
Novel Selective IDO1 Inhibitors with Isoxazolo[5,4-d]pyrimidin-4(5H)-one Scaffold

 , ,
, , 

Abstract
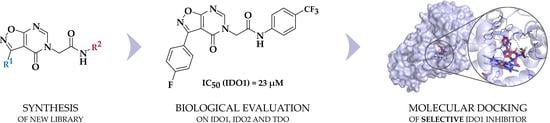
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. hIDO1 Expression
2.3. Determination of Inhibitory Potency
3. Materials and Methods
3.1. Chemistry and Chemical Characterization of Compounds
3.2. General Procedure for the Synthesis of Aldehyde Oximes (6a–k)
3.3. General Procedure for the Synthesis of N-Hydroxyimidoyl Chlorides (7a–k)
3.4. General Procedure for the Synthesis of 5-Aminoisoxazole-4-Carboxamides (8a–k)
3.4.1. Procedure I
3.4.2. Procedure II
3.5. General Procedure for the Synthesis of Isoxazolo[5,4-d]pyrimidin-4(5H)-ones (9a–k)
3.6. General Procedures for the N-Acylation (Preparation of Alkyl Halogenides 11a–y)
3.6.1. Procedure III
3.6.2. Procedure IV
3.6.3. Procedure V
3.7. General Procedure for the Synthesis of Final Compounds (12–53)
3.8. Cloning, Expression and Isolation of rhIDO1
3.9. hIDO1 Inhibitory Potency Evaluation
3.10. hIDO2 Inhibitory Potency Evaluation
3.11. hTDO Inhibitory Potency Evaluation
3.12. Cytotoxicity
3.13. Cell-Based IDO1 Inhibitory Assay
3.14. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Hu, N.; Yang, D.; Oxenkrug, G.; Yang, Q. Regulating the balance between the kynurenine and serotonin pathways of tryptophan metabolism. FEBS J. 2017, 284, 948–966. [Google Scholar] [CrossRef]
- Yan, D.; Lin, Y.-W.; Tan, X. Heme-containing enzymes and inhibitors for tryptophan metabolism. Metallomics 2017, 9, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y. Studien über den intermediären stoffwechsel des tryptophans xviii–xxiv. Hoppe-Seyler´s Z. Physiol. Chem. 1936, 243, 237–265. [Google Scholar] [CrossRef]
- Knox, W.E.; Mehler, A.H. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J. Biol. Chem. 1950, 187, 419–430. [Google Scholar] [CrossRef]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? JOS 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Jusof, F.F.; Bakmiwewa, S.M.; Weiser, S.; Too, L.K.; Metz, R.; Prendergast, G.C.; Fraser, S.T.; Hunt, N.H.; Ball, H.J. Investigation of the tissue distribution and physiological roles of indoleamine 2,3-dioxygenase-2. Int. J. Tryptophan. Res. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An important immunotherapy target in cancer treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef]
- Van Baren, N.; van den Eynde, B.J. Tumoral immune resistance mediated by enzymes that degrade tryptophan. Cancer Immunol. Res. 2015, 3, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Schultze, J.L. New insights into IDO biology in bacterial and viral infections. Front. Immunol. 2014, 5, 384. [Google Scholar] [CrossRef]
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Variation of genes encoding KAT1, AADAT and IDO1 as a potential risk of depression development. Eur. Psychiatry 2018, 52, 95–103. [Google Scholar] [CrossRef]
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Białek, K.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Association between single nucleotide polymorphisms of TPH1 and TPH2 genes, and depressive disorders. J. Cell. Mol. Med. 2018, 22, 1778–1791. [Google Scholar] [CrossRef]
- Ogbechi, J.; Clanchy, F.I.; Huang, Y.-S.; Topping, L.M.; Stone, T.W.; Williams, R.O. IDO activation, inflammation and musculoskeletal disease. Exp. Gerontol. 2020, 131, 110820. [Google Scholar] [CrossRef] [PubMed]
- Dolšak, A.; Gobec, S.; Sova, M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol. Ther. 2020, 107746. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, K.; Pu, Q.; Achab, A.; Ardolino, M.J.; Cheng, M.; Deng, Y.; Doty, A.C.; Ferguson, H.; Fradera, X.; et al. Discovery of amino-cyclobutarene-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors for cancer immunotherapy. ACS Med. Chem. Lett. 2019, 10, 1530–1536. [Google Scholar] [CrossRef]
- Steeneck, C.; Kinzel, O.; Anderhub, S.; Hornberger, M.; Pinto, S.; Morschhaeuser, B.; Braun, F.; Kleymann, G.; Hoffmann, T. Discovery of hydroxyamidine based inhibitors of IDO1 for cancer immunotherapy with reduced potential for glucuronidation. ACS Med. Chem. Lett. 2020, 11, 179–187. [Google Scholar] [CrossRef]
- Wen, H.; Liu, Y.; Wang, S.; Wang, T.; Zhang, G.; Chen, X.; Li, Y.; Cui, H.; Lai, F.; Sheng, L. Design and synthesis of indoleamine 2,3-dioxygenase 1 inhibitors and evaluation of their use as anti-tumor agents. Molecules 2019, 24, 2124. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [PubMed]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef]
- Pham, K.N.; Yeh, S.-R. Mapping the binding trajectory of a suicide inhibitor in human indoleamine 2,3-dioxygenase 1. J. Am. Chem. Soc. 2018, 140, 14538–14541. [Google Scholar] [CrossRef] [PubMed]
- Fraunhoffer, K.J.; DelMonte, A.J.; Beutner, G.L.; Bultman, M.S.; Camacho, K.; Cohen, B.; Dixon, D.D.; Fan, Y.; Fanfair, D.; Freitag, A.J.; et al. Rapid development of a commercial process for linrodostat, an indoleamine 2,3-dioxygenase (IDO) inhibitor. Org. Process. Res. Dev. 2019, 23, 2482–2498. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolušić, E.; Frédérick, R.; Plaen, E.D.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Sova, M.; Dolšak, A.; Proj, M.; Knez, D.; Lešnik, S.; Konc, J.; Gobec, S. Silico Design, Synthesis and Biochemical Evaluation of Novel Small-Molecule Indoleamine 2,3-Dioxygenase 1 Inhibitors with a Pyrimidin-4(3H)-One Scaffold; Slovensko Farmacevtsko Društvo: Ljubljana, Slovenia, 2018; p. 202, P155. [Google Scholar]
- Tomek, P.; Palmer, B.D.; Flanagan, J.U.; Fung, S.-P.S.; Bridewell, D.J.A.; Jamie, J.F.; Ching, L.-M. Formation of an N-formylkynurenine-derived fluorophore and its use for measuring indoleamine 2,3-dioxygenase 1 activity. Anal. Bioanal. Chem. 2013, 405, 2515–2524. [Google Scholar] [CrossRef]
- Yu, G.J.; Yang, B.; Verkman, A.S.; Kurth, M.J. Isoxazolopyrimidines as novel δF508-CFTR correctors. Synlett 2010, 2010, 1063–1066. [Google Scholar] [CrossRef]
- Tran, N.C.; Dhondt, H.; Flipo, M.; Deprez, B.; Willand, N. Synthesis of functionalized 2-isoxazolines as three-dimensional fragments for fragment-based drug discovery. Tetrahedron. Lett. 2015, 56, 4119–4123. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Pajouhesh, H.; Ding, Y.; Tan, J.; Grimwood, M.; Belardetti, F.; Kaul, R. Bicyclic Pyrimidine Derivatives as Calcium Channel Blockers. U.S. Patent US 8,133,998, 13 March 2012. [Google Scholar]
- Anderluh, M.; Mravljak, J.; Sova, M.; Perdih, A.; Pečar, S. Medicinal Chemistry III: Laboratory Practice and Seminars; Faculty of Pharmacy, University of Ljubljana: Ljubljana, Slovenia, 2011. [Google Scholar]
- Yang, D.; Zhang, S.; Fang, X.; Guo, L.; Hu, N.; Guo, Z.; Li, X.; Yang, S.; He, J.C.; Kuang, C.; et al. N-benzyl/aryl substituted tryptanthrin as dual inhibitors of indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase. J. Med. Chem. 2019, 62, 9161–9174. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Wang, R.; Cui, M.; Liu, W.; Yang, Q.; Kuang, C. Synthesis of novel tryptanthrin derivatives as dual inhibitors of indoleamine 2,3-dioxygenase 1 and tryptophan 2,3-dioxygenase. Bioorg. Med. Chem. Lett. 2020, 127159. [Google Scholar] [CrossRef]
- Pan, S.; Zhou, Y.; Wang, Q.; Wang, Y.; Tian, C.; Wang, T.; Huang, L.; Nan, J.; Li, L.; Yang, S. Discovery and structure-activity relationship studies of 1-aryl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione derivatives as potent dual inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) and trytophan 2,3-dioxygenase (TDO). Eur. J. Med. Chem. 2020, 207, 112703. [Google Scholar] [CrossRef]
- Feng, X.; Shen, P.; Wang, Y.; Li, Z.; Bian, J. Synthesis and in vivo antitumor evaluation of an orally active potent phosphonamidate derivative targeting IDO1/IDO2/TDO. Biochem. Pharmacol. 2019, 168, 214–223. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.-R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Pantouris, G.; Serys, M.; Yuasa, H.J.; Ball, H.J.; Mowat, C.G. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino. Acids 2014, 46, 2155–2163. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3-(4-fluorophenyl)isoxazolo[5,4-d]pyrimidin-4(5H)-one scaffold | ||||
|---|---|---|---|---|
 | ||||
| Compound | R2 | % of Inhibition @ 100 µM or IC50 a,b | ||
| IDO1 | IDO2 | TDO | ||
| 12 |  | IC50, 50 ± 1 μM | n.a. c | n.a. |
| 13 |  | 21 ± 5% | n.a. | n.a. |
| 14 |  | 27 ± 4% | n.a. | n.a. |
| 15 |  | IC50, 88 ± 4 µM | n.a. | n.a. |
| 16 |  | 15 ± 4% | 28 ± 9% | n.a. |
| 17 |  | 33 ± 2% | n.a. | n.a. |
| 18 |  | 32 ± 4% | 15 ± 1% | n.a. |
| 19 |  | IC50, 55 ± 2 µM | n.a. | n.a. |
| 20 |  | IC50, 27 ± 1 µM | n.a. | n.a. |
| 21 |  | 27 ± 3% | n.a. | n.a. |
| 22 |  | IC50, 55 ± 2 µM | n.a. | n.a. |
| 23 |  | IC50, 23 ± 1 µM | n.a. | n.a. |
| 24 |  | 30 ± 2% | 39 ± 5% | 25 ± 8% |
| 25 |  | IC50, 98 ± 5 µM | 28 ± 0% | 24 ± 7% |
| 26 |  | 11 ± 2% | n.a. | n.a. |
| 27 |  | 11 ± 4% | n.a. | 17 ± 2% |
| 28 |  | 31 ± 3% | n.a. | 18 ± 4% |
| 29 |  | IC50, 30 ± 1 µM | n.a. | 29 ± 3% |
| 30 |  | n.a. | n.a. | 17 ± 2% |
| 31 |  | IC50, 87 ± 3 µM | n.a. | n.a. |
| 32 |  | IC50, 22 ± 1 µM | n.a. | n.a. |
| 33 |  | n.a. | n.a. | 22 ± 4% |
| 34 |  | n.a. | n.a. | n.a. |
| 35 |  | 20 ± 6% | n.a. | n.a. |
| 36 |  | IC50, 46 ± 2 µM | 13 ± 2% | n.a. |
| 37 |  | 17 ± 5% | n.a. | n.a. |
| 3-arylisoxazolo[5,4-d]pyrimidin-4(5H)-one scaffold | |||||
|---|---|---|---|---|---|
 | |||||
| Compound | R1 | R2 | % of Inhibition @ 100 µM or IC50 a,b | ||
| IDO1 | IDO2 | TDO | |||
| 38 |  |  | n.a. c | 16 ± 4% | n.a. |
| 39 |  |  | IC50, 22 ± 2 µM | 11 ± 7% | n.a. |
| 40 |  |  | IC50, 127 ± 5 µM | 16 ± 5% | n.a. |
| 41 |  |  | 32 ± 1% | n.a. | n.a. |
| 42 |  |  | 15 ± 4% | n.d. d | n.d. |
| 43 |  |  | n.a. | n.d. | n.d. |
| 44 |  |  | n.a. | n.a. | n.a. |
| 45 |  |  | n.a. | n.a. | n.a. |
| 46 |  |  | 14 ± 3% | n.a. | n.a. |
| 47 |  |  | 34 ± 1% | n.a. | n.a. |
| 48 |  |  | IC50, 75 ± 4 µM | n.a. | 10 ± 6% |
| 49 |  |  | IC50, 96 ± 5 µM | n.a. | 10 ± 2% |
| 50 |  |  | n.a. | n.a. | n.a. |
| 51 |  |  | IC50, 53 ± 3 µM | n.a. | n.a. |
| 52 |  |  | 23 ± 5% | n.a. | n.a. |
| 53 |  |  | IC50, 40 ± 2 µM | 21 ± 6% | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolšak, A.; Bratkovič, T.; Mlinarič, L.; Ogorevc, E.; Švajger, U.; Gobec, S.; Sova, M. Novel Selective IDO1 Inhibitors with Isoxazolo[5,4-d]pyrimidin-4(5H)-one Scaffold. Pharmaceuticals 2021, 14, 265. https://doi.org/10.3390/ph14030265
Dolšak A, Bratkovič T, Mlinarič L, Ogorevc E, Švajger U, Gobec S, Sova M. Novel Selective IDO1 Inhibitors with Isoxazolo[5,4-d]pyrimidin-4(5H)-one Scaffold. Pharmaceuticals. 2021; 14(3):265. https://doi.org/10.3390/ph14030265
Chicago/Turabian StyleDolšak, Ana, Tomaž Bratkovič, Larisa Mlinarič, Eva Ogorevc, Urban Švajger, Stanislav Gobec, and Matej Sova. 2021. "Novel Selective IDO1 Inhibitors with Isoxazolo[5,4-d]pyrimidin-4(5H)-one Scaffold" Pharmaceuticals 14, no. 3: 265. https://doi.org/10.3390/ph14030265
APA StyleDolšak, A., Bratkovič, T., Mlinarič, L., Ogorevc, E., Švajger, U., Gobec, S., & Sova, M. (2021). Novel Selective IDO1 Inhibitors with Isoxazolo[5,4-d]pyrimidin-4(5H)-one Scaffold. Pharmaceuticals, 14(3), 265. https://doi.org/10.3390/ph14030265