
Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats

 , , ,
, , , 
Abstract
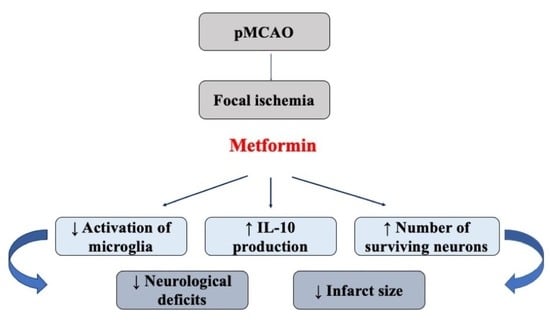
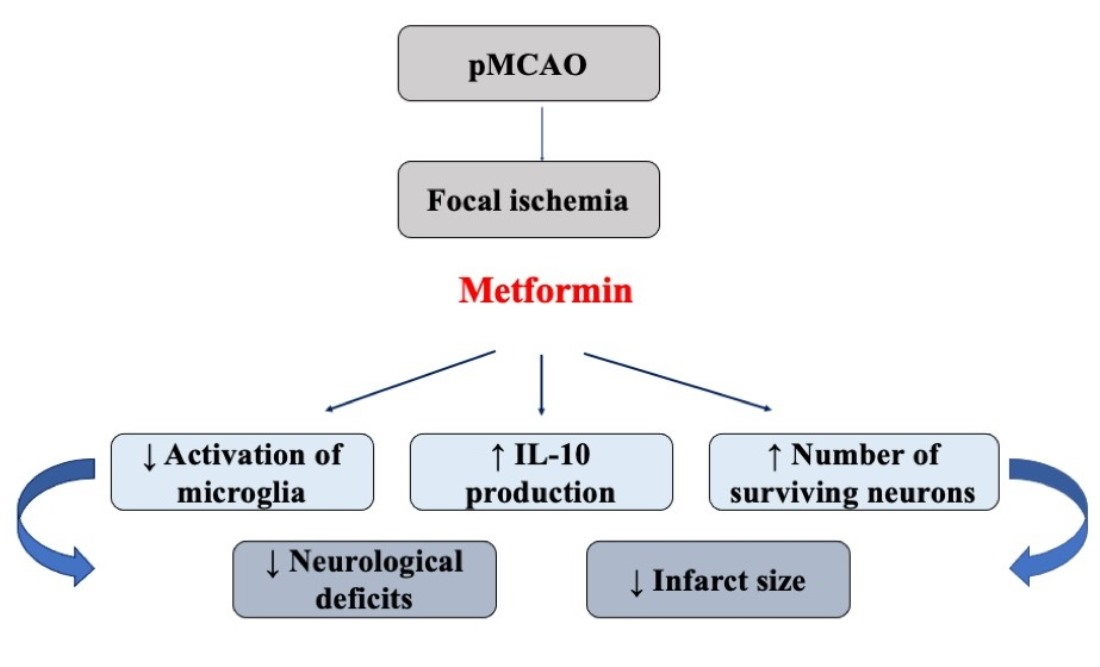
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
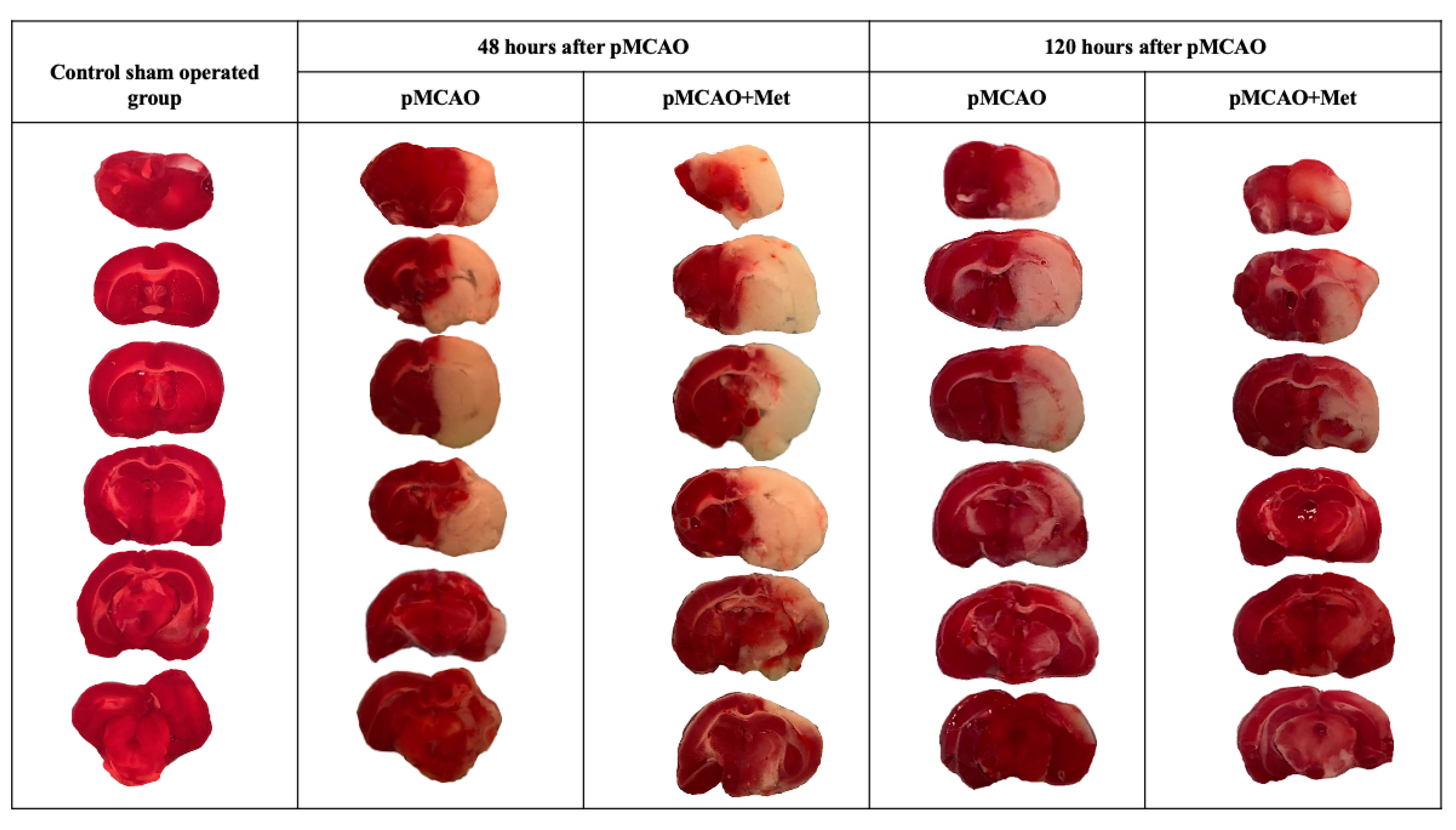
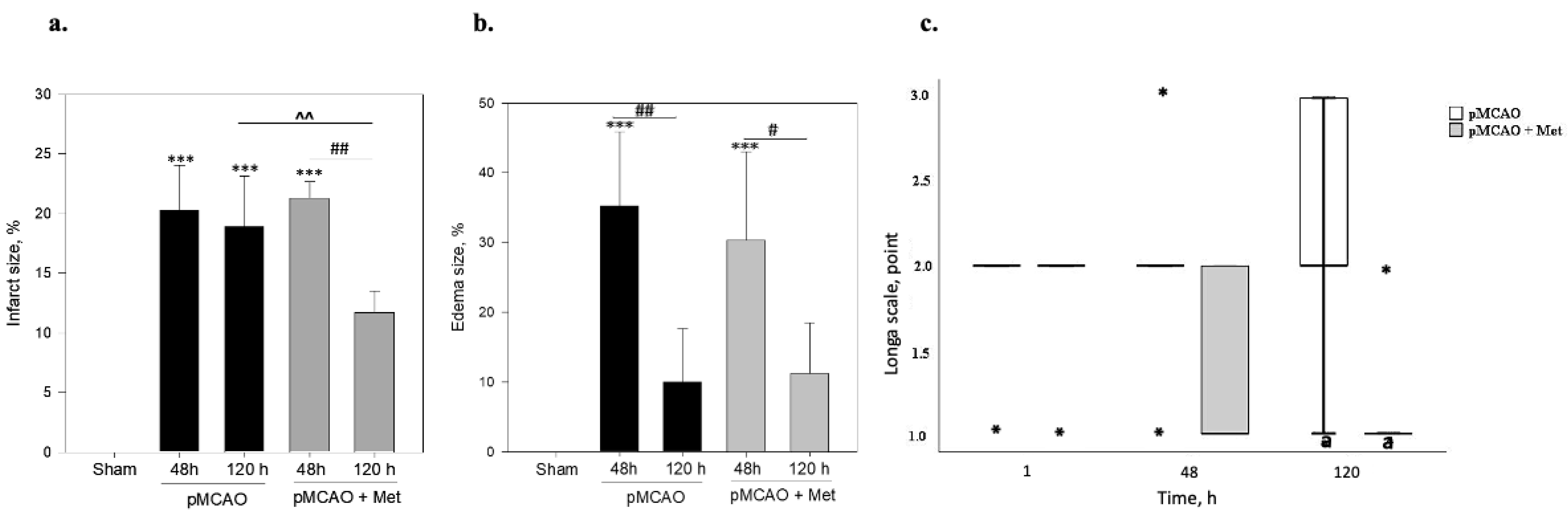
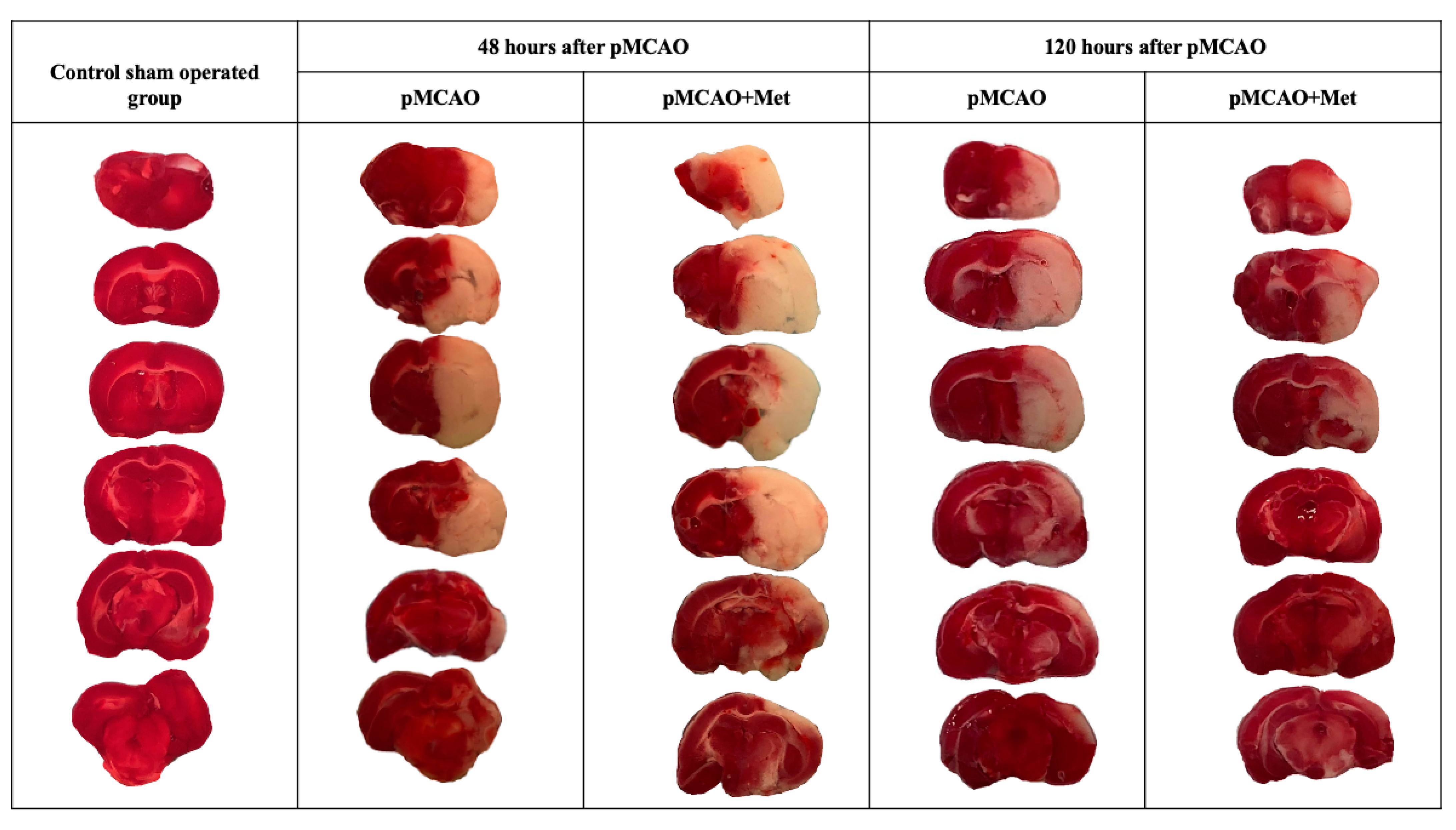
2.1. The Effect of Metformin on Infarct Size, Brain Oedema and Neurological State after pMCAO
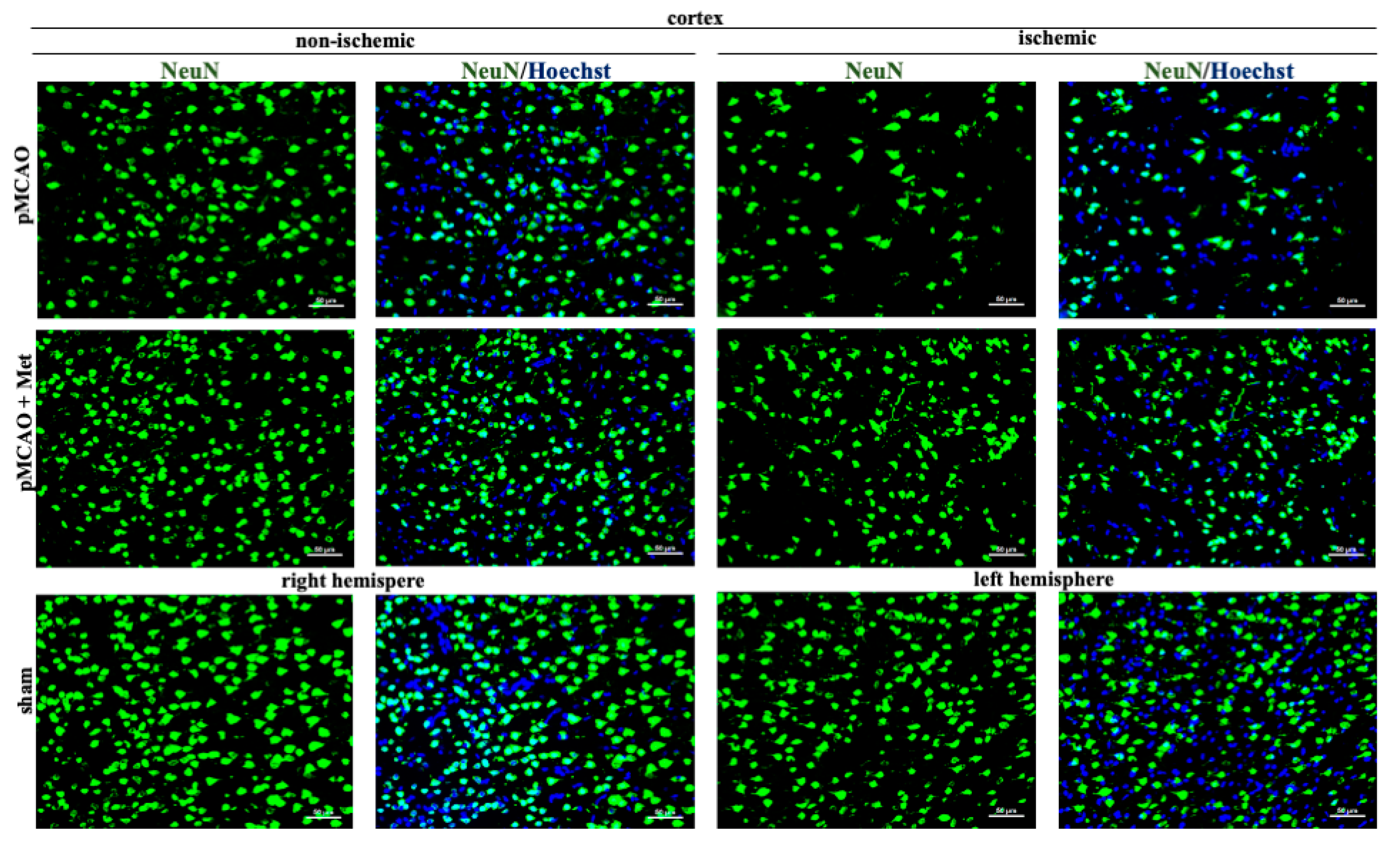
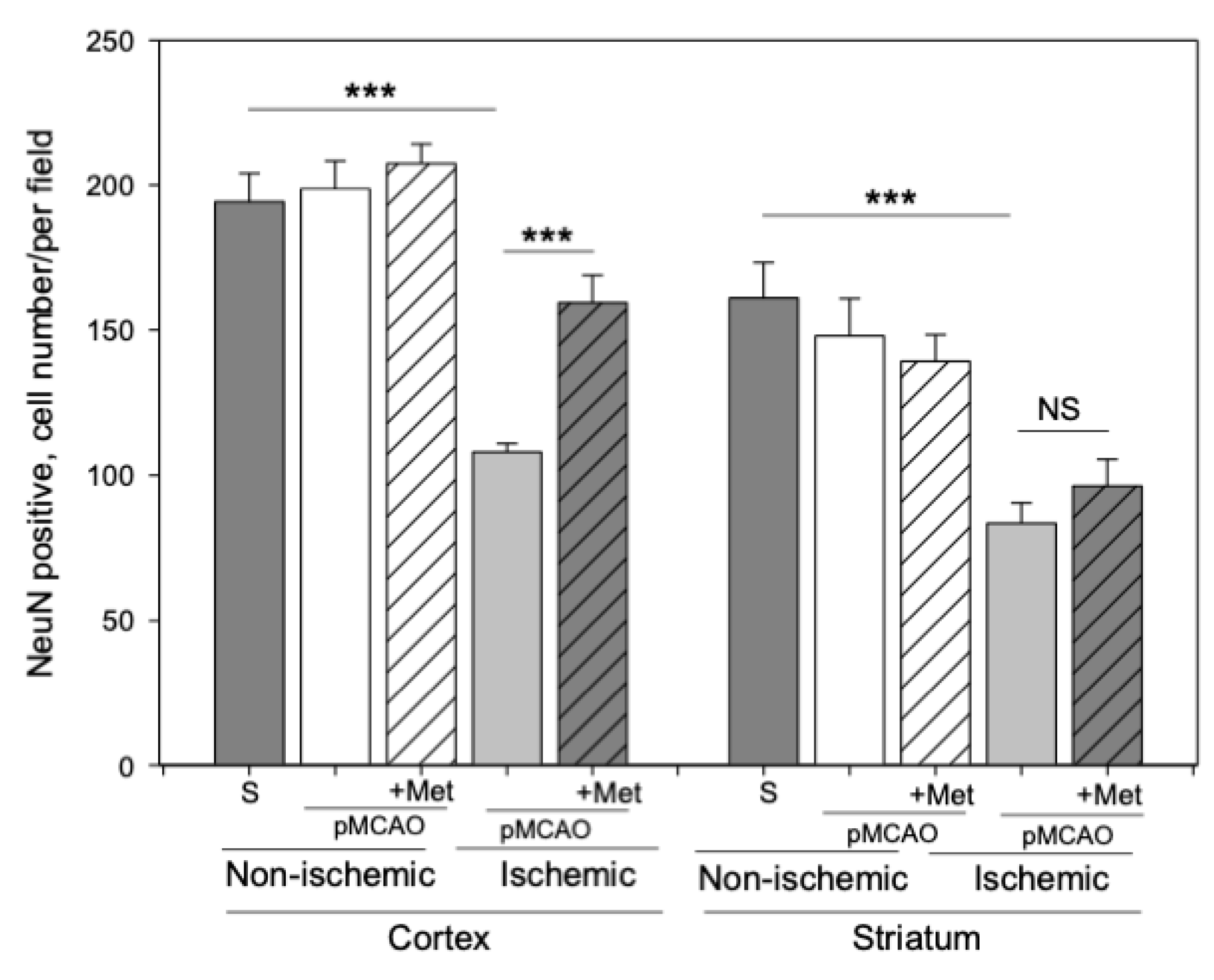
2.2. The Effect of Metformin on Neuronal Densities in Brain Sections after pMCAO
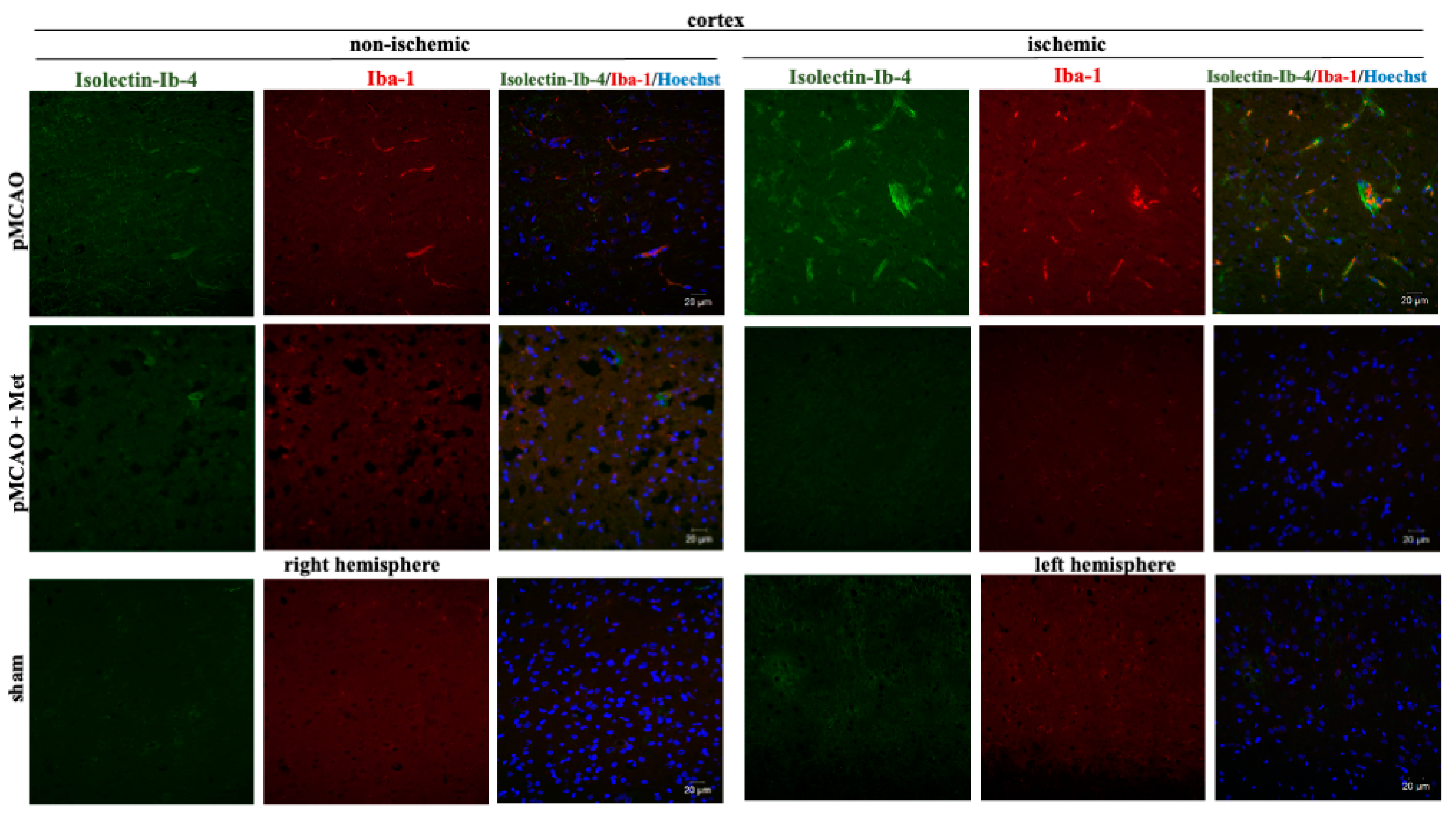
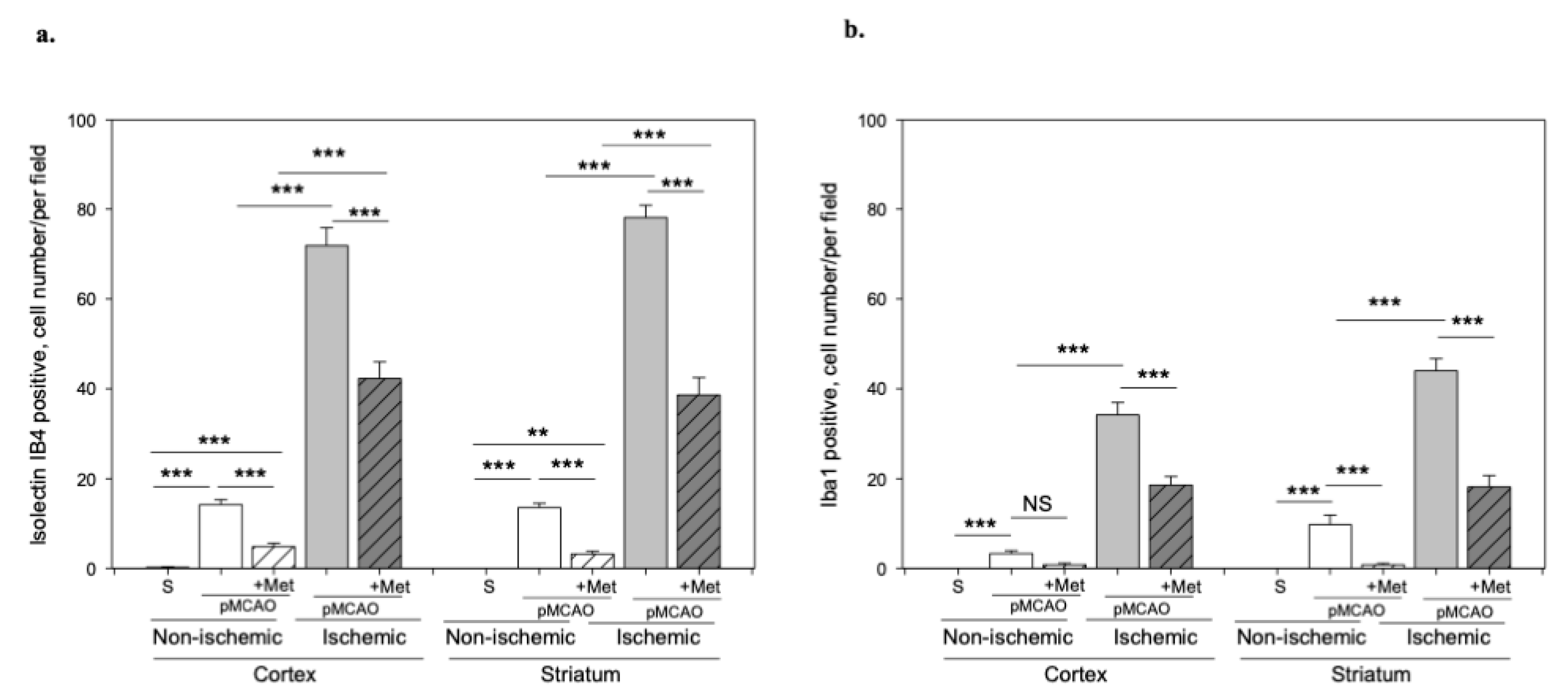
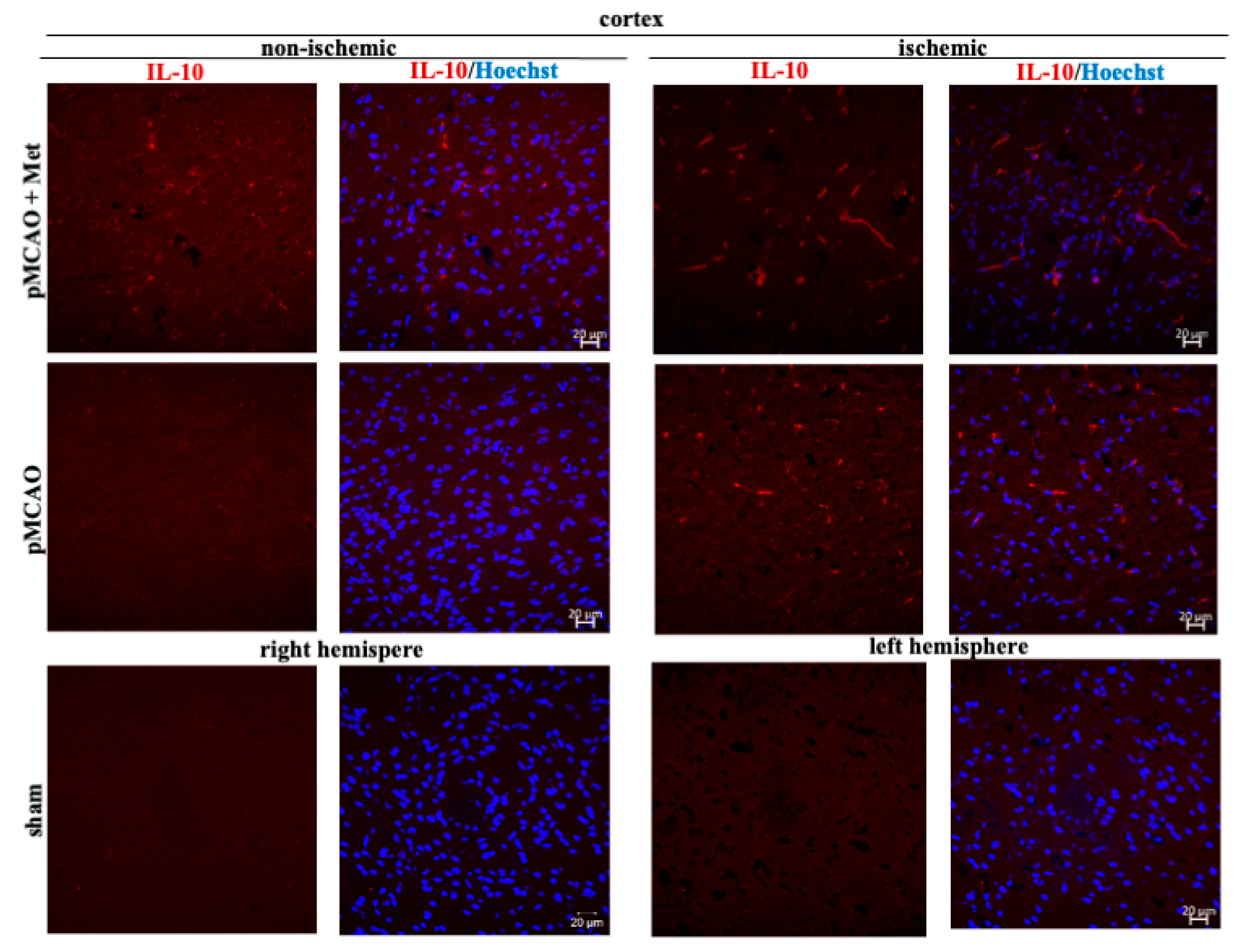
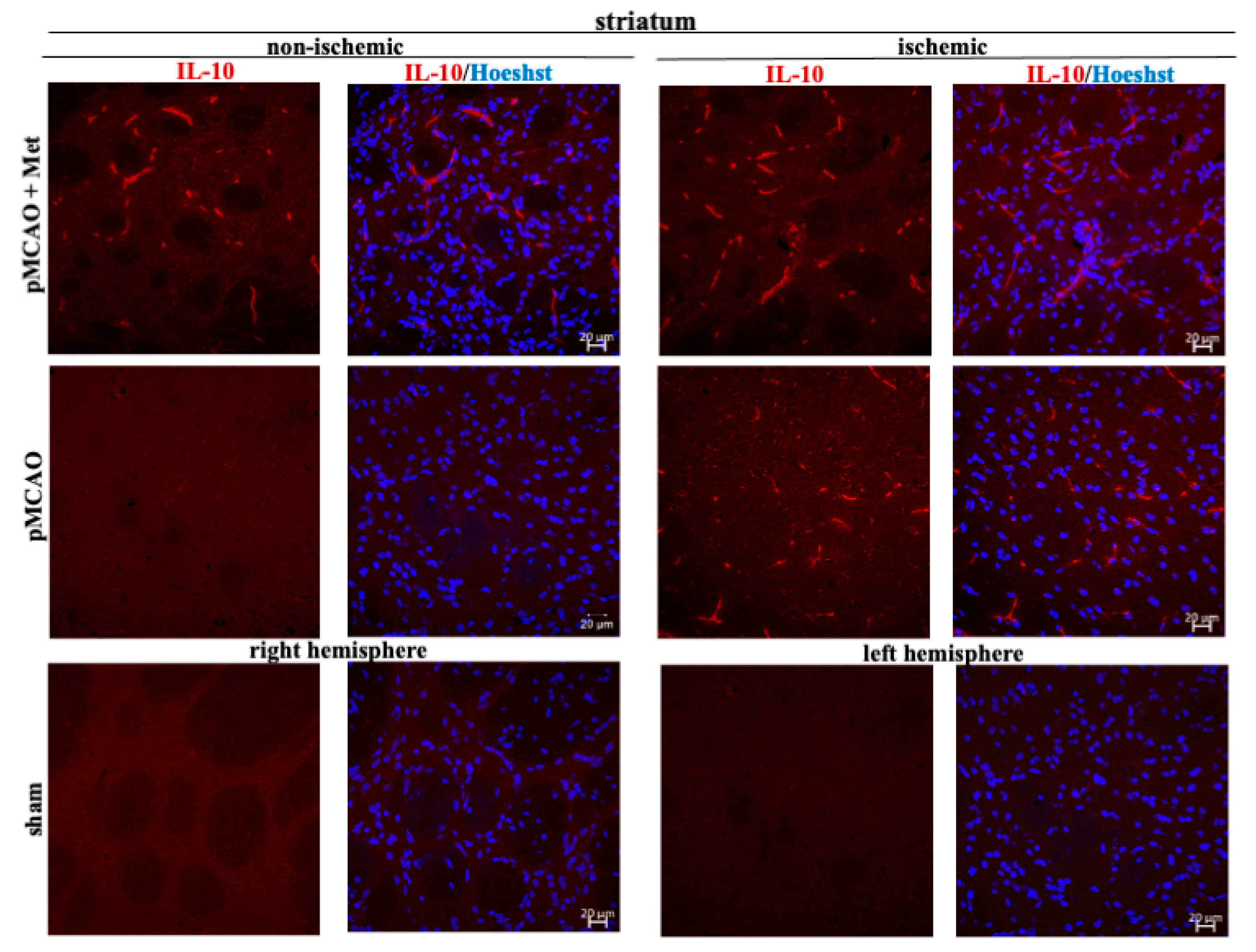
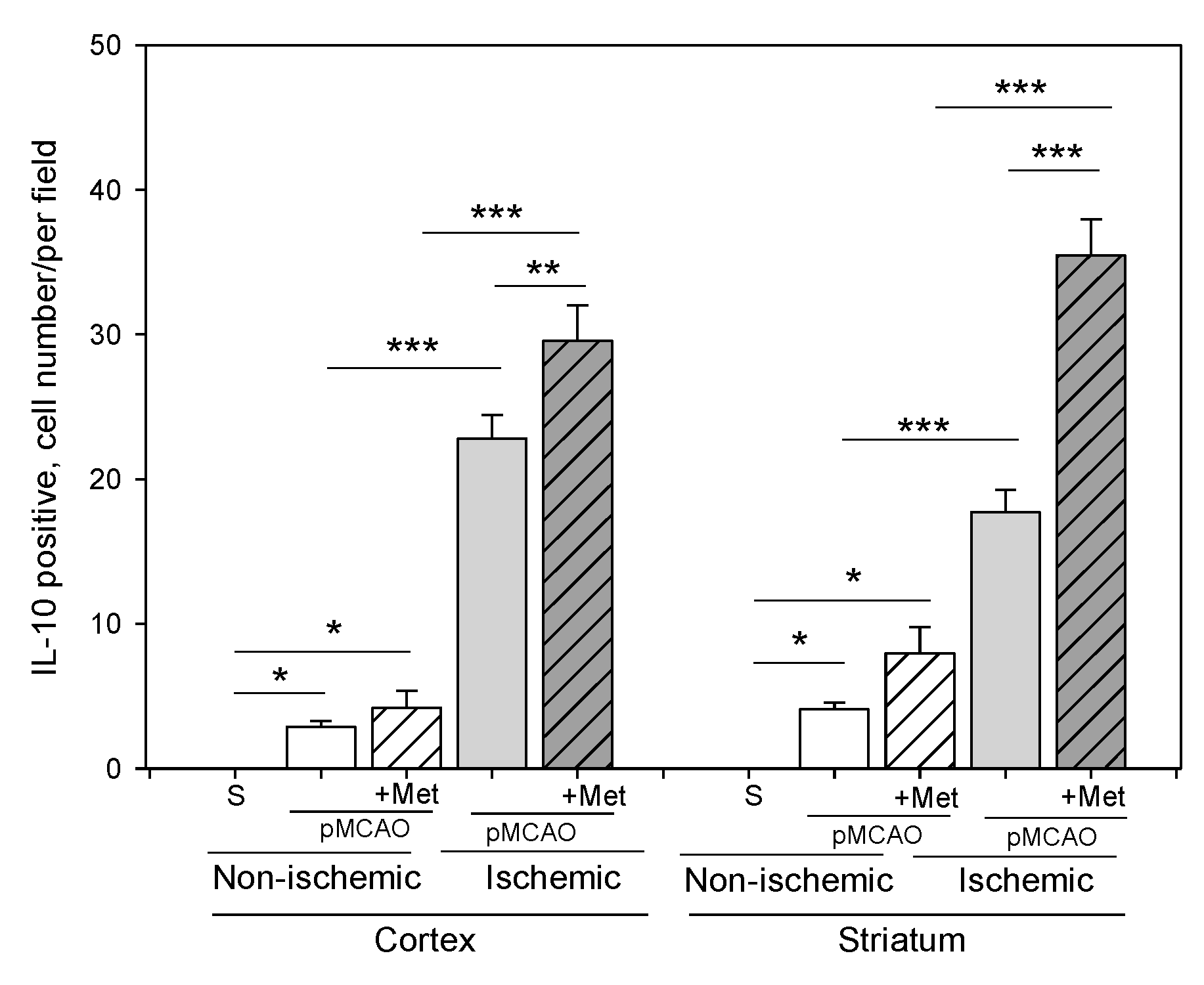
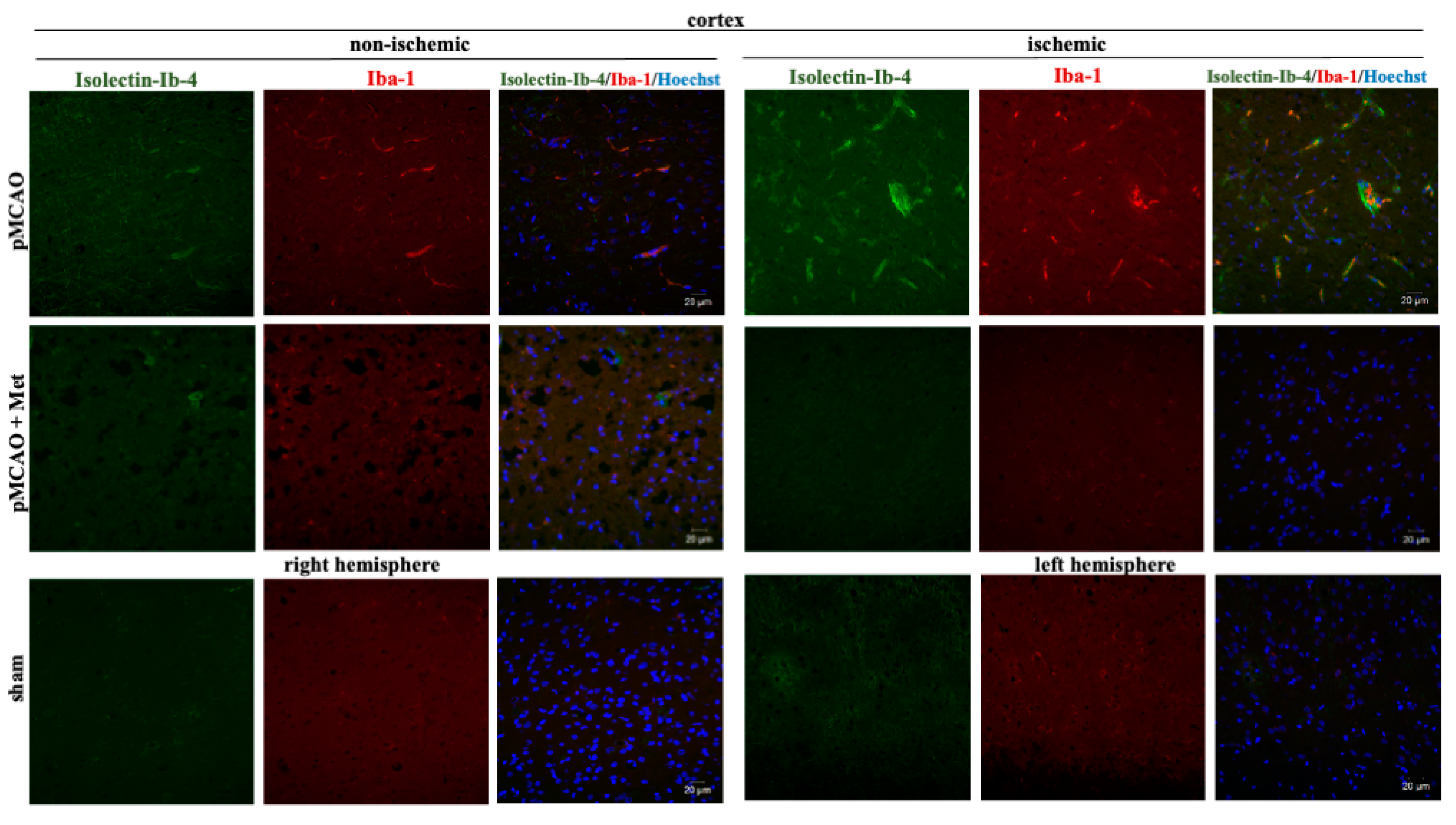
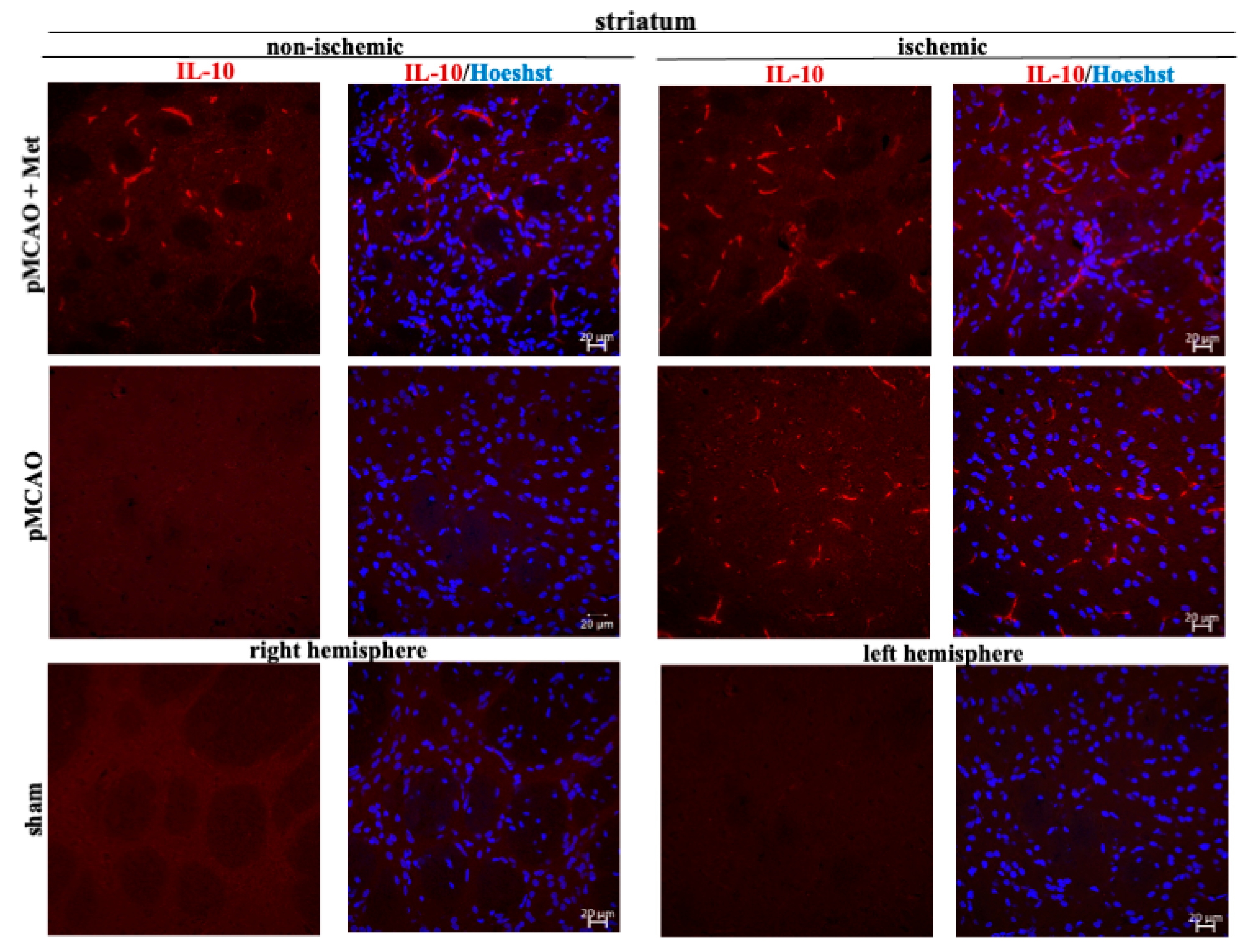
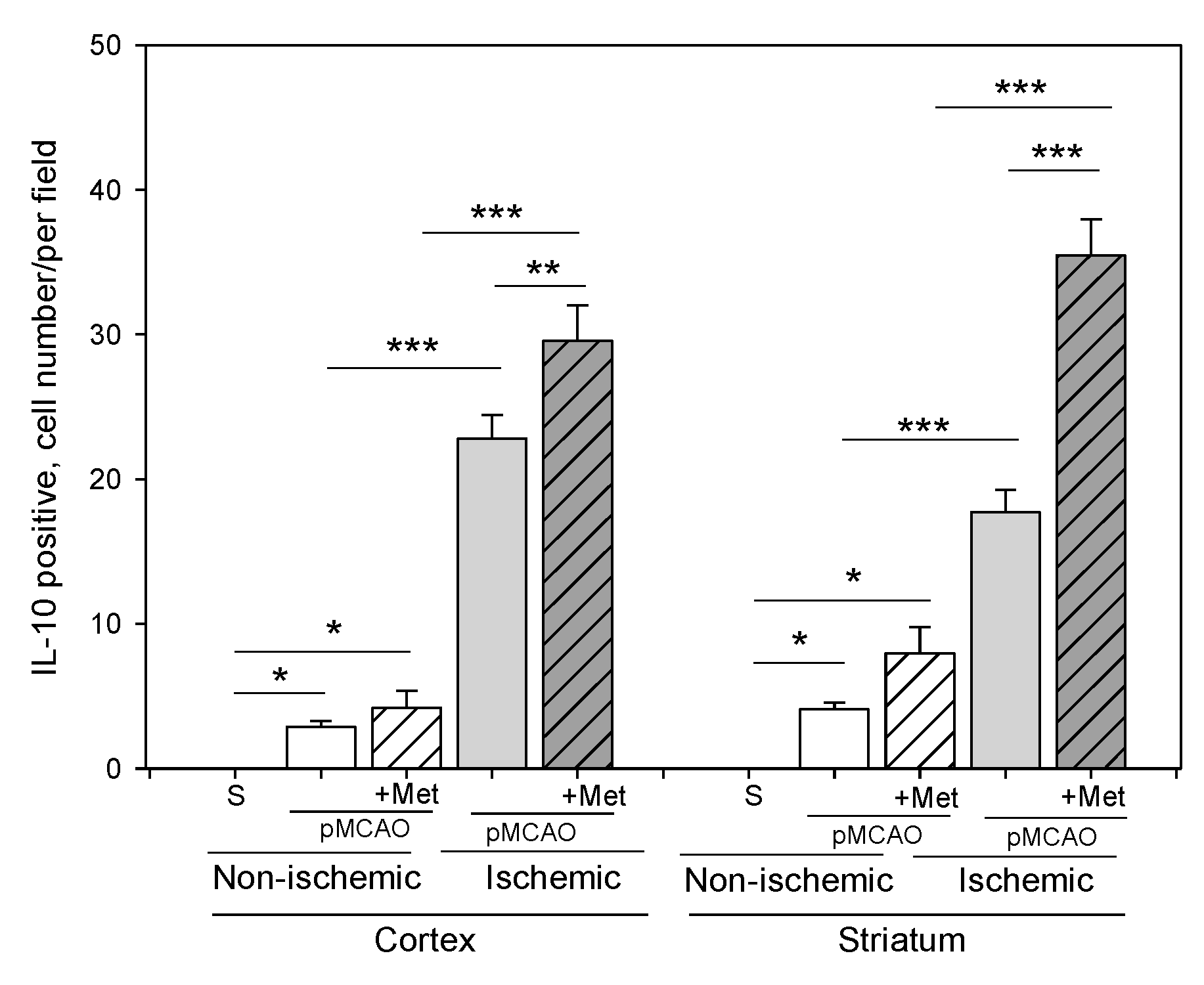
2.3. The Effect of Metformin on Microglial Activation and IL-10 Production after pMCAO
3. Discussion
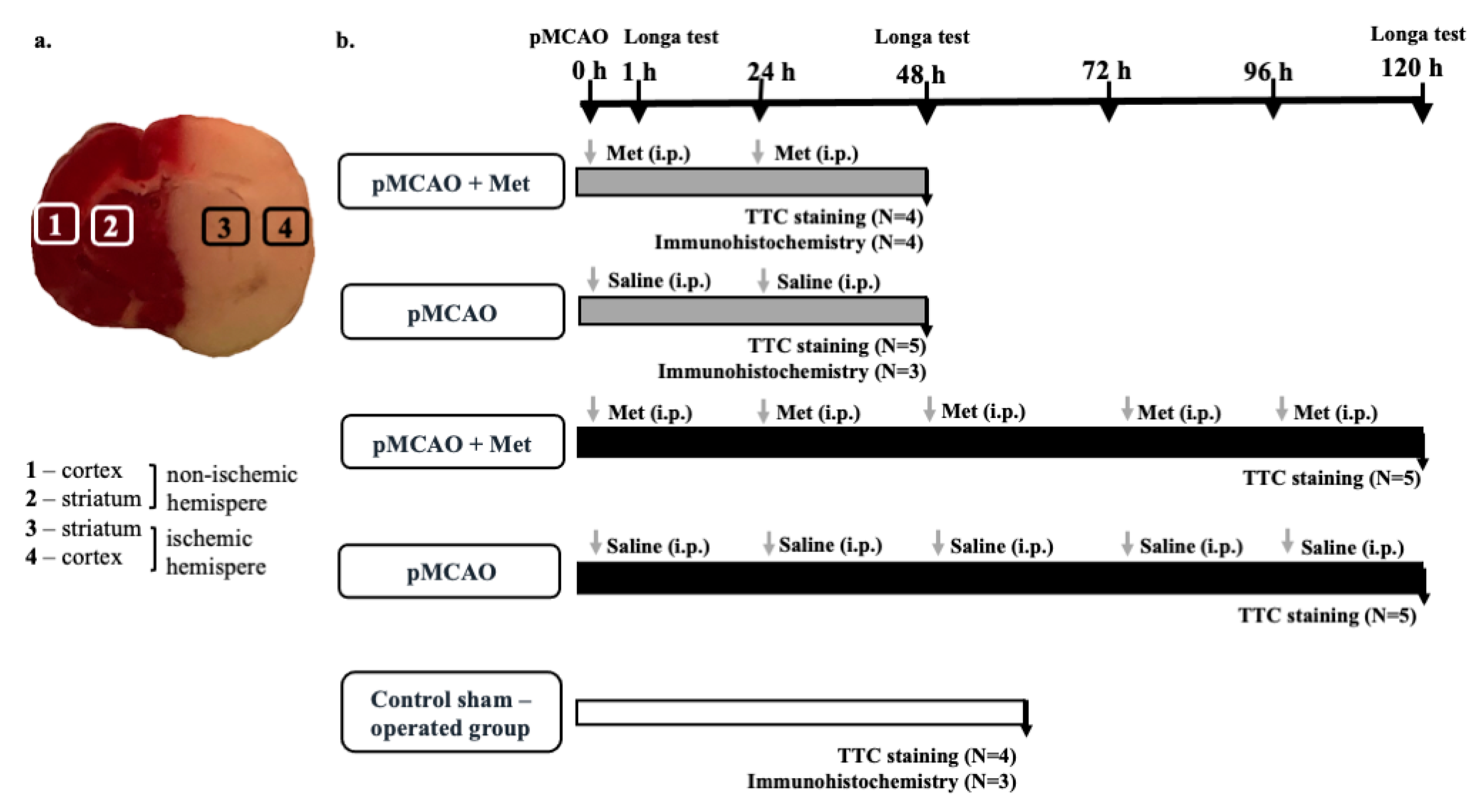
4. Methods and Materials
4.1. Middle Cerebral Artery Occlusion
4.2. Animal Groups
4.3. Measurement of Infarct Size
4.4. Measurement of Brain Oedema Area
4.5. Behavioural Testing
4.6. Immunohistochemistry
4.7. Fluorescence and Cell Quantification
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| CCA | common carotid artery |
| ECA | external carotid artery |
| IBA-1 | ionized calcium-binding adaptor molecule 1 |
| ICA | internal carotid artery |
| IL-10 | interleukin—10 |
| MCAO | middle cerebral artery occlusion |
| Met | metformin, |
| NeuN | neuronal nuclear antigen |
| PBS | phosphate-buffered saline |
| pMCAO | permanent middle cerebral artery occlusion |
| TTC | 2,3,5-triphenyltetrazolium chloride |
References
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.J.; Pulli, B.; Gonzalez, R.G. Imaging-based treatment selection for intravenous and intra-arterial stroke therapies: A comprehensive review. Expert Rev. Cardiovasc. Ther. 2011, 9, 857–876. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, H.; Bluhmki, E.; Bendszus, M.; Eschenfelder, C.C.; Donnan, G.A.; Leys, D.; Molina, C.; Ringleb, P.A.; Schellinger, P.D.; Schwab, S.; et al. European Cooperative Acute Stroke Study-4: Extending the time for thrombolysis in emergency neurological deficits ECASS-4: ExTEND. Int. J. Stroke 2016, 11, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.G.; McMullen, P.W. Neuroprotection in the Treatment of Acute Ischemic Stroke. Prog. Cardiovasc. Dis. 2017, 59, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Saver, J.L. Citicoline—The first effective neuroprotectant to be combined with thrombolysis in acute ischemic stroke? J. Neurol. Sci. 2008, 247, 119–120. [Google Scholar] [CrossRef]
- Muir, K.W.; Lees, K.R. Excitatory amino acid antagonists for acute stroke. Cochrane Database Syst. Rev. 2003, 2003, CD001244. [Google Scholar] [CrossRef]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital Use of Magnesium Sulfate as Neuroprotection in Acute Stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, J.; Li, D.; Zhang, C.; Liu, M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, J.; Yoshida, Y.; Nakazawa, T.; Ooneda, G. Experimental studies of ischemic brain edema. Jpn. J. Stroke 1986, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- McBride, D.W.; Zhang, J.H. Precision Stroke Animal Models: The Permanent MCAO Model Should Be the Primary Model, Not Transient MCAO. Transl. Stroke Res. 2017, 8, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.A.; Li, T.; Kury LTAl Zeb, A.; Khatoon, S.; Liu, G.; Yang, X.; Liu, F.; Yao, H.; Khan, A.U.; Koh, P.O.; et al. Pathological Comparisons of the Hippocampal Changes in the Transient and Permanent Middle Cerebral Artery Occlusion Rat Models. Front. Neurol 2019, 10, 1178. [Google Scholar] [CrossRef]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.H.; Dhawan, J.; Kovoor, J.A.; Sullivan, J.; Zhang, W.X.; Choi, D.; Biegon, A. Aromatase and neuroinflammation in rat focal brain ischemia. J. Steroid Biochem. Mol. Biol. 2017, 174, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, W.N.; Matei, N.; Li, X.; Pang, J.W.; Mo, J.; Chen, S.P.; Tang, J.P.; Yan, M.; Zhang, J.H. Ezetimibe Attenuates Oxidative Stress and Neuroinflammation via the AMPK/Nrf2/TXNIP Pathway after MCAO in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 4717258. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.; Drummond, G.R.; et al. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Zhu, L.; Liu, J.; Zhu, T.; Xie, Z.; Sun, X.; Zhang, H. Metformin Protects against Oxidative Stress Injury Induced by Ischemia/Reperfusion via Regulation of the lncRNA-H19/miR-148a-3p/Rock2 Axis. Oxid. Med. Cell. Longev. 2019, 2019, 8768327. [Google Scholar] [CrossRef] [PubMed]
- Leech, T.; Chattipakorn, N.; Chattipakorn, S.C. The beneficial roles of metformin on the brain with cerebral ischaemia/reperfusion injury. Pharmacol. Res. 2019, 146, 104261. [Google Scholar] [CrossRef]
- Jin, Q.; Cheng, J.; Liu, Y.; Wu, J.; Wang, X.; Wei, S.; Zhou, X.; Qin, Z.; Jia, J.; Zhen, X. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav. Immun. 2014, 40, 131–142. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, K.; Huang, K.; Gu, Y.; Hu, Y.; Pan, S.; Ji, Z. Metformin improves neurologic outcome Via AMP-activated protein kinase-mediated autophagy activation in a rat model of cardiac arrest and resuscitation. J. Am. Heart Assoc. 2018, 7, e008389. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Zhu, X.C.; Jiang, T.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Wang, H.F.; Ding, Z.Z.; Tan, L.; Yu, J.T. Chronic Metformin Preconditioning Provides Neuroprotection via Suppression of NF-κB-Mediated Inflammatory Pathway in Rats with Permanent Cerebral Ischemia. Mol. Neurobiol. 2015, 52, 375–385. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Wang, H.F.; Tan, M.S.; Cao, L.; Zhang, Q.Q.; Gao, L.; Shi, J.Q.; Zhang, Y.D.; et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br. J. Pharmacol. 2014, 171, 3146–3157. [Google Scholar] [CrossRef]
- Deng, T.; Zheng, Y.R.; Hou, W.W.; Yuan, Y.; Shen, Z.; Wu, X.L.; Chen, Y.; Zhang, L.S.; Hu, W.W.; Chen, Z.; et al. Pre-stroke Metformin Treatment is Neuroprotective Involving AMPK Reduction. Neurochem. Res. 2016, 41, 2719–2727. [Google Scholar] [CrossRef]
- Li, J.; McCullough, L.D. Effects of AMP-activated protein kinase in cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Manwani, B.; Mccullough, L.D. Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J. Neurosci. Res. 2013, 91, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Vosler, P.S.; Graham, S.H.; Wechsler, L.R.; Chen, J. Mitochondrial targets for stroke: Focusing basic science research toward development of clinically translatable therapeutics. Stroke 2009, 40, 3149–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mir, M.Y.; Detaille, D.; R-Villanueva, G.; Delgado-Esteban, M.; Guigas, B.; Attia, S.; Fontaine, E.; Almeida, A.; Leverve, X. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 2008, 34, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Skemiene, K.; Rekuviene, E.; Jekabsone, A.; Cizas, P.; Morkuniene, R.; Borutaite, V. Comparison of effects of metformin, phenformin, and inhibitors of mitochondrial complex i on mitochondrial permeability transition and ischemic brain injury. Biomolecules 2020, 10, 1400. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; McGrath, J.C.; Catterall, W.A.; Spedding, M.; Peters, J.A.; Harmar, A.J. The concise guide to PHARMACOLOGY 2013/14: Overview. Br. J. Pharmacol. 2013, 170, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsameea, A.; Moustaf, A.A.; Mohamed, A.M. Modulation of the oxidative stress by metformin in the cerebrum of rats exposed to global cerebral ischemia and ischemia/reperfusion. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2387–2392. [Google Scholar]
- Pan, Y.; Sun, X.; Jiang, L.; Hu, L.; Kong, H.; Han, Y.; Qian, C.; Song, C.; Qian, Y.; Liu, W. Metformin Reduces Morphine Tolerance by Inhibiting Microglial-Mediated Neuroinflammation. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saber, S.; Ghanim, A.M.H.; El-Ahwany, E.; El-Kader, E.M.A. Novel Complementary Antitumour Effects of Celastrol and Metformin by Targeting IκBκB, Apoptosis and NLRP3 Inflammasome Activation in Diethylnitrosamine-Induced Murine Hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343. [Google Scholar] [CrossRef]
- Hu, Y.; Young, A.J.; Ehli, E.A.; Nowotny, D.; Davies, P.S.; Droke, E.A.; Soundy, T.J.; Davies, G.E. Metformin and Berberine Prevent Olanzapine-Induced Weight Gain in Rats. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Tajima, K.; Nakamura, A.; Shirakawa, J.; Togashi, Y.; Orime, K.; Sato, K.; Inoue, H.; Kaji, M.; Sakamoto, E.; Ito, Y.; et al. Metformin Prevents Liver Tumorigenesis Induced by High-Fat Diet in C57Bl/6 Mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin Improves Obesity-Associated Inflammation by Altering Macrophages Polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin Inhibits the Production of Reactive Oxygen Species from NADH: Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-Activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin Increases Antitumor Activity of MEK Inhibitors through GLI1 Downregulation in LKB1 Positive Human NSCLC Cancer Cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fifield, K.E.; Vanderluit, J.L. Rapid degeneration of neurons in the penumbra region following a small, focal ischemic stroke. Eur. J. Neurosci. 2020, 52, 3196–3214. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Mima, Y.; Kuwashiro, T.; Yasaka, M.; Tsurusaki, Y.; Nakamura, A.; Wakugawa, Y.; Okada, Y. Impact of Metformin on the Severity and Outcomes of Acute Ischemic Stroke in Patients with Type 2 Diabetes Mellitus. J. Stroke Cerebrovasc. Dis. 2016, 25, 436–446. [Google Scholar] [CrossRef]
- Westphal, L.P.; Widmer, R.; Held, U.; Steigmiller, K.; Hametner, C.; Ringleb, P.; Curtze, S.; Martinez-Majander, N.; Tiainen, M.; Nolte, C.H.; et al. Association of Prestroke Metformin Use, Stroke Severity, and Thrombolysis Outcome. Neurology 2020, 95, E362–E373. [Google Scholar] [CrossRef]
- Sardu, C.; Paolisso, P.; Sacra, C.; Mauro, C.; Minicucci, F.; Portoghese, M.; Rizzo, M.R.; Barbieri, M.; Sasso, F.C.; D’Onofrio, N.; et al. Effects of Metformin Therapy on Coronary Endothelial Dysfunction in Patients with Prediabetes with Stable Angina and Nonobstructive Coronary Artery Stenosis: The Codyce Multicenter Prospective Study. Diabetes Care 2019, 42, 1946–1955. [Google Scholar] [CrossRef] [Green Version]
- Boyko, M.; Ohayon, S.; Goldsmith, T.; Novack, L.; Novack, V.; Perry, Z.H.; Gruenbaum, B.F.; Gruenbaum, S.E.; Steiner, O.; Shapira, Y.; et al. Morphological and neuro-behavioral parallels in the rat model of stroke. Behav. Brain Res. 2011, 223, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kuts, R.; Frank, D.; Gruenbaum, B.F.; Grinshpun, J.; Melamed, I.; Knyazer, B.; Tarabrin, O.; Zvenigorodsky, V.; Shelef, I.; Zlotnik, A.; et al. A Novel Method for Assessing Cerebral Edema, Infarcted Zone and Blood-Brain Barrier Breakdown in a Single Post-stroke Rodent Brain. Front. Neurosci. 2019, 13, 1105. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, J.P.; Watson, S.J. The Rat Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: Cambridge, MA, USA, 1987. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zemgulyte, G.; Tanaka, S.; Hide, I.; Sakai, N.; Pampuscenko, K.; Borutaite, V.; Rastenyte, D. Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats. Pharmaceuticals 2021, 14, 312. https://doi.org/10.3390/ph14040312
Zemgulyte G, Tanaka S, Hide I, Sakai N, Pampuscenko K, Borutaite V, Rastenyte D. Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats. Pharmaceuticals. 2021; 14(4):312. https://doi.org/10.3390/ph14040312
Chicago/Turabian StyleZemgulyte, Gintare, Shigeru Tanaka, Izumi Hide, Norio Sakai, Katryna Pampuscenko, Vilmante Borutaite, and Daiva Rastenyte. 2021. "Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats" Pharmaceuticals 14, no. 4: 312. https://doi.org/10.3390/ph14040312
APA StyleZemgulyte, G., Tanaka, S., Hide, I., Sakai, N., Pampuscenko, K., Borutaite, V., & Rastenyte, D. (2021). Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats. Pharmaceuticals, 14(4), 312. https://doi.org/10.3390/ph14040312