
Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer



Abstract
:
1. Introduction
2. Results and Discussions
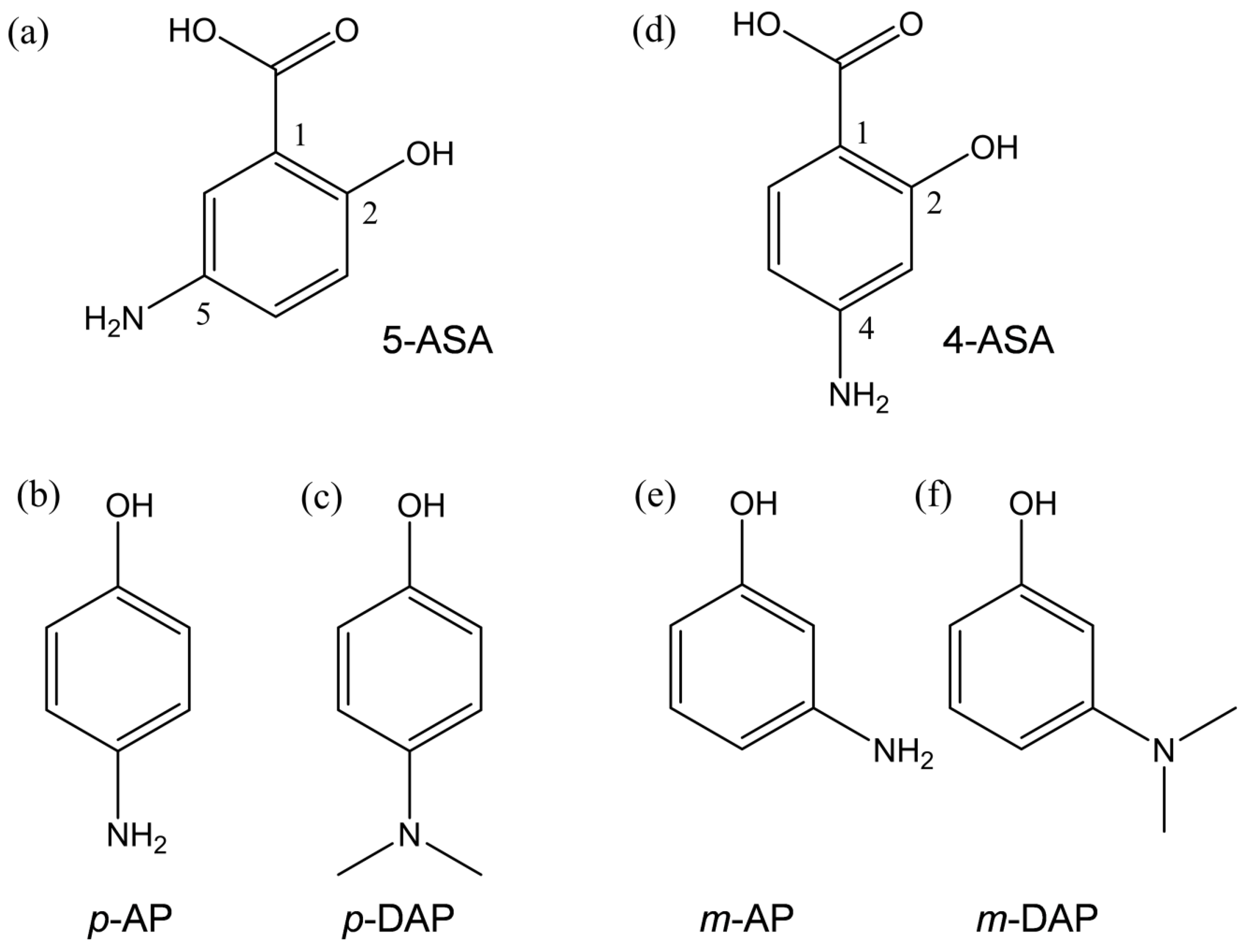
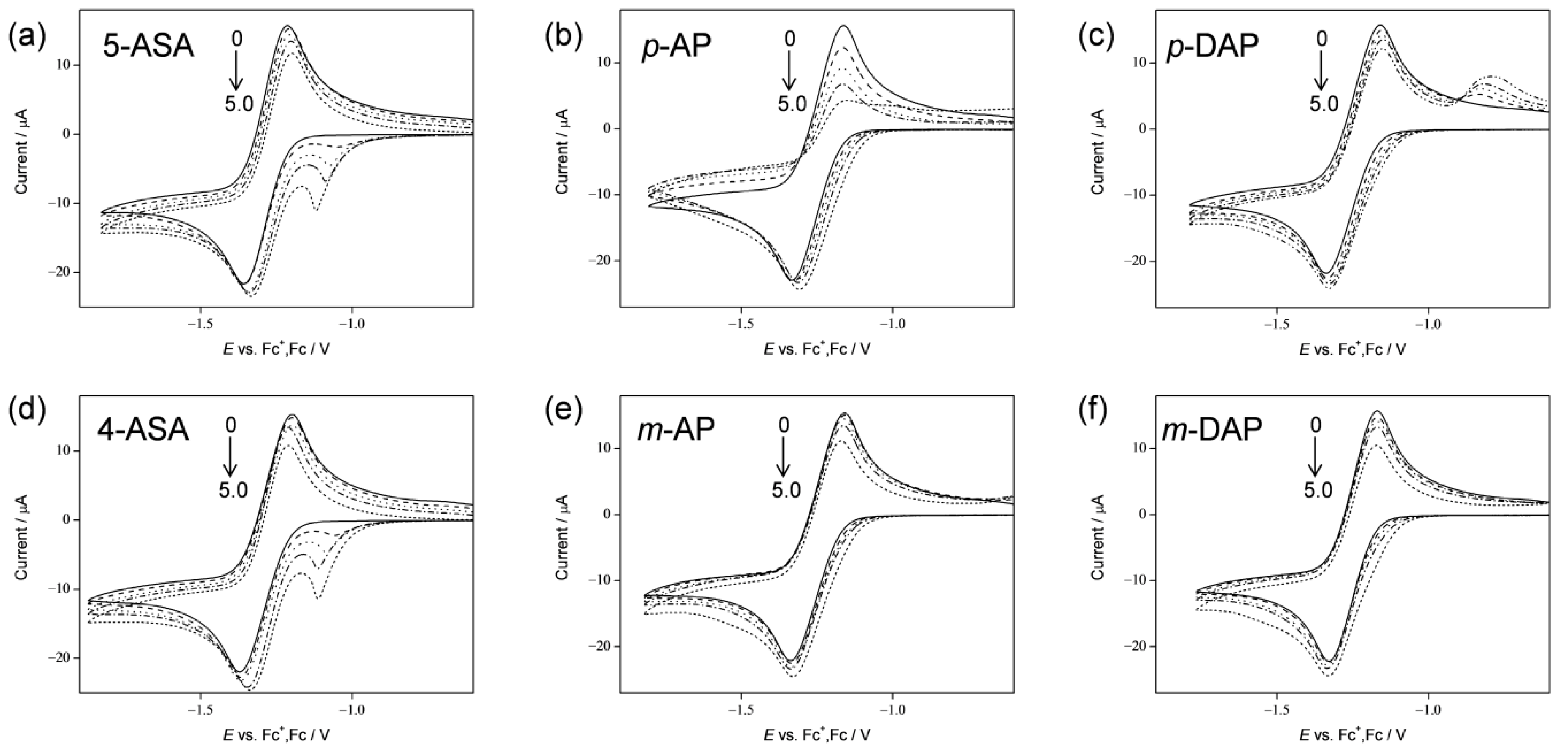
2.1. Cyclic Voltammetry Analysis of O2 in the Presence of 5-ASA and 4-ASA
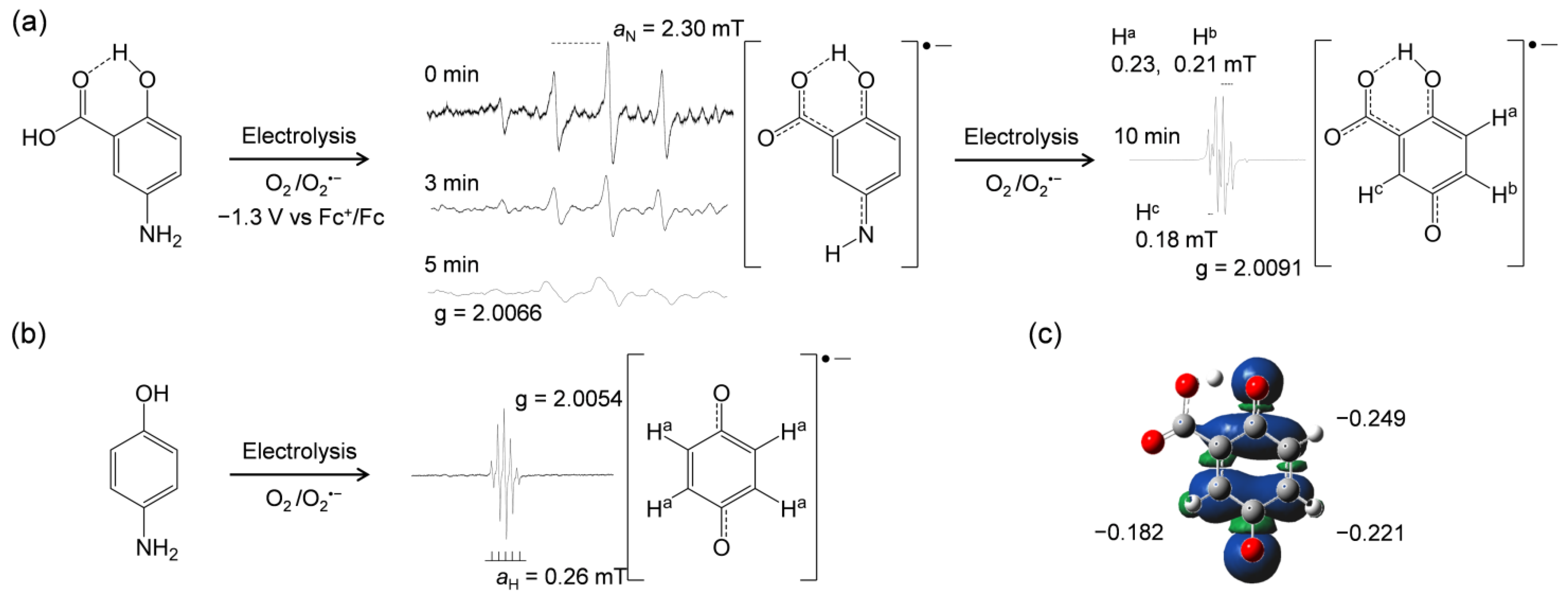
2.2. In Situ Electrolytic ESR Analyis of O2/O2•− in the Presence of 5-ASA and the Related Compounds
2.3. Stable-Structure Optimization for 5-ASA and Its Anion by DFT Calculation
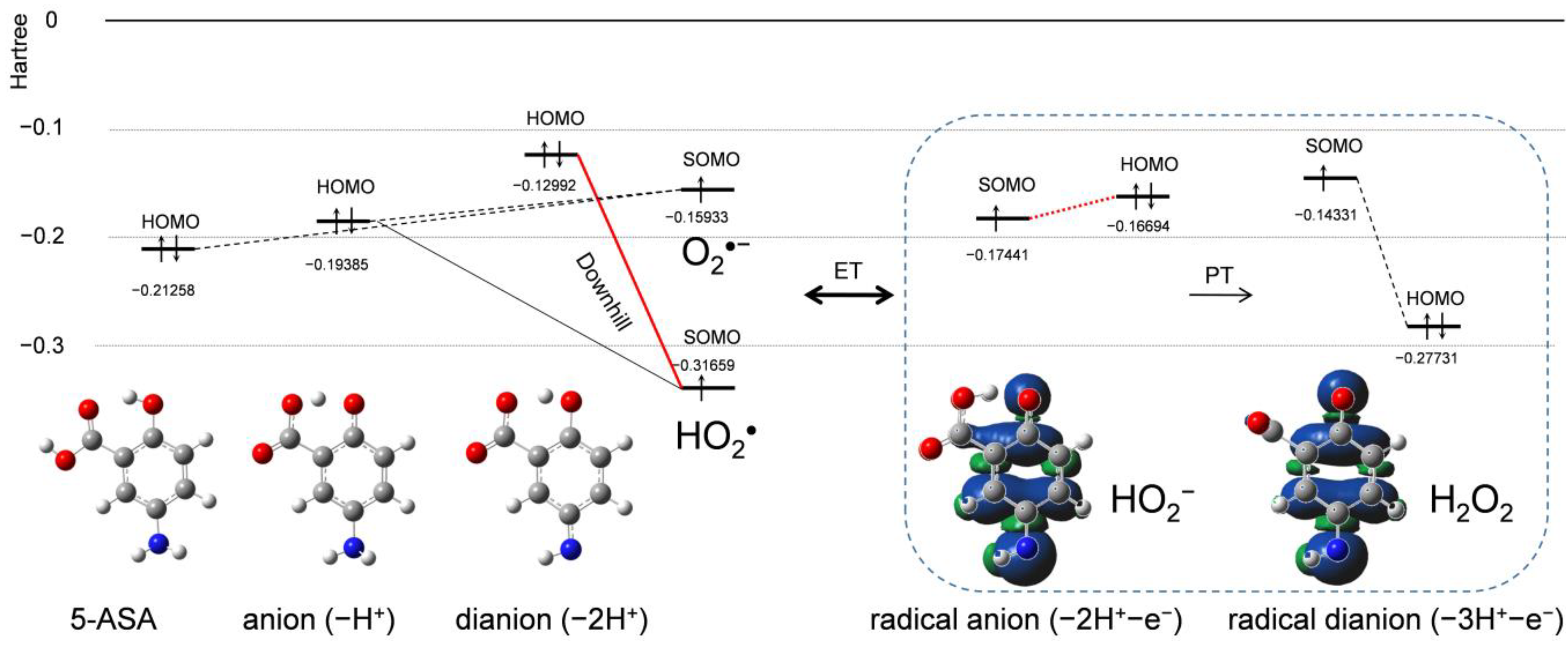
2.4. Change in HOMO–LUMO Energies upon PCET Between O2•− and 5-ASA
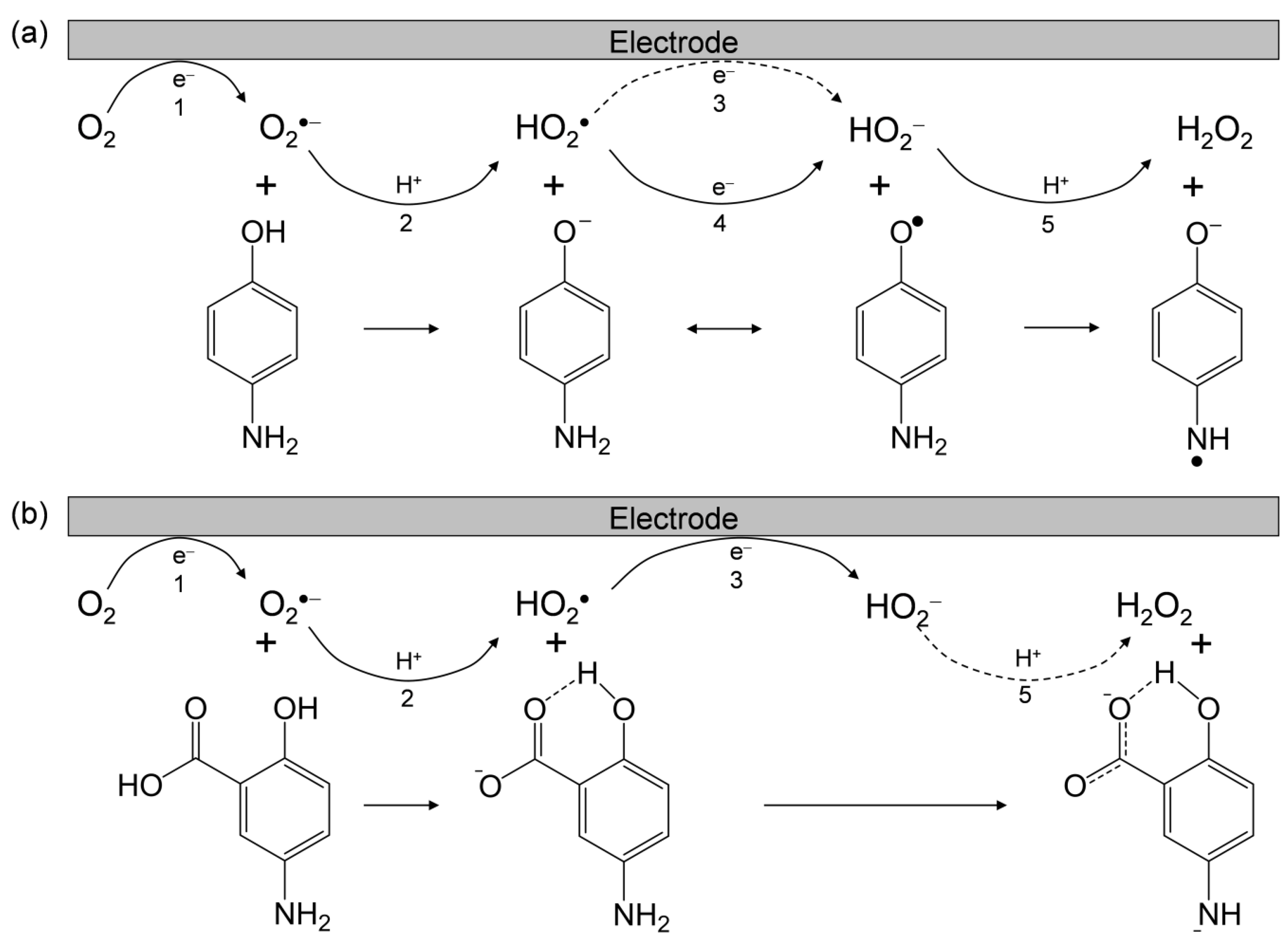
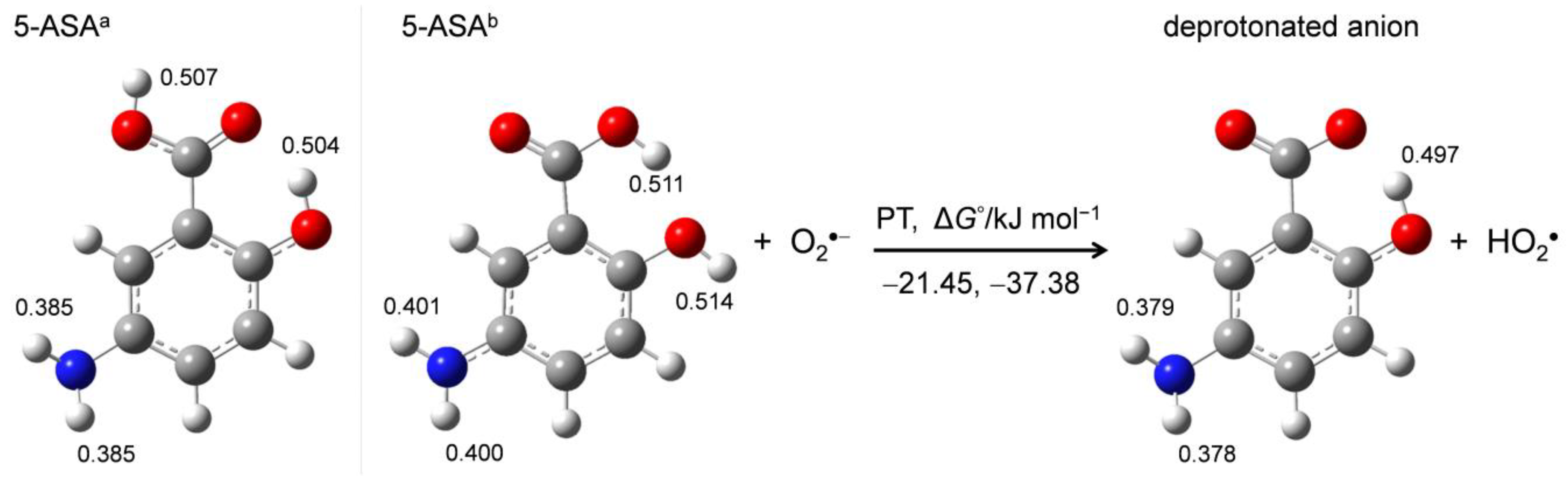
2.5. Free-Energy Analysis of PCET between Electrogenerated O2•− and the Deprotonated 5-ASA Anion
3. Materials and Methods
3.1. Chemicals
3.2. Electrochemical and In Situ Electrolytic ESR Measurements
3.3. Calculations
4. Conclusions
- 5-ASA eliminates O2•− through PCET involving three PTs and one ET;
- the net reaction mechanism involves three steps: initial deprotonation of the 1-carboxyl group of 5-ASA, subsequent PCET between the p-AP moiety of the deprotonated 5-ASA anion and O2•−, and subsequent exchange of a nitrogen atom with an oxygen atom, forming a 1,4-benzoquinone radical via an intermediate 1,4-benzoquinone imine radical;
- the 1-carboxyl group plays a protective role against the oxidative decomposition of 5-ASA under the action of O2•− in acidic solutions.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rachmilewitz, D.; Barbier, F.; Defrance, P.; De Reuck, M.; Devis, G.; DeVos, M.; Elewaut, A.; Potvin, P.H.; Rutgeerts, P.; Vanheuverzwyn, R.; et al. Coated mesalazine (5-aminosalicylic acid) versus sulphasalazine in the treatment of active ulcerative colitis: A randomised trial. Br. Med. J. 1989, 298, 82–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.A.; Ingram, M.J.; Naughton, D.P. A novel anti-oxidant and anti-cancer strategy: A peptoid anti-inflammatory drug conjugate with SOD mimic activity. Biochem. Biophys. Res. Commun. 2004, 317, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Kumar, S.; Unnikrishnan, M.; Mukherjee, T. Free radical scavenging reactions of sulfasalazine, 5-aminosalicylic acid and sulfapyridine: Mechanistic aspects and antioxidant activity. Free Radic. Res. 2005, 39, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, H.; Höfer, P.; Schmidt, M.; Böhne, P.; Kruis, W.; Gugler, R. Superoxide, hydroxyl and fatty acid radical scavenging by aminosalicylates. Direct evaluation with electron spin resonance spectroscopy. Biochem. Pharmacol. 1992, 43, 259–262. [Google Scholar] [CrossRef]
- Langmead, L.; Dawson, C.; Hawkins, C.; Banna, N.; Loo, S.; Rampton, D.S. Antioxidant effects of herbal therapies used by patients with inflammatory bowel disease: An in vitro study. Aliment. Pharmacol. Ther. 2002, 16. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Superoxide dismutase. In Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 352–354. ISBN 9780123786319. [Google Scholar]
- Singh, P.S.; Evans, D.H. Study of the electrochemical reduction of dioxygen in acetonitrile in the presence of weak acids. J. Phys. Chem. B 2006, 110, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Uno, B. Quinone-hydroquinone π-conjugated redox reaction involving proton-coupled electron transfer plays an important role in scavenging superoxide by polyphenolic antioxidants. Chem. Lett. 2010, 39, 162–164. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Importance of proton-coupled electron transfer from natural phenolic compounds in superoxide scavenging. Chem. Pharm. Bull. 2015, 63, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, T.; Uno, B. Structural properties of 4-substituted phenols capable of proton-coupled electron transfer to superoxide. Int. J. Adv. Res. Chem. Sci. 2016, 3, 11–19. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Concerted two-proton-coupled electron transfer from catechols to superoxide via hydrogen bonds. Electrochim. Acta 2016, 208, 304–309. [Google Scholar] [CrossRef]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J. Electrocatalytic oxygen reduction reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications; Springer: Berlin, Germany, 2008; pp. 89–134. ISBN 9781848009356. [Google Scholar]
- Fischer, V.; Mason, R.P. Stable free radical and benzoquinone imine metabolites of an acetaminophen analogue. J. Biol. Chem. 1984, 259, 10284–10288. [Google Scholar] [CrossRef]
- Capelle, S.; Pellegrini, S.; Cotelle, P.; Imbenotte, M.; Pommery, J.; Catteau, J.P. Autoxidation of gentisic acid in aqueous media. Water Res. 1996, 30, 1299–1303. [Google Scholar] [CrossRef]
- Nakayama, T.; Okumura, N.; Uno, B. Complementary effect of intra- and intermolecular hydrogen bonds on electron transfer in β–hydroxy-anthraquinone derivatives. J. Phys. Chem. B 2020, 124, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Gilman, H.; Avakian, S.; Benkeser, R.A.; Broadbent, H.S.; Clark, R.M.; Karmas, G.; Marshall, F.J.; Massie, S.M.; Shirley, D.A.; Woods, L.A. The synthesis of N-methyl-3-isopropyl-4-dimethylaminophenyl carbamate and some related derivatives. J. Org. Chem. 1954, 19, 1067–1079. [Google Scholar] [CrossRef]
- Okumura, N.; Uno, B. Electronic spectra of the electrogenerated 1,4-benzoquinone π-dianion and the strongly hydrogen-bonded charge-transfer complex with methanol. Bull. Chem. Soc. Jpn. 1999, 72, 1213–1217. [Google Scholar] [CrossRef]
- Frisch, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | PT1 | PT2 | PT3 | PT4 | ET1 | ET2 | ET3 | Total 1 |
|---|---|---|---|---|---|---|---|---|
| 5-ASA anion | 155.8 | −198.7 | 428.4 | −4.4 | 267.8 | −86.7 | −519.6 | 64.6 |
| p-AP | 64.5 | −302.4 | 447.6 | −10.6 | 332.9 | −44.0 | −502.3 | 9.7 |
| 4-ASA anion | 133.8 | −218.0 | 435.6 | 118.3 | 314.0 | −37.7 | −355.1 | 214.4 |
| m-AP | 47.9 | −294.9 | 438.7 | −7.0 | 358.1 | 15.2 | −430.5 | 56.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, T.; Honda, R. Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer. Pharmaceuticals 2021, 14, 120. https://doi.org/10.3390/ph14020120
Nakayama T, Honda R. Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer. Pharmaceuticals. 2021; 14(2):120. https://doi.org/10.3390/ph14020120
Chicago/Turabian StyleNakayama, Tatsushi, and Ryo Honda. 2021. "Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer" Pharmaceuticals 14, no. 2: 120. https://doi.org/10.3390/ph14020120
APA StyleNakayama, T., & Honda, R. (2021). Electrochemical and Mechanistic Study of Superoxide Elimination by Mesalazine through Proton-Coupled Electron Transfer. Pharmaceuticals, 14(2), 120. https://doi.org/10.3390/ph14020120