
Citicoline: A Candidate for Adjunct Treatment of Multiple Sclerosis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
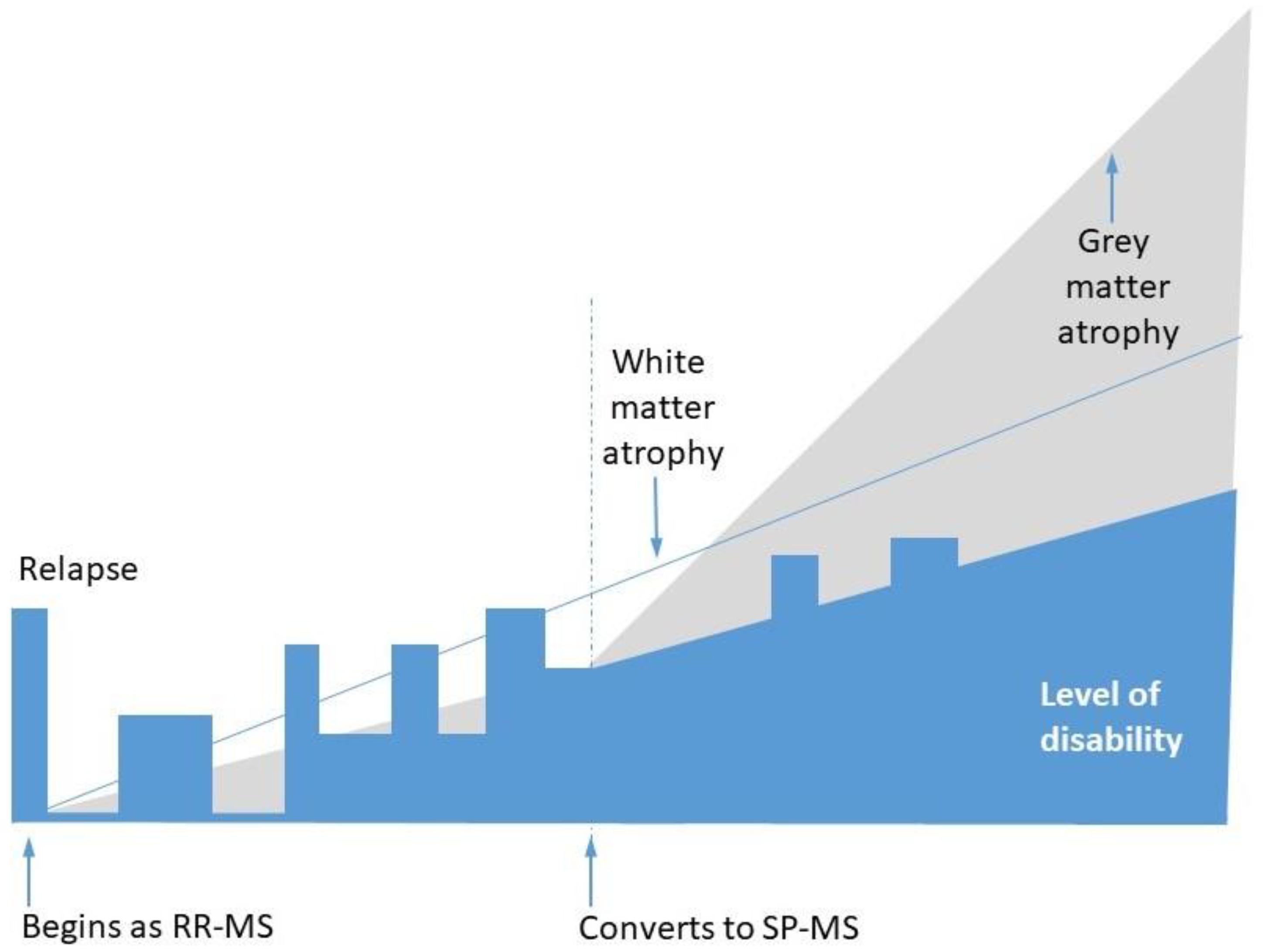
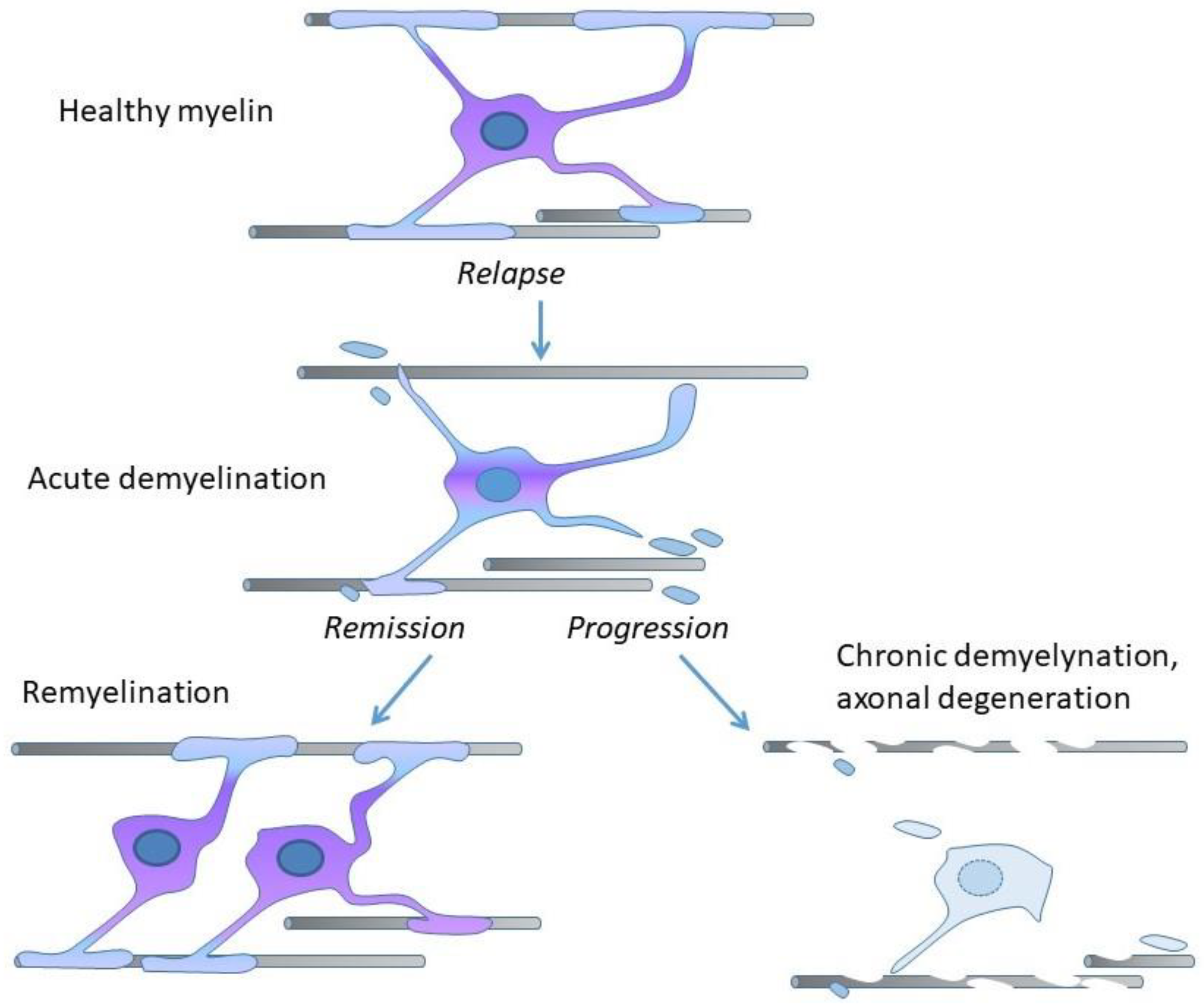
2. Demyelination, Remyelination, and Neurodegeneration in MS
3. Preclinical Models of MS
4. Disease-Modifying Therapies for MS
5. Visual Evoked Potentials for Monitoring Remyelination
6. Pharmacodynamics of Citicoline
7. Assessment of Citicoline in Preclinical Models of MS
8. Citicoline Should Be Evaluated in MS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallin, M.T.; Culpepper, W.J.; Nichols, E.; Bhutta, Z.A.; Gebrehiwot, T.T.; Hay, S.I.; Khalil, I.A.; Krohn, K.J.; Liang, X.; Naghavi, M.; et al. Global, regional and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Scalfari, A.; Knappertz, V.; Cutter, G.; Goodin, D.S.; Ashton, R.; Ebers, G.C. Mortality in patients with multiple sclerosis. Neurology 2013, 81, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Larochelle, C.; Uphaus, T.; Prat, A.; Zipp, F. Secondary Progression in Multiple Sclerosis: Neuronal Exhaustion or Distinct Pathology? Trends Neurosci. 2016, 39, 325–339. [Google Scholar] [CrossRef]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Göttle, P.; Förster, M.; Weyers, V.; Küry, P.; Rejdak, K.; Hartung, H.-P.; Kremer, D. An unmet clinical need: Roads to remyelination in MS. Neurol. Res. Pract. 2019, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Morrell, P.; Quarles, R.H. Myelin Formation, Structure and Biochemistry. In Basic Neurochemistry, 6th ed.; Siegel, G.J., Agranoff, B.W., Eds.; Raven Press: New York, NY, USA, 1999; pp. 70–93. [Google Scholar]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, S.; Tanaka, Y.; Toyoda, Y.; Kon, K.; Ono, Y.T. Turnover of Myelin Lipids in Aging Brain. Neurochem. Res. 2003, 28, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.C.; Zhao, C.; Franklin, R.J. Remyelination—An effective means of neuroprotection. Horm. Behav. 2010, 57, 56–62. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Luchicchi, A.; Hart, B.T.; Frigerio, I.; Van Dam, A.; Perna, L.; Offerhaus, H.L.; Stys, P.K.; Schenk, G.J.; Geurts, J.J.G. Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Ann. Neurol. 2021, 89, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14, 599717. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.-P.; Zettl, U.K. Animal models of multiple sclerosis—Potentials and limitations. Prog. Neurobiol. 2010, 92, 386–404. [Google Scholar] [CrossRef]
- Storch, M.K.; Stefferl, A.; Brehm, U.; Weissert, R.; Wallström, E.; Kerschensteiner, M.; Olsson, T.; Linington, C.; Lassmann, H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998, 8, 681–694. [Google Scholar] [CrossRef]
- Schneider, C.; Schuetz, G.; Zollner, T.M. Acute neuroinflammation in Lewis rats—A model for acute multiple sclerosis relapses. J. Neuroimmunol. 2009, 213, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Amor, S. Mouse models of multiple sclerosis: Lost in translation? Curr. Pharm. Des. 2015, 21, 2440–2452. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced de- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, T.; Prat, A. A roadmap to precision medicine for multiple sclerosis. Mult. Scler. J. 2020, 26, 522–532. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Carter, J.L. Multiple Sclerosis: Current and Emerging Disease-Modifying Therapies and Treatment Strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Winkelmann, A.; Loebermann, M.; Reisinger, M.L.E.C.; Hartung, H.-P.; Zettl, A.W.U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 217–233. [Google Scholar] [CrossRef]
- Jakimovski, D.; Vaughn, C.B.; Eckert, S.; Zivadinov, R.; Weinstock-Guttman, B. Long-term drug treatment in multiple sclerosis: Safety success and concerns. Expert Opin. Drug Saf. 2020, 19, 1121–1142. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, N.; Coles, A. Promoting remyelination in multiple sclerosis. J. Neurol. 2021, 268, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lapiscina, E.H.; Sanchez-Dalmau, B.F.; Fraga-Pumar, E.; Ortiz-Perez, S.I.; Tercero-Uribe, A.; Torres-Torres, R.; Villoslada, P. The visual pathway as a model to understand brain damage in multiple sclerosis. Mult. Scler. J. 2014, 20, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, J.L.; Garber, J.Y.; Klistorner, A.; Barnett, M.H. The electrophysiological assessment of visual function in Multiple Sclerosis. Clin. Neurophysiol. Pract. 2019, 4, 90–96. [Google Scholar] [CrossRef]
- You, Y.; Klistorner, A.; Thie, J.; Graham, S.L. Latency Delay of Visual Evoked Potential Is a Real Measurement of Demyelination in a Rat Model of Optic Neuritis. Investig. Opthalmol. Vis. Sci. 2011, 52, 6911–6918. [Google Scholar] [CrossRef] [Green Version]
- Farley, B.J.; Morozova, E.; Dion, J.; Wang, B.; Harvey, B.D.; Gianni, D.; Wipke, B.; Cadavid, D.; Wittmann, M.; Hajos, M. Evoked potentials as a translatable biomarker to track functional remyelination. Mol. Cell. Neurosci. 2019, 99, 103393. [Google Scholar] [CrossRef]
- Leocani, L.; Guerrieri, S.; Comi, G. Visual Evoked Potentials as a Biomarker in Multiple Sclerosis and Associated Optic Neuritis. J. Neuroophthalmol. 2018, 38, 350–357. [Google Scholar] [CrossRef]
- Canham, L.; Kane, N.; Cottrell, D. The potential of visual physiology: An instrument with a place in MS translation. Clin. Neurophysiol. Pract. 2019, 4, 112–113. [Google Scholar] [CrossRef]
- Anderson, D.C.; Slater, G.E.; Sherman, R.; Ettinger, M.G. Evoked Potentials to Test a Treatment of Chronic Multiple Sclerosis. Arch. Neurol. 1987, 44, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Sühs, K.-W.; Hein, K.; Sättler, M.B.; Görlitz, A.; Ciupka, C.; Scholz, K.; Käsmann-Kellner, B.; Papanagiotou, P.; Schäffler, N.; Restemeyer, C.; et al. A randomized, double-blind, phase 2 study of erythropoietin in optic neuritis. Ann. Neurol. 2012, 72, 199–210. [Google Scholar] [CrossRef]
- Tsakiri, A.; Kallenbach, K.; Fuglø, D.; Wanscher, B.; Larsson, H.; Frederiksen, J. Simvastatin improves final visual outcome in acute optic neuritis: A randomized study. Mult. Scler. 2012, 18, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, R.; Hickman, S.J.; Toosy, A.; Sharrack, B.; Mallik, S.; Paling, D.; Altmann, D.R.; Yiannakas, M.C.; Malladi, P.; Sheridan, R.; et al. Phenytoin for neuroprotection in patients with acute optic neuritis: A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.A.; Calabresi, P.; Drulović, J.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- Albert, C.; Mikolajczak, J.; Liekfeld, A.; Piper, S.K.; Scheel, M.; Zimmermann, H.G.; Nowak, C.; Dörr, J.; Bellmann-Strobl, J.; Chien, C.; et al. Fingolimod after a first unilateral episode of acute optic neuritis (MOVING)—Preliminary results from a randomized, rater-blind, active-controlled, phase 2 trial. BMC Neurol. 2020, 20, 75. [Google Scholar] [CrossRef] [Green Version]
- Teather, L.A. Dietary CDP-choline supplementation prevents memory impairment caused by impoverished environmental conditions in rats. Learn. Mem. 2005, 12, 39–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareri, P.; Cotroneo, A.M.; Castagna, A.; Putignano, S.; Lacava, R.; Monteleone, F.; Rocca, F.; Malara, A.; Fanto, F. Effectiveness and safety of citicoline in mild vascular cognitive impairment: The IDEALE study. Clin. Interv. Aging 2013, 8, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Giurgea, C. Vers une pharmacologie de l’active integrative du cerveau: Tentative du concept nootrope en psychopharmacol-ogie. Actual. Pharmacol. 1972, 25, 115–156. [Google Scholar]
- Petkov, V.D.; Stancheva, S.L.; Tocuschieva, L.; Petkov, V.V. Changes in brain biogenic monoamines induced by the nootropic drugs adafenoxate and meclofenoxate and by citicholine (experiments on rats). Gen. Pharmacol. Vasc. Syst. 1990, 21, 71–75. [Google Scholar] [CrossRef]
- Grieb, P. Neuroprotective Properties of Citicoline: Facts, Doubts and Unresolved Issues. CNS Drugs 2014, 28, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Grieb, P.; Juenemann, A.; Rekas, M.; Rejdak, R. Citicoline: A Food Beneficial for Patients Suffering from or Threated with Glaucoma. Front. Aging Neurosci. 2016, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faiq, M.A.; Wollstein, G.; Schuman, J.S.; Chan, K.C. Cholinergic nervous system and glaucoma: From basic science to clinical applications. Prog. Retin. Eye Res. 2019, 72, 100767. [Google Scholar] [CrossRef]
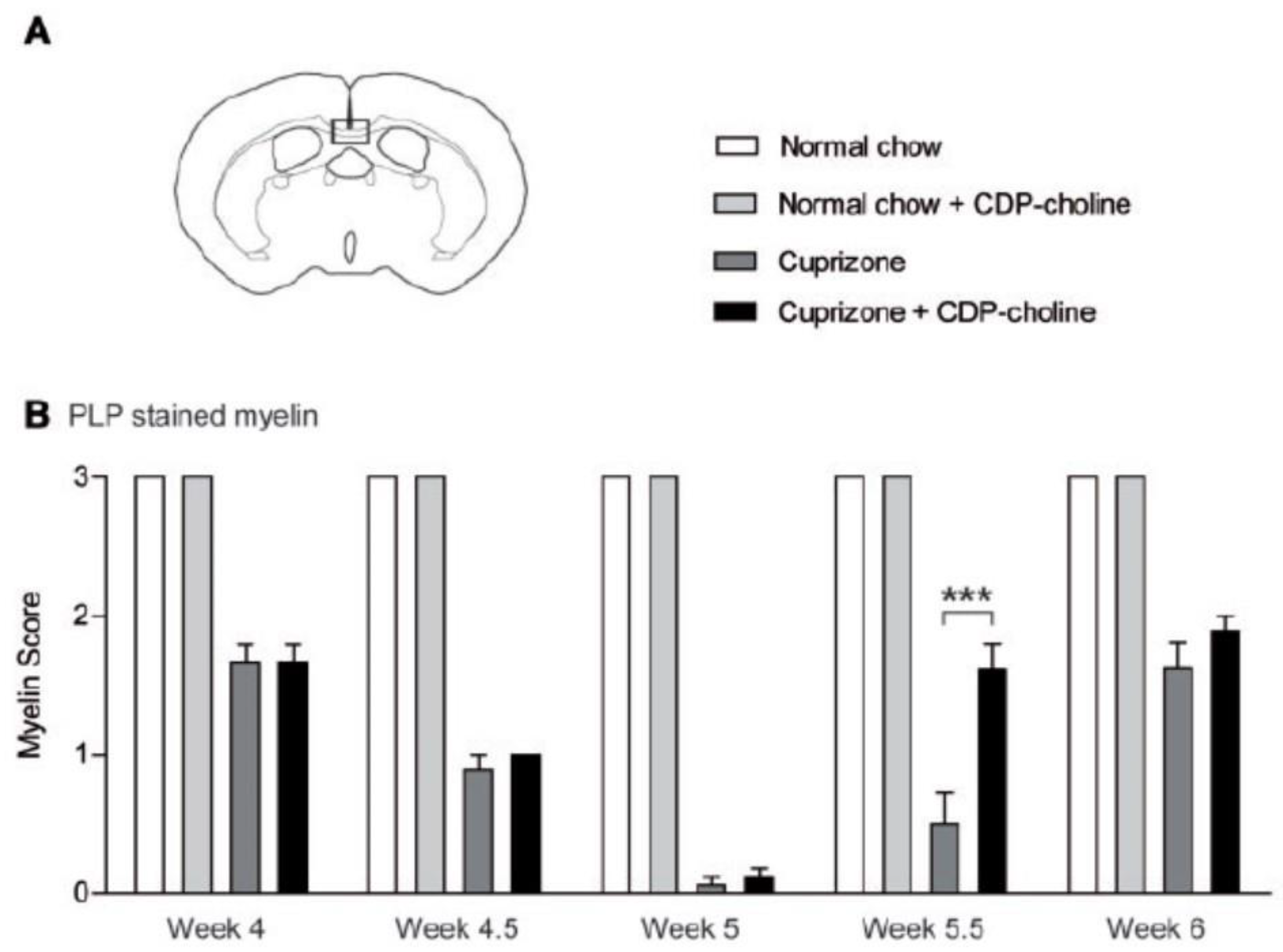
- Skripuletz, T.; Manzel, A.; Gropengießer, K.; Schäfer, N.; Gudi, V.; Singh, V.; Tejedor, L.S.; Jörg, S.; Hammer, A.; Voss, E.; et al. Pivotal role of choline metabolites in remyelination. Brain 2014, 138, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M. The best basic science paper in multiple sclerosis in 2014: Citicoline, remyelination and neuroprotection: Commentary. Mult. Scler. J. 2015, 21, 374–375. [Google Scholar] [CrossRef]
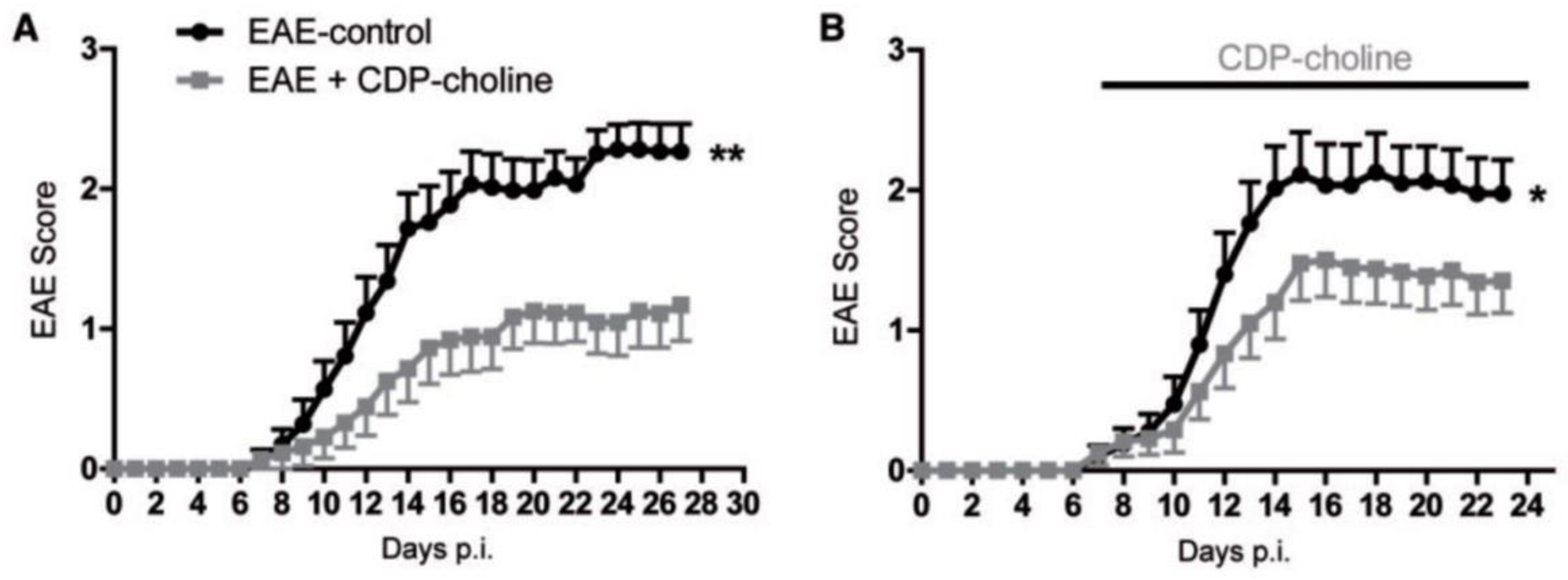
- Clayton, N.; Locke, K.W.; Wu, T.H.; Maher, T.J. Beneficial effects of citicoline in experimental allergic encephalomyelitis (EAE) in rats and mice. FASEB J. 1999, 13, A1104. [Google Scholar]
- Grieb, P.; Kwiatkowska-Patzer, B.; Gadamski, R. Citicoline attenuates brain and spinal cord inflammation in rat EAE. Neurology 2004, 62 (Suppl. 5), A113–A114. [Google Scholar]
- Kremer, D.; Göttle, P.; Hartung, H.-P.; Küry, P. Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci. 2016, 39, 246–263. [Google Scholar] [CrossRef]
- Mullard, A. Remyelination researchers regroup after proof-of-concept setback in multiple sclerosis. Nat. Rev. Drug Discov. 2016, 15, 519–521. [Google Scholar] [CrossRef]
- Senger, C.; Moreto, R.; Watanabe, S.E.; Matos, A.G.; Paula, J.S. Electrophysiology in Glaucoma. J. Glaucoma 2020, 29, 147–153. [Google Scholar] [CrossRef]
- Parisi, V.; Manni, G.; Colacino, G.; Bucci, M.G. Cytidine-5′-diphosphocholine (citicoline) improves retinal and cortical responses in patients with glaucoma. Ophthalmology 1999, 106, 1126–1134. [Google Scholar] [CrossRef]
- Rejdak, R.; Toczołowski, J.; Kurkowski, J.; Kamiński, M.L.; Rejdak, K.; Stelmasiak, Z.; Grieb, P. Oral citicoline treatment improves visual pathway function in glaucoma. Med. Sci. Monit. 2003, 9, PI24–PI28. [Google Scholar] [PubMed]
- Grieb, P. Citicoline and Eye Health. In Handbook of Nutrition, Diet, and the Eye, 2nd ed.; Preedy, V.R., Watson, R.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 585–603. [Google Scholar] [CrossRef]
- Lanza, M.; Carnevale, U.A.G.; Mele, L.; Sconocchia, M.B.; Bartollino, S.; Costagliola, C. Morphological and Functional Evaluation of Oral Citicoline Therapy in Chronic Open-Angle Glaucoma Patients: A Pilot Study with a 2-Year Follow-Up. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lapiscina, E.H.; Arnow, S.A.; Wilson, J.; Saidha, S.; Preiningerova, J.L.; Oberwahrenbrock, T.; Brandt, A.U.E.; Pablo, L.; Guerrieri, S.; Gonzalez, I.; et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: A cohort study. Lancet Neurol. 2016, 15, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Schurz, N.; Sariaslani, L.; Altmann, P.; Leutmezer, F.; Mitsch, C.; Pemp, B.; Rommer, P.; Zrzavy, T.; Berger, T.; Bsteh, G. Evaluation of Retinal Layer Thickness Parameters as Biomarkers in a Real-World Multiple Sclerosis Cohort. Eye Brain 2021, 13, 59–69. [Google Scholar] [CrossRef]
- Synoradzki, K.; Grieb, P. Citicoline: A Superior Form of Choline? Nutrients 2019, 11, 1569. [Google Scholar] [CrossRef] [Green Version]
- Piamonte, B.L.C.; Espiritu, A.I.; Anlacan, V.M.M. Effects of Citicoline as an Adjunct Treatment for Alzheimer’s Disease: A Systematic Review. J. Alzheimer’s Dis. 2020, 76, 725–732. [Google Scholar] [CrossRef]
- Zweifler, R.M. Membrane stabilizer: Citicoline. Curr. Med. Res. Opin. 2002, 18, s14–s17. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Lee, S.H.; Sohn, M.; Ryu, O.H.; Choi, M.-G.; Suh, S.W. Acetylcholine precursor, citicoline (cytidine 5′-diphosphocholine), reduces hypoglycaemia-induced neuronal death in rats. J. Neuroendocr. 2018, 30, e12567. [Google Scholar] [CrossRef]
- Feng, L.; Jiang, H.; Li, Y.; Teng, F.; He, Y. Effects of citicoline therapy on the network connectivity of the corpus callosum in patients with leukoaraiosis. Medicine 2017, 96, e5931. [Google Scholar] [CrossRef]
- Hachinski, V.; Potter, P.; Merskey, H. Leuko-Araiosis: An Ancient Term for a New Problem. Can. J. Neurol. Sci. 1986, 13, 533–534. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Petrovic, K.; Ropele, S.; Enzinger, C.; Fazekas, F. Progression of Leukoaraiosis and Cognition. Stroke 2007, 38, 2619–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.-K.; Yoshino, J.; Le, T.Q.; Lin, S.-J.; Sun, S.-W.; Cross, A.H.; Armstrong, R.C. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 2005, 26, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Timmler, S.; Simons, M. Grey matter myelination. Glia 2019, 67, 2063–2070. [Google Scholar] [CrossRef]
- Del Boccio, P.; Pieragostino, D.; Di Ioia, M.; Petrucci, F.; Lugaresi, A.; De Luca, G.; Gambi, D.; Onofrj, M.; Di Ilio, C.; Sacchetta, P.; et al. Lipidomic investigations for the characterization of circulating serum lipids in multiple sclerosis. J. Proteom. 2011, 74, 2826–2836. [Google Scholar] [CrossRef]
- Ferreira, H.B.; Melo, T.; Monteiro, A.; Paiva, A.; Domingues, P.; Domingues, M.R. Serum phospholipidomics reveals altered lipid profile and promising biomarkers in multiple sclerosis. Arch. Biochem. Biophys. 2021, 697, 108672. [Google Scholar] [CrossRef] [PubMed]
- Penkert, H.; Lauber, C.; Gerl, M.J.; Klose, C.; Damm, M.; Fitzner, D.; Flierl-Hecht, A.; Kümpfel, T.; Kerschensteiner, M.; Hohlfeld, R.; et al. Plasma lipidomics of monozygotic twins discordant for multiple sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 2461–2466. [Google Scholar] [CrossRef]
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An emerging potential of metabolomics in multiple sclerosis: A comprehensive overview. Cell. Mol. Life Sci. 2021, 1–23. [Google Scholar] [CrossRef]
- Swanberg, K.M.; Landheer, K.; Pitt, D.; Juchem, C. Quantifying the Metabolic Signature of Multiple Sclerosis by in vivo Proton Magnetic Resonance Spectroscopy: Current Challenges and Future Outlook in the Translation from Proton Signal to Diagnostic Biomarker. Front. Neurol. 2019, 10, 1173. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grieb, P.; Świątkiewicz, M.; Kamińska, A.; Jünemann, A.; Rejdak, R.; Rejdak, K. Citicoline: A Candidate for Adjunct Treatment of Multiple Sclerosis. Pharmaceuticals 2021, 14, 326. https://doi.org/10.3390/ph14040326
Grieb P, Świątkiewicz M, Kamińska A, Jünemann A, Rejdak R, Rejdak K. Citicoline: A Candidate for Adjunct Treatment of Multiple Sclerosis. Pharmaceuticals. 2021; 14(4):326. https://doi.org/10.3390/ph14040326
Chicago/Turabian StyleGrieb, Paweł, Maciej Świątkiewicz, Agnieszka Kamińska, Anselm Jünemann, Robert Rejdak, and Konrad Rejdak. 2021. "Citicoline: A Candidate for Adjunct Treatment of Multiple Sclerosis" Pharmaceuticals 14, no. 4: 326. https://doi.org/10.3390/ph14040326
APA StyleGrieb, P., Świątkiewicz, M., Kamińska, A., Jünemann, A., Rejdak, R., & Rejdak, K. (2021). Citicoline: A Candidate for Adjunct Treatment of Multiple Sclerosis. Pharmaceuticals, 14(4), 326. https://doi.org/10.3390/ph14040326