int. J. Mol. Sci. 2017, 18, 415. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Baroud, M.; Lepeltier, E.; Thepot, S.; El-Makhour, Y.; Duval, O. The evolution of nucleosidic analogues: Self-assembly of prodrugs into nanoparticles for cancer drug delivery. Nanoscale Adv. 2021, 3, 2157–2179. [Google Scholar] [CrossRef]
- Kordella, C.; Lamprianidou, E.; Kotsianidis, I. Mechanisms of Action of Hypomethylating Agents: Endogenous Retroelements at the Epicenter. Front. Oncol. 2021, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Gil-Perez, A.; Montalban-Bravo, G. Management of myelodysplastic syndromes after failure of response to hypomethylating agents. Ther. Adv. Hematol. 2019, 10, 2040620719847059. [Google Scholar] [CrossRef]
- Prebet, T.; Thepot, S.; Gore, S.D.; Dreyfus, F.; Fenaux, P.; Vey, N. Outcome of patients with low-risk myelodysplasia after azacitidine treatment failure. Haematologica 2013, 98, e18–e19. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of High-Risk Myelodysplastic Syndrome After Azacitidine Treatment Failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Balouzet, C.; Chanat, C.; Jobard, M.; Brandely-Piat, M.-L.; Chast, F. Stability of 25 mg/mL Azacitidine Suspensions Kept in Fridge after Freezing. Pharm. Technol. Hosp. Pharm. 2017, 2, 11–16. [Google Scholar] [CrossRef]
- Walker, S.E.; Charbonneau, L.F.; Law, S.; Earle, C. Stability of Azacitidine in Sterile Water for Injection. Can. J. Hosp. Pharm. 2012, 65, 352–359. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Mowles, D.; Yao, S.; Ng, A.; Young, J.D.; Cass, C.E.; Tong, Z. Role of Human Nucleoside Transporters in the Uptake and Cytotoxicity of Azacitidine and Decitabine. Nucleosides Nucleotides Nucleic Acids 2012, 31, 236–255. [Google Scholar] [CrossRef]
- Fanciullino, R.; Mercier, C.; Serdjebi, C.; Berda, Y.; Fina, F.; Ouafik, L.; Lacarelle, B.; Ciccolini, J.; Costello, R. Lethal toxicity after administration of azacytidine: Implication of the Cytidine Deaminase-Deficiency Syndrome. Pharm. Genom. 2015, 25, 317–321. [Google Scholar] [CrossRef]
- Chabner, B.A.; Drake, J.C.; Johns, D.G. Deamination of 5-azacytidine by a human leukemia cell cytidine deaminase. Biochem. Pharmacol. 1973, 22, 2763–2765. [Google Scholar] [CrossRef]
- Fanciullino, R.; Mercier, C.; Serdjebi, C.; Venton, G.; Colle, J.; Fina, F.; Ouafik, L.; Lacarelle, B.; Ciccolini, J.; Costello, R. Yin and yang of cytidine deaminase roles in clinical response to azacitidine in the elderly: A pharmacogenetics tale. Pharmacogenomics 2015, 16, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Hu, C.; Wu, Y.; Zhong, D.; Xu, X.; Gu, Z. Engineering Anticancer Amphipathic Peptide-Dendronized Compounds for Highly-Efficient Plasma/Organelle Membrane Perturbation and Multidrug Resistance Reversal. ACS Appl. Mater. Interfaces 2018, 10, 30952–30962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef]
- Zhong, Y.-J.; Shao, L.-H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef]
- Xu, Y.; Geng, J.; An, P.; Xu, Y.; Huang, J.; Lu, W.; Liu, S.; Yu, J. Cathepsin B-sensitive cholesteryl hemisuccinate–gemcitabine prodrug nanoparticles: Enhanced cellular uptake and intracellular drug controlled release. RSC Adv. 2014, 5, 6985–6992. [Google Scholar] [CrossRef]
- Ni, Y.; Hai, Z.; Zhang, T.; Wang, Y.; Yang, Y.; Zhang, S.; Liang, G. Cathepsin B Turning Bioluminescence “On” for Tumor Imaging. Anal. Chem. 2019, 91, 14834–14837. [Google Scholar] [CrossRef]
- Luo, C.; Sun, J.; Sun, B.; He, Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol. Sci. 2014, 35, 556–566. [Google Scholar] [CrossRef]
- Mura, S.; Bui, D.T.; Couvreur, P.; Nicolas, J. Lipid prodrug nanocarriers in cancer therapy. J. Control. Release 2015, 208, 25–41. [Google Scholar] [CrossRef]
- Sivakova, S.; Rowan, S.J. Nucleobases as supramolecular motifs. Chem. Soc. Rev. 2005, 34, 9–21. [Google Scholar] [CrossRef]
- Gong, X.; Moghaddam, M.J.; Sagnella, S.M.; Conn, C.E.; Danon, S.J.; Waddington, L.J.; Drummond, C.J. Lamellar crystalline self-assembly behaviour and solid lipid nanoparticles of a palmityl prodrug analogue of Capecitabine—A chemotherapy agent. Colloids Surf. B Biointerfaces 2011, 85, 349–359. [Google Scholar] [CrossRef]
- Lepeltier, E.; Bourgaux, C.; Maksimenko, A.; Meneau, F.; Rosilio, V.; Sliwinski, E.; Zouhiri, F.; Desmaële, D.; Couvreur, P. Self-Assembly of Polyisoprenoyl Gemcitabine Conjugates: Influence of Supramolecular Organization on Their Biological Activity. Langmuir 2014, 30, 6348–6357. [Google Scholar] [CrossRef]
- Picou, F.; Debeissat, C.; Bourgeais, J.; Gallay, N.; Ferrié, E.; Foucault, A.; Ravalet, N.; Maciejewski, A.; Vallet, N.; Ducrocq, E.; et al. n-3 Polyunsaturated fatty acids induce acute myeloid leukemia cell death associated with mitochondrial glycolytic switch and Nrf2 pathway activation. Pharmacol. Res. 2018, 136, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yamagami, T.; Porada, C.D.; Pardini, R.; Zanjani, E.D.; Almeida-Porada, G.D. Docosahexanoic acid induces dose dependent cell death in an early undifferentiated subtype of acute myeloid leukemia cell line. Cancer Biol. Ther. 2009, 8, 331–337. [Google Scholar] [CrossRef]
- Aires, V.; Hichami, A.; Filomenko, R.; Plé, A.; Rébé, C.; Bettaieb, A.; Khan, N.A. Docosahexaenoic Acid Induces Increases in [Ca2+]ivia Inositol 1,4,5-Triphosphate Production and Activates Protein Kinase Cγ and -δ via Phosphatidylserine Binding Site: Implication in Apoptosis in U937 Cells. Mol. Pharmacol. 2007, 72, 1545–1556. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Racanicchi, S.; Martelli, M.P.; Nocentini, G.; Fettucciari, K.; Riccardi, C.; Marconi, P.; DI Nardo, P.; Grignani, F.; Binaglia, L.; et al. Eicosapentaenoic Acid Demethylates a Single CpG That Mediates Expression of Tumor Suppressor CCAAT/Enhancer-binding Protein δ in U937 Leukemia Cells. J. Biol. Chem. 2011, 286, 27092–27102. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Nocentini, G.; Billi, M.; Racanicchi, S.; Riccardi, C.; Roberti, R.; Grignani, F.; Binaglia, L.; Vecchini, A. Eicosapentaenoic Acid Activates RAS/ERK/C/EBPβ Pathway through H-Ras Intron 1 CpG Island Demethylation in U937 Leukemia Cells. PLoS ONE 2014, 9, e85025. [Google Scholar] [CrossRef][Green Version]
- Varney, M.E.; Hardman, W.E.; Sollars, V.E. Omega 3 fatty acids reduce myeloid progenitor cell frequency in the bone marrow of mice and promote progenitor cell differentiation. Lipids Health Dis. 2009, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Desmaële, D.; Gref, R.; Couvreur, P. Squalenoylation: A generic platform for nanoparticular drug delivery. J. Control. Release 2012, 161, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Lepeltier, E.; Bourgaux, C.; Rosilio, V.; Poupaert, J.H.; Meneau, F.; Zouhiri, F.; Lepêtre-Mouelhi, S.; Desmaële, D.; Couvreur, P. Self-Assembly of Squalene-Based Nucleolipids: Relating the Chemical Structure of the Bioconjugates to the Architecture of the Nanoparticles. Langmuir 2013, 29, 14795–14803. [Google Scholar] [CrossRef]
- Maksimenko, A.; Caron, J.; Mougin, J.; Desmaële, D.; Couvreur, P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int. J. Pharm. 2015, 482, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, F.; Chen, X.; Wan, J.; Wang, Y.; Li, T.; Wang, H. Self-Assembled Gemcitabine Prodrug Nanoparticles Show Enhanced Efficacy against Patient-Derived Pancreatic Ductal Adenocarcinoma. ACS Appl. Mater. Interfaces 2020, 12, 3327–3340. [Google Scholar] [CrossRef]
- Bui, D.T.; Nicolas, J.; Maksimenko, A.; Desmaële, D.; Couvreur, P. Multifunctional squalene-based prodrug nanoparticles for targeted cancer therapy. Chem. Commun. 2014, 50, 5336–5338. [Google Scholar] [CrossRef]
- Vandana, M.; Sahoo, S.K. Long circulation and cytotoxicity of PEGylated gemcitabine and its potential for the treatment of pancreatic cancer. Biomaterials 2010, 31, 9340–9356. [Google Scholar] [CrossRef]
- Coppens, E.; Desmaële, D.; Mougin, J.; Tusseau-Nenez, S.; Couvreur, P.; Mura, S. Gemcitabine Lipid Prodrugs: The Key Role of the Lipid Moiety on the Self-Assembly into Nanoparticles. Bioconjug. Chem. 2021, 32, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Castelli, F.; Sarpietro, M.G.; Ceruti, M.; Rocco, F.; Cattel, L. Characterization of Lipophilic Gemcitabine Prodrug−Liposomal Membrane Interaction by Differential Scanning Calorimetry. Mol. Pharm. 2006, 3, 737–744. [Google Scholar] [CrossRef]
- Sun, B.; Luo, C.; Cui, W.; Sun, J.; He, Z. Chemotherapy agent-unsaturated fatty acid prodrugs and prodrug-nanoplatforms for cancer chemotherapy. J. Control. Release 2017, 264, 145–159. [Google Scholar] [CrossRef]
- Naguib, Y.W.; Lansakara-P., D.; Lashinger, L.M.; Rodriguez, B.L.; Valdes, S.; Niu, M.; Aldayel, A.M.; Peng, L.; Hursting, S.D.; Cui, Z. Synthesis, Characterization, and In Vitro and In Vivo Evaluations of 4-(N)-Docosahexaenoyl 2′, 2′-Difluorodeoxycytidine with Potent and Broad-Spectrum Antitumor Activity. Neoplasia 2016, 18, 33–48. [Google Scholar] [CrossRef]
- Khoury, H.; Reddy, L.H.; Bildstein, L.; Deroussent, A.; Dubernet, C.; Couvreur, P.; Vassal, G.; Paci, A. Squalenoylation a New Concept in Drug Targeting: Evidence of a Potent Anti-Cancer Activity of Squalenoyl-Gemcitabine. Cancer Res. 2008, 68 (Suppl. 9), 5621. [Google Scholar]
- Huang, C.-W.; Mohamed, M.G.; Zhu, C.-Y.; Kuo, S.-W. Functional Supramolecular Polypeptides Involving π–π Stacking and Strong Hydrogen-Bonding Interactions: A Conformation Study toward Carbon Nanotubes (CNTs) Dispersion. Macromolecules 2016, 49, 5374–5385. [Google Scholar] [CrossRef]
- Huang, L.; Wan, J.; Wu, H.; Chen, X.; Bian, Q.; Shi, L.; Jiang, X.-C.; Yuan, A.-R.; Gao, J.-Q.; Wang, H. Quantitative Self-Assembly of Photoactivatable Small Molecular Prodrug Cocktails for Safe and Potent Cancer Chemo-Photodynamic Therapy. Nano Today 2021, 36, 101030. [Google Scholar] [CrossRef]
- Tucci, S.T.; Seo, J.W.; Kakwere, H.; Kheirolomoom, A.; Ingham, E.S.; Mahakian, L.M.; Tam, S.; Tumbale, S.; Baikoghli, M.; Cheng, H.; et al. A Scalable Method for Squalenoylation and Assembly of Multifunctional 64Cu-Labeled Squalenoylated Gemcitabine Nanoparticles. Nanotheranostics 2018, 2, 387–402. [Google Scholar] [CrossRef]
- Xie, B.; Wan, J.; Chen, X.; Han, W.; Wang, H. Preclinical Evaluation of a Cabazitaxel Prodrug Using Nanoparticle Delivery for the Treatment of Taxane-Resistant Malignancies. Mol. Cancer Ther. 2020, 19, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, Z.; Wang, L.; Guo, T.; Wu, J.; Wan, J.; Zhou, L.; Li, H.; Li, Z.; Jiang, D.; et al. New Generation Nanomedicines Constructed from Self-Assembling Small-Molecule Prodrugs Alleviate Cancer Drug Toxicity. Cancer Res. 2017, 77, 6963–6974. [Google Scholar] [CrossRef]
- Gaudin, A.; Yemisci, M.; Eroğlu, H.; Lepetre-Mouelhi, S.; Turkoglu, O.F.; Dönmez-Demir, B.; Caban-Toktas, S.; Sargon, M.F.; Garcia-Argote, S.; Pieters, G.; et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat. Nanotechnol. 2014, 9, 1054–1062. [Google Scholar] [CrossRef]
- Fan, Q.; Ji, Y.; Wang, J.; Wu, L.; Li, W.; Chen, R.; Chen, Z. Self-assembly behaviours of peptide–drug conjugates: Influence of multiple factors on aggregate morphology and potential self-assembly mechanism. R. Soc. Open Sci. 2018, 5, 172040. [Google Scholar] [CrossRef]
- Buettner, C.J.; Wallace, A.J.; Ok, S.; Manos, A.A.; Nicholl, M.J.; Ghosh, A.; Tweedle, M.F.; Goldberger, J.E. Balancing the intermolecular forces in peptide amphiphiles for controlling self-assembly transitions. Org. Biomol. Chem. 2017, 15, 5220–5226. [Google Scholar] [CrossRef]
- Lepeltier, E.; Bourgaux, C.; Amenitsch, H.; Rosilio, V.; Lepêtre-Mouelhi, S.; Zouhiri, F.; Desmaele, D.; Couvreur, P. Influence of the nanoprecipitation conditions on the supramolecular structure of squalenoyled nanoparticles. Eur. J. Pharm. Biopharm. 2015, 96, 89–95. [Google Scholar] [CrossRef]
- Panahi, Y.; Farshbaf, M.; Mohammadhosseini, M.; Mirahadi, M.; Khalilov, R.; Saghfi, S.; Akbarzadeh, A. Recent advances on liposomal nanoparticles: Synthesis, characterization and biomedical applications. Artif. Cells Nanomed. Biotechnol. 2017, 45, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Rius, M.; Markelova, M.R.; Fichtner, I.; Hals, P.-A.; Sandvold, M.L.; Lyko, F. Delivery of 5-Azacytidine to Human Cancer Cells by Elaidic Acid Esterification Increases Therapeutic Drug Efficacy. Mol. Cancer Ther. 2010, 9, 1256–1264. [Google Scholar] [CrossRef]
- Tsume, Y.; Amidon, G.L. Selection of Suitable Prodrug Candidates for in Vivo Studies via in Vitro Studies; The Correlation of Prodrug Stability in Between Cell Culture Homogenates and Human Tissue Homogenates. J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Soc. Can. Sci. Pharm. 2012, 15, 433–446. [Google Scholar] [CrossRef]
- Arora, M.; Pandey, G.; Chauhan, S.S. Cysteine Cathepsins and Their Prognostic and Therapeutic Relevance in Leukemia. Ann. Natl. Acad. Med. Sci. India 2021, 57, 108–116. [Google Scholar] [CrossRef]
- Pandey, G.; Bakhshi, S.; Kumar, M.; Thakur, B.; Jain, P.; Kaur, P.; Chauhan, S.S. Prognostic and Therapeutic Relevance of Cathepsin B in Pediatric Acute Myeloid Leukemia. Am. J. Cancer Res. 2019, 9, 2634–2649. [Google Scholar] [PubMed]
- Mihalik, R.; Imre, G.; Peták, I.; Szende, B.; Kopper, L. Cathepsin B-independent abrogation of cell death by CA-074-OMe upstream of lysosomal breakdown. Cell Death Differ. 2004, 11, 1357–1360. [Google Scholar] [CrossRef][Green Version]
- Abraham, A.; Varatharajan, S.; Abbas, S.; Zhang, W.; Shaji, R.V.; Ahmed, R.; Abraham, A.; George, B.; Srivastava, A.; Chandy, M.; et al. Cytidine Deaminase Genetic Variants Influence RNA Expression and Cytarabine Cytotoxicity in Acute Myeloid Leukemia. Pharmacogenomics 2012, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Days | Hydrodynamic Diameter (nm) | Zeta Potential (mV) | PDI | Attenuator |
|---|---|---|---|---|---|
| AzaEPA | 1 | 235.8 ± 7.3% | 16.1 ± 8.54% | 0.121 ± 24% | 7 |
| 2 | 206.7 ± 1.57% | −9.89 ± 2.02% | 0.169 ± 12.8% | 7 | |
| 3 | 190.1 ± 1.37% | 25.8 ± 2.33% | 0.16 ± 16.6% | 7 | |
| 4 | 190.5 ± 0.963% | 25.9 ± 2.8% | 0.149 ± 14.4% | 7 | |
| 5 | 194.5 ± 0.119% | 35.7 ± 7.12% | 0.137 ± 22.7% | 7 | |
| 6 | 213 ± 1.76% | 33.2 ± 8.79% | 0.15 ± 16.4% | 7 | |
| 7 | 217 ± 2.89 % | 35.8 ± 4.86% | 0.184 ± 13% | 7 | |
| AzaDHA | 1 | 229.5 ± 0.575% | 18 ± 6.74% | 0.082 ± 23.9% | 7 |
| 2 | 232.5 ± 1.37% | 21.9 ± 4.63% | 0.094 ± 16.4% | 7 | |
| 3 | 217.7 ± 0.87% | −2.63 ± 28.6% | 0.075 ± 21.8% | 7 | |
| 4 | 189.2 ± 0.242% | 22.7 ± 6.68% | 0.77 ± 47.2% | 7 | |
| 5 | 185 ± 0.952% | 17.8 ± 4.05% | 0.055 ± 67.5% | 7 | |
| 6 | 189.7 ± 0.777% | 37.8 ± 2.97% | 0.138 ± 14.3% | 7 | |
| 7 | 182.5 ± 3.91% | 33 ± 6.09% | 0.155 ± 13.2% | 7 |
| IC50 | Azacitidine | DHA | EPA | AzaDHA | AzaEPA |
|---|---|---|---|---|---|
| 6 h | 6.5 µM | 100.2 µM | 196.7 µM | 27.5 µM | 33.7 µM |
| 24 h | 1.0 µM | 92.8 µM | 115.9 µM | 11.3 µM | 16.6 µM |
| 48 h | 1.4 µM | 44.5 µM | 103.8 µM | 10.2 µM | 13.7 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baroud, M.; Lepeltier, E.; El-Makhour, Y.; Lautram, N.; Bejaud, J.; Thepot, S.; Duval, O. Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes. Pharmaceuticals 2021, 14, 1317. https://doi.org/10.3390/ph14121317
Baroud M, Lepeltier E, El-Makhour Y, Lautram N, Bejaud J, Thepot S, Duval O. Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes. Pharmaceuticals. 2021; 14(12):1317. https://doi.org/10.3390/ph14121317
Chicago/Turabian StyleBaroud, Milad, Elise Lepeltier, Yolla El-Makhour, Nolwenn Lautram, Jerome Bejaud, Sylvain Thepot, and Olivier Duval. 2021. "Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes" Pharmaceuticals 14, no. 12: 1317. https://doi.org/10.3390/ph14121317
APA StyleBaroud, M., Lepeltier, E., El-Makhour, Y., Lautram, N., Bejaud, J., Thepot, S., & Duval, O. (2021). Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes. Pharmaceuticals, 14(12), 1317. https://doi.org/10.3390/ph14121317