
Discovery and Optimization of Selective Inhibitors of Meprin α (Part II)

 , ,
, ,
Abstract
:1. Introduction
2. Results
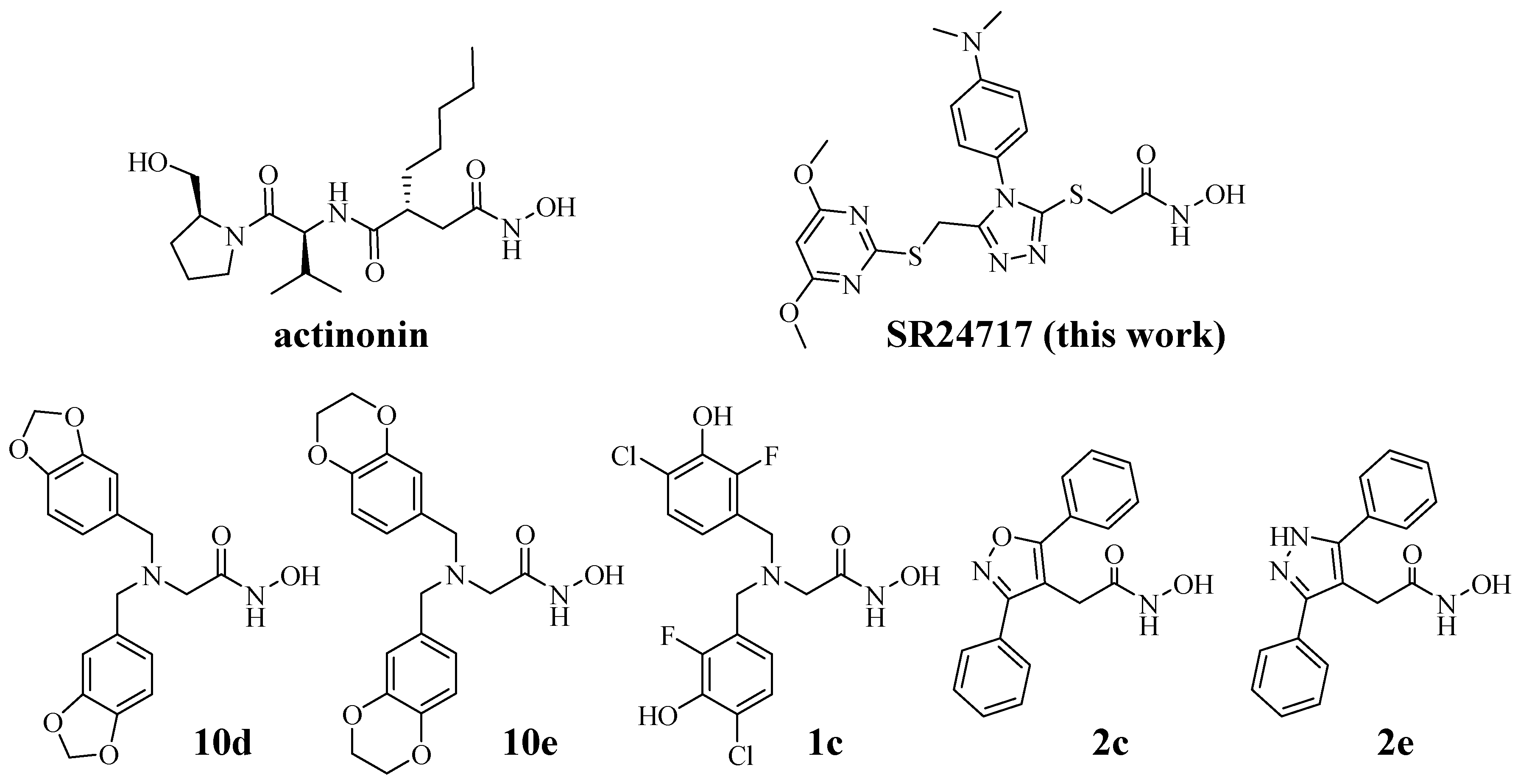
2.1. Modeling of Lead Series
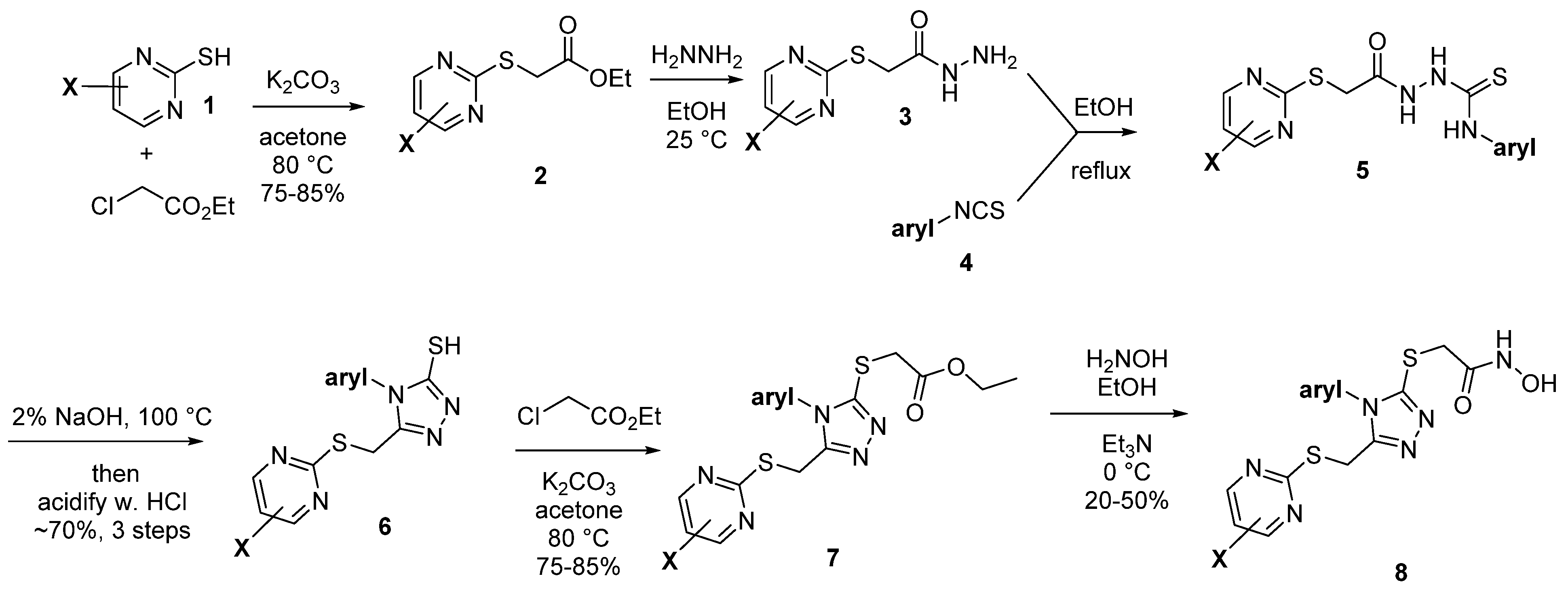
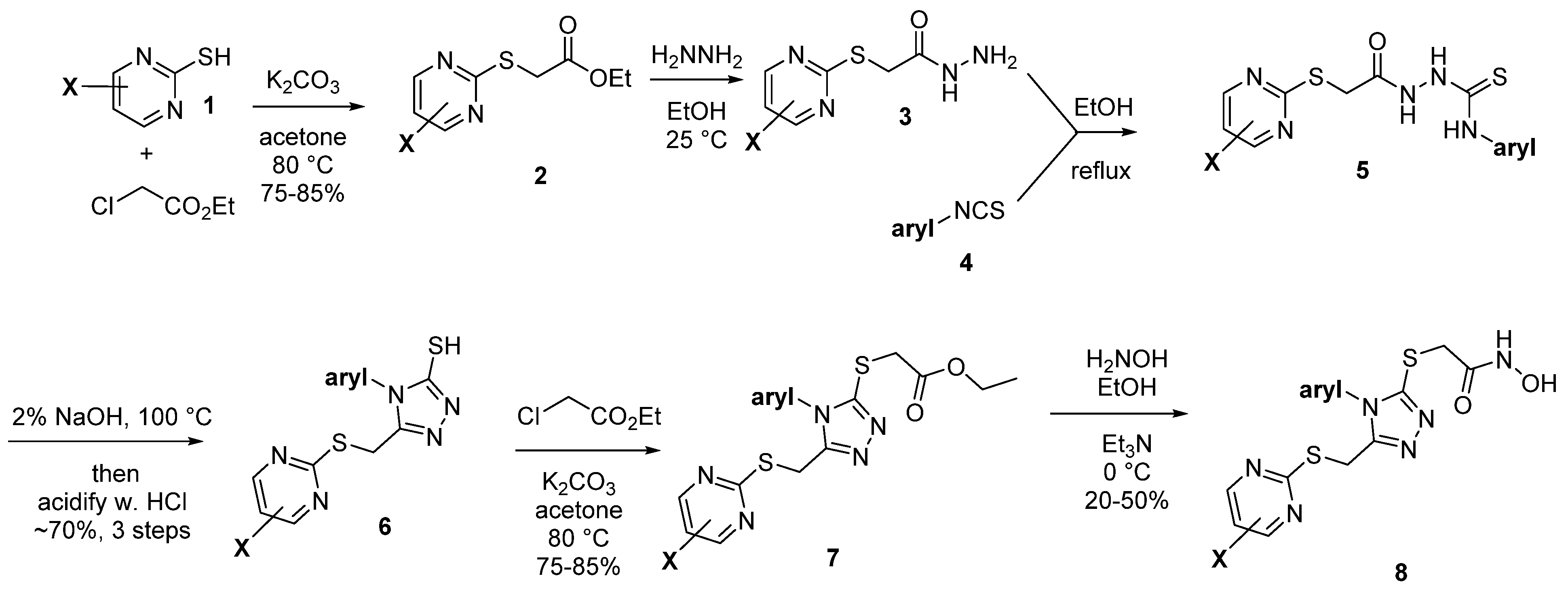
2.2. Synthetic Strategy
2.2.1. A. Pyrimidine Substitution
2.2.2. Phenyl Substitution
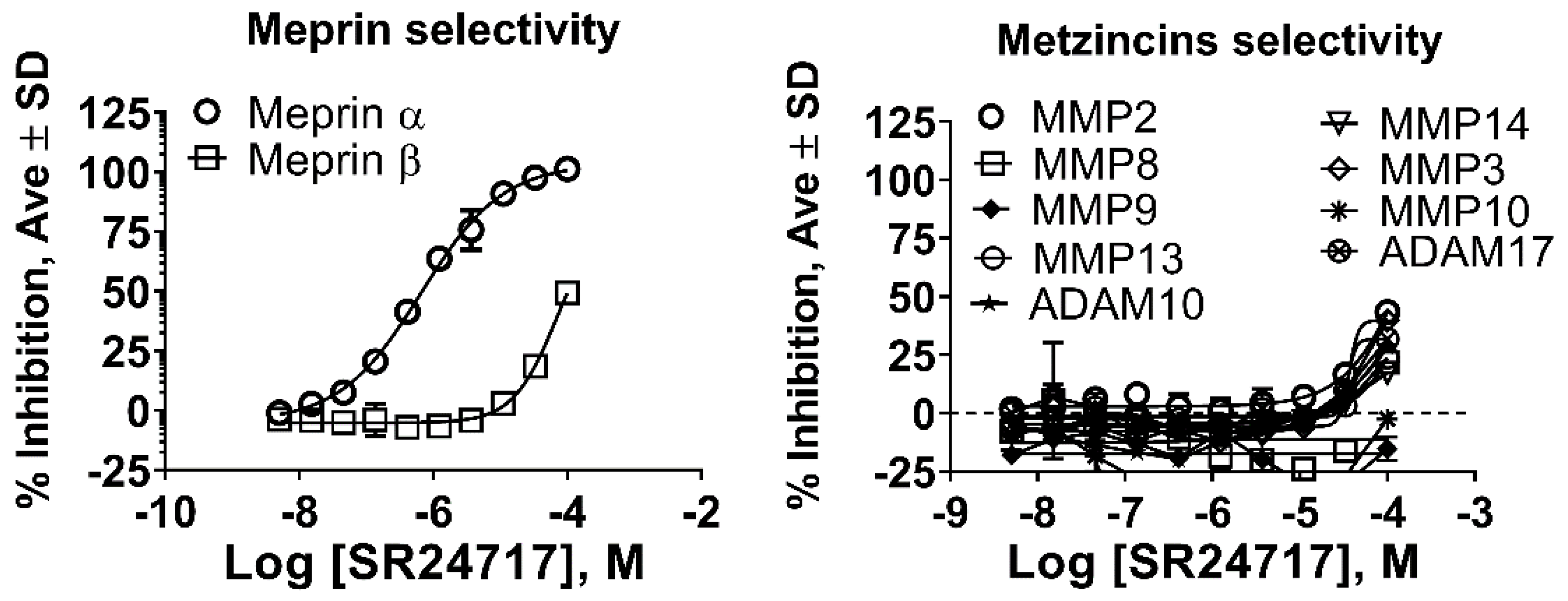
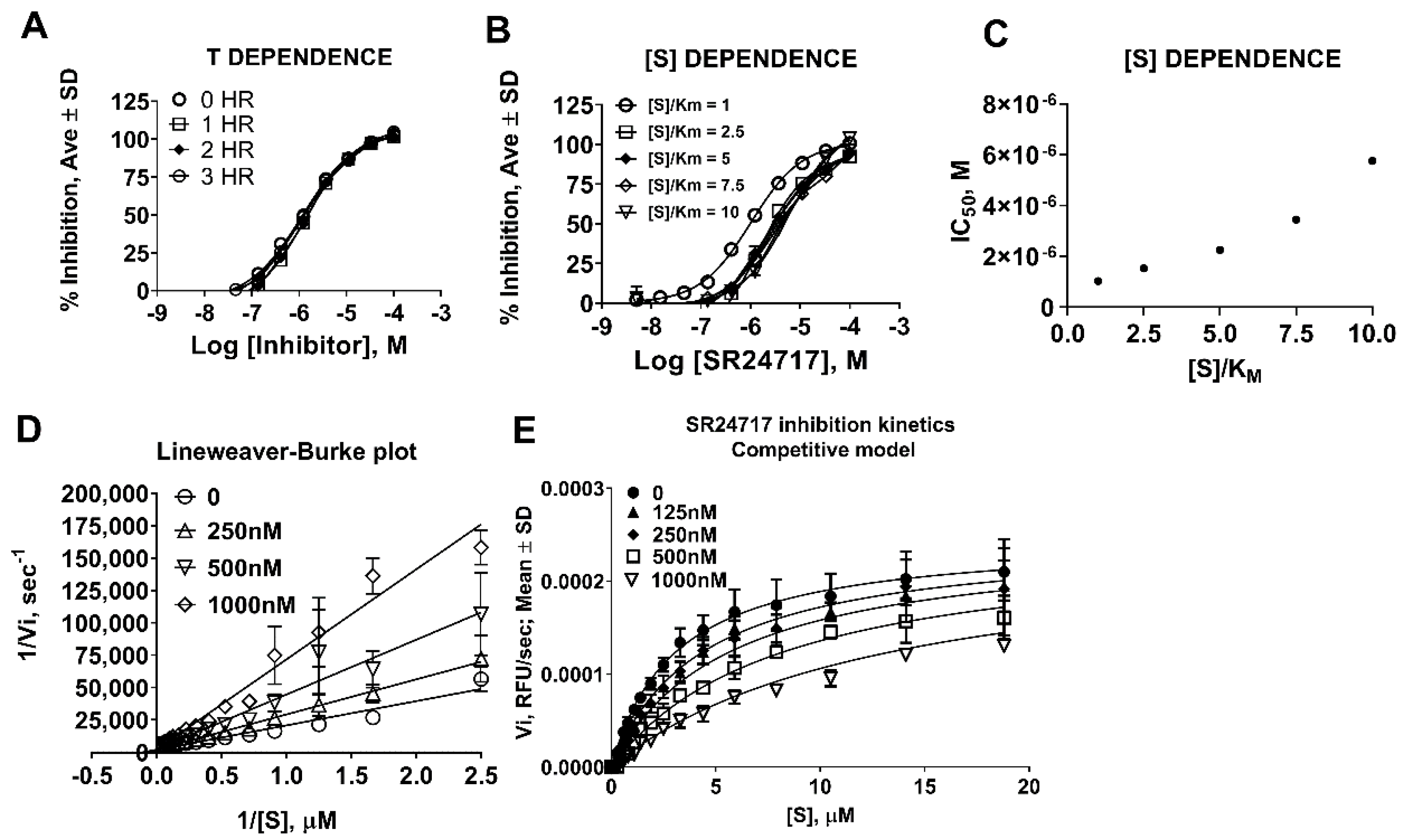
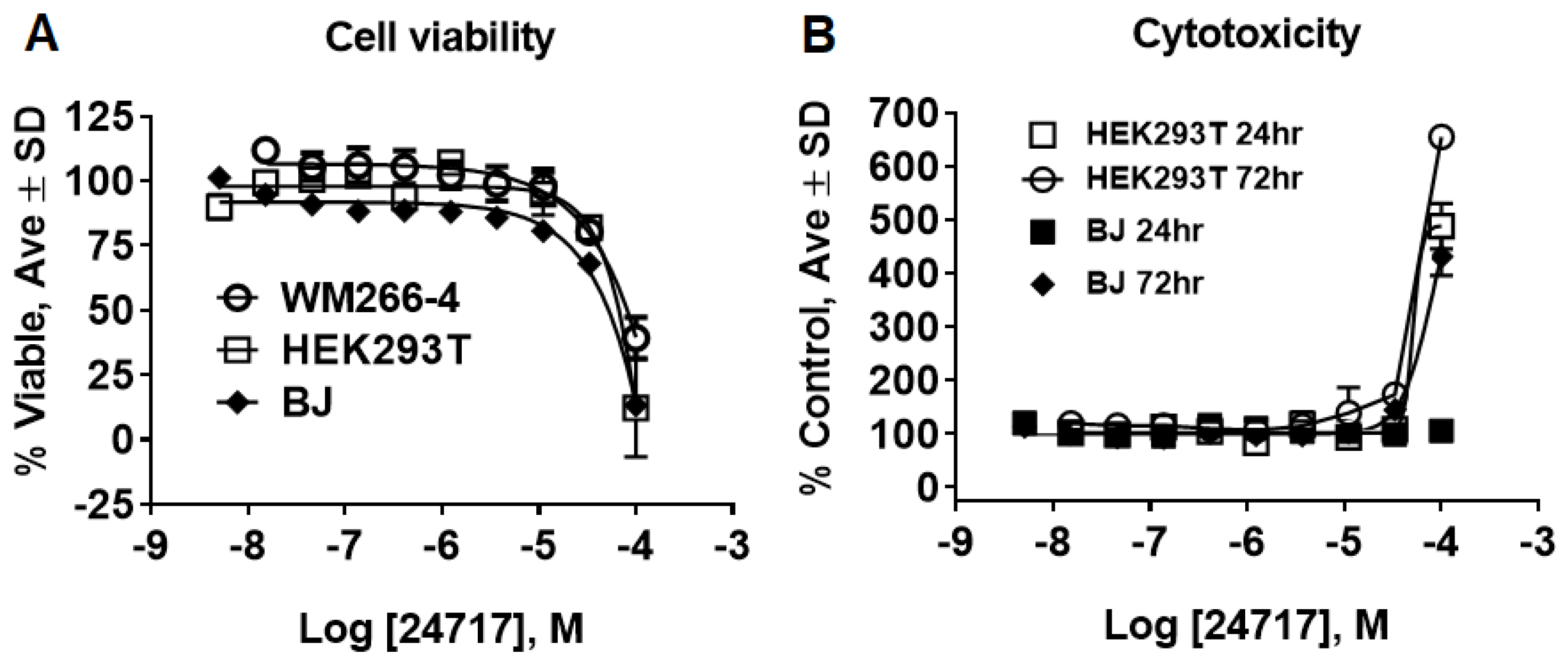
2.3. SR24717 Characterization in In Vitro Assays
3. Discussion
4. Materials and Methods
4.1. Assay Reagents
4.2. Meprin α and Meprin β Substrate Synthesis
4.3. Meprins Expression Protocol
4.4. Meprin α and Meprin β Assays in a 384-Well Plate
4.5. Determination of Kinetic Parameters of Meprin α Mediated Proteolysis in the Presence of Potential Probe SR24717
4.6. ADAM10 and ADAM17 Assays
4.7. MMP Assays
4.8. Cell Toxicity Studies
4.9. Molecular Modeling Studies
4.10. Compound Synthesis and Characterization
4.11. Data for Other Test Compounds
4.11.1. SR19855, N-hydroxy-2-((4-(4-methoxyphenyl)-5-((pyrimidin-2-ylthio)methyl)-4H-1,2,4-triazol-3-yl)thio)acetamide
4.11.2. SR24139, 2-((5-(((4,6-dimethylpyrimidin-2-yl)thio)methyl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.3. SR24144, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.4. SR24460, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(3-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.5. SR24459, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(2-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.6. SR24718, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(pyridin-3-yl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.7. SR24003, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(4-(trifluoromethyl)phenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.8. SR24462, 2-((4-(4-bromophenyl)-5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.9. SR24463, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.10. SR24467, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(m-tolyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.11. SR26467, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(naphthalen-1-yl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.12. SR26466, 2-((5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4-(4-morpholinophenyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
4.11.13. SR26465, 2-((4-(4-(diethylamino)phenyl)-5-(((4,6-dimethoxypyrimidin-2-yl)thio)methyl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyacetamide
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MMP | matrix metalloprotease |
| ADAM | a disintegrin and metalloprotease |
References
- Prox, J.; Arnold, P.; Becker-Pauly, C. Meprin alpha and meprin beta: Procollagen proteinases in health and disease. Matrix Biol. 2015, 44–46, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Broder, C.; Arnold, P.; Vadon-Le Goff, S.; Konerding, M.A.; Bahr, K.; Müller, S.; Overall, C.M.; Bond, J.S.; Koudelka, T.; Tholey, A.; et al. Metalloproteases meprin alpha and meprin beta are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc. Natl. Acad. Sci. USA 2013, 110, 14219–14224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, F.; Becker-Pauly, C. Role of meprin metalloproteases in metastasis and tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.N.; Becker, C.; Lottaz, D.; Köhler, D.; Yiallouros, I.; Krell, H.W.; Sterchi, E.E.; Stöcker, W. Human meprin alpha and beta homo-oligomers: Cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem. J. 2004, 378 Pt 2, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Madoux, F.; Tredup, C.; Spicer, T.P.; Scampavia, L.; Chase, P.S.; Hodder, P.S.; Fields, G.B.; Becker-Pauly, C.; Minond, D. Development of high throughput screening assays and pilot screen for inhibitors of metalloproteases meprin alpha and beta. Biopolymers 2014, 102, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsbeck, D.; Hamann, A.; Schlenzig, D.; Schilling, S.; Buchholz, M. First insight into structure-activity relationships of selective meprin beta inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2428–2431. [Google Scholar] [CrossRef] [PubMed]
- Ramsbeck, D.; Hamann, A.; Richter, G.; Schlenzig, D.; Geissler, S.; Nykiel, V.; Cynis, H.; Schilling, S.; Buchholz, M. Structure-Guided Design, Synthesis, and Characterization of Next-Generation Meprin beta Inhibitors. J. Med. Chem. 2018, 61, 4578–4592. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Jäger, C.; Schlenzig, D.; Schilling, S.; Buchholz, M.; Ramsbeck, D. Tertiary-Amine-Based Inhibitors of the Astacin Protease Meprin alpha. ChemMedChem 2018, 13, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Diez, J.; Wang, C.; Becker-Pauly, C.; Fields, G.B.; Bannister, T.D.; Spicer, T.P.; Scampavia, L.D.; Minond, D.A. Discovery and Optimization of Selective Inhibitors of Meprin α (Part I). Pharmaceuticals 2021. Accepted. [Google Scholar]
- Altintop, M.D.; Kaplancikli, Z.A.; Ḉiftḉi, G.A.; Demirel, R. Synthesis and biological evaluation of thiazoline derivatives as newantimicrobial and anticancer agents. Eur. J. Med. Chem. 2014, 74, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Jäger, C.; Körschgen, H.; Geissler, S.; Schlenzig, D.; Buchholz, M.; Stöcker, W.; Ramsbeck, D. Heteroaromatic Inhibitors of the Astacin Proteinases Meprin a, Meprin b and Ovastacin Discovered by a Scaffold-Hopping Approach. ChemMedChem 2020. [Google Scholar] [CrossRef]
- Broder, C.; Becker-Pauly, C. The metalloproteases meprin alpha and meprin beta: Unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem. J. 2013, 450, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, G.B.; Lauer-Fields, J.L.; Liu, R.-Q.; Barany, G. Principles and Practice of Solid-Phase Peptide Synthesis. In Synthetic Peptides: A User’s Guide, 2nd ed.; Grant, G.A., Ed.; W.H. Freeman & Co.: New York, NY, USA, 2001; pp. 93–219. [Google Scholar]
- de Jong, G.I.; Buwalda, B.; Schuurman, T.; Luiten, P.G. Synaptic plasticity in the dentate gyrus of aged rats is altered after chronic nimodipine application. Brain Res. 1992, 596, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Becker-Pauly, C.; Höwel, M.; Walker, T.; Vlad, A.; Aufenvenne, K.; Oji, V.; Lottaz, D.; Sterchi, E.E.; Debela, M.; Magdolen, V.; et al. The alpha and beta subunits of the metalloprotease meprin are expressed in separate layers of human epidermis, revealing different functions in keratinocyte proliferation and differentiation. J. Investig. Dermatol. 2007, 127, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kaplancıklıa, Z.A.; Yurttaas, L.; Turan-Zitouni, G.; Ã-zdemir, A.; Göger, G.; Demirci, F.; Mohsen, U. Synthesis and Antimicrobial Activity of New Pyrimidine-Hydrazones. Lett. Drug Des. Discov. 2014, 11, 76–81. [Google Scholar] [CrossRef]
- Lokwani, D.; Azad, R.; Sarkate, A.; Reddanna, P.; Shinde, D. Structure Based Library Design (SBLD) for new 1,4-dihydropyrimidine scaffold as simultaneous COX-1/COX-2 and 5-LOX inhibitors. Bioorg. Med. Chem. 2015, 23, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
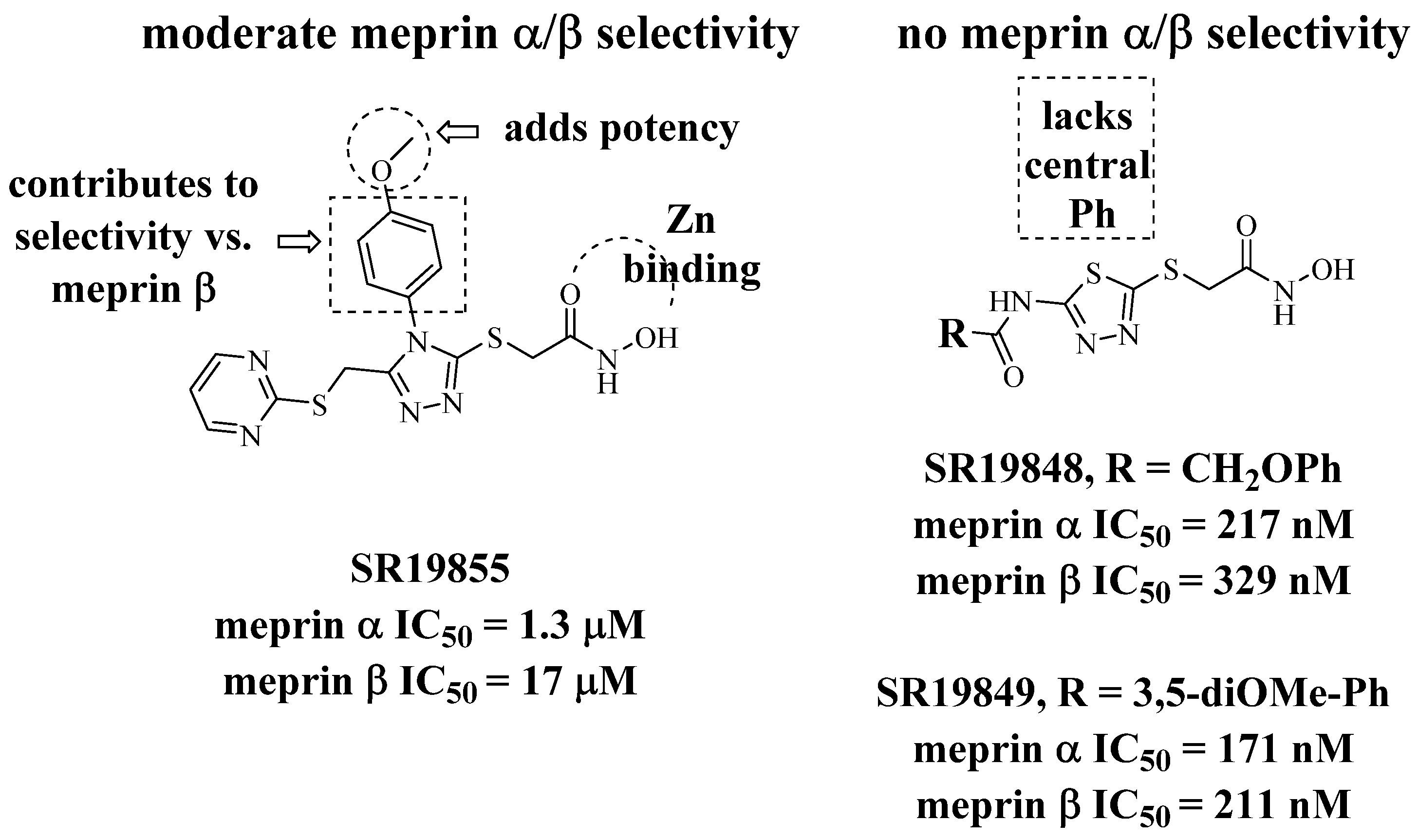






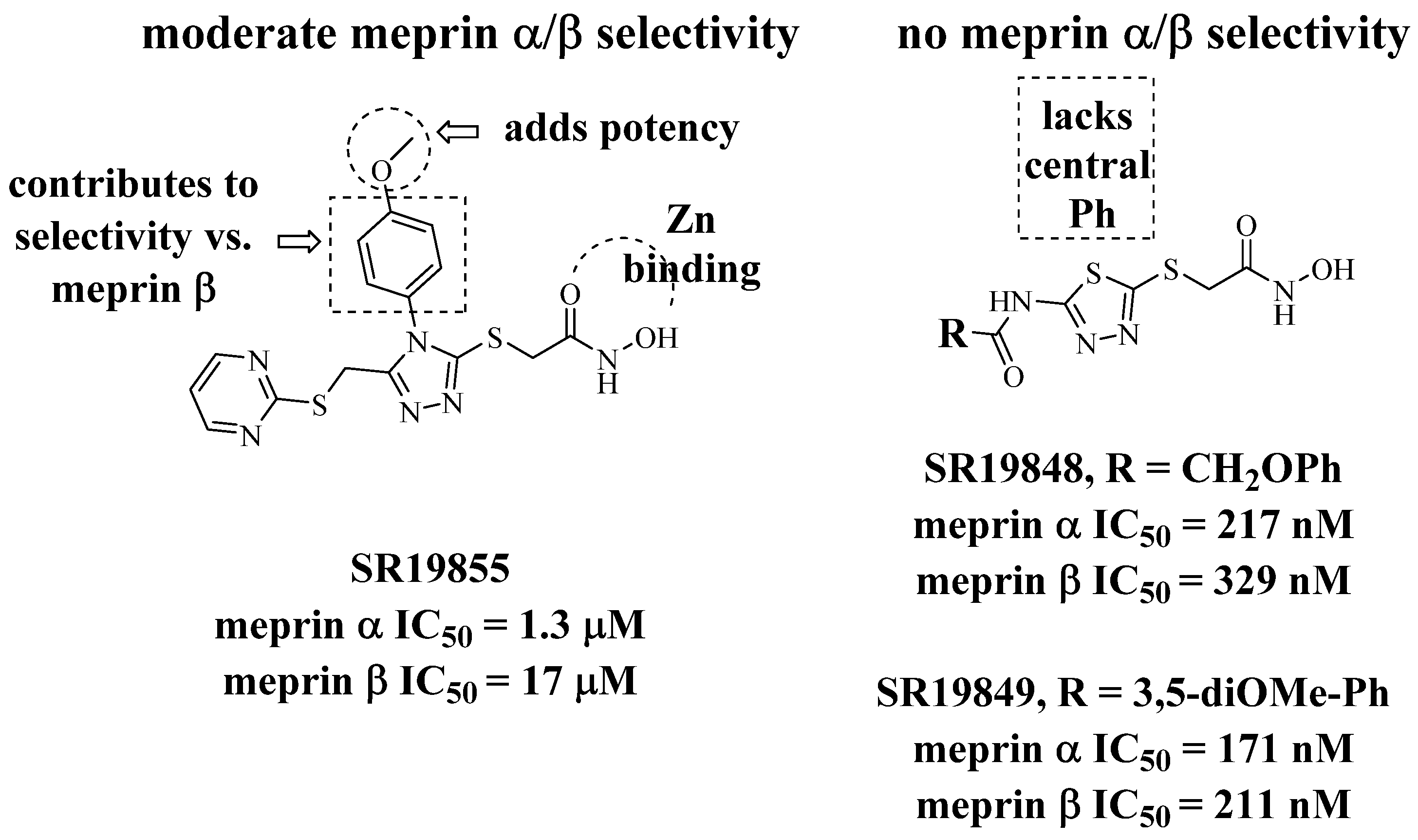{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | X | ID | Meprin α IC50, μM | Meprin β IC50, μM |
| H | SR19855 | 1.3 ± 0.1 | 17 ± 3 | |
| Me | SR24139 | 4.1 ± 0.8 | 60 ± 7 | |
| OMe | SR24144 | 8.7 ± 1.1 | >100 |
 | |||
| aryl | ID | Meprin α IC50, μM | Meprin β IC50, μM |
|---|---|---|---|
 | SR19855 | 1.3 ± 0.1 | 17 ± 3 |
 | SR24460 | 10.3 ± 1.1 | 59 ± 10 |
 | SR24459 | 12.1 ± 3.9 | 52 ± 1 |
 | SR24718 | 10.1 ± 2.3 | >100 |
 | SR24003 | 5.4 ± 1.2 | >100 |
 | SR24462 | 11.2 ± 2.6 | >100 |
 | SR24463 | 10.4 ± 2.8 | >100 |
 | SR24467 | 10.3 ± 2.2 | >100 |
 | SR26467 | 2.0 ± 0.3 | >100 |
 | SR26466 | 1.1 ± 0.1 | >100 |
 | SR26465 | 0.60 ± 0.06 | 75 ± 15 |
 | SR24717 | 0.66 ± 0.03 | 70 ± 5 |
| ID | Meprin α | Meprin β | MMP2 | MMP3 | MMP8 | MMP9 | MMP10 | MMP13 | MMP14 | ADAM10 | ADAM17 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SR24717 a | 0.66 ± 0.03 | 70 ± 5 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| Actinonin b | 0.004 | 4.8 ± 0.5 | 0.09 | NR | 0.19 | 0.1 ± 0.01 | NR | 0.1 | >1000 | 2.2 ± 0.2 | 0.2 ± 0.02 |
| 10d [8] c | 0.16 ± 0.001 | 2.95 ± 0.35 | 95% 26% | 91% 61% | 86% 55% | 86% 61% | 86% 57% | ||||
| 10e [8] | 0.40 ± 0.03 | 7.59 ± 0.01 | 94% 65% | 91% 21% | 98% 47% | 80% 56% | 80% 41% | ||||
| 1c [11] d | 0.19 ± 0.001 | 0.03 ± 0.35 | 95% | 91% | 86% | 86% | 86% | ||||
| 2c [11] e | 0.004 ± 0.001 | 0.813 ± 0.001 | 87% | 86% | 80% | 80% | 62% | ||||
| 2e [11] f | 0.003 ± 0.001 | 0.199 ± 0.001 | 104% | 81% | 75% | 73% | 61% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Diez, J.; Park, H.; Becker-Pauly, C.; Fields, G.B.; Spicer, T.P.; Scampavia, L.D.; Minond, D.; Bannister, T.D. Discovery and Optimization of Selective Inhibitors of Meprin α (Part II). Pharmaceuticals 2021, 14, 197. https://doi.org/10.3390/ph14030197
Wang C, Diez J, Park H, Becker-Pauly C, Fields GB, Spicer TP, Scampavia LD, Minond D, Bannister TD. Discovery and Optimization of Selective Inhibitors of Meprin α (Part II). Pharmaceuticals. 2021; 14(3):197. https://doi.org/10.3390/ph14030197
Chicago/Turabian StyleWang, Chao, Juan Diez, Hajeung Park, Christoph Becker-Pauly, Gregg B. Fields, Timothy P. Spicer, Louis D. Scampavia, Dmitriy Minond, and Thomas D. Bannister. 2021. "Discovery and Optimization of Selective Inhibitors of Meprin α (Part II)" Pharmaceuticals 14, no. 3: 197. https://doi.org/10.3390/ph14030197
APA StyleWang, C., Diez, J., Park, H., Becker-Pauly, C., Fields, G. B., Spicer, T. P., Scampavia, L. D., Minond, D., & Bannister, T. D. (2021). Discovery and Optimization of Selective Inhibitors of Meprin α (Part II). Pharmaceuticals, 14(3), 197. https://doi.org/10.3390/ph14030197