
Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods

 , ,
, ,  , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
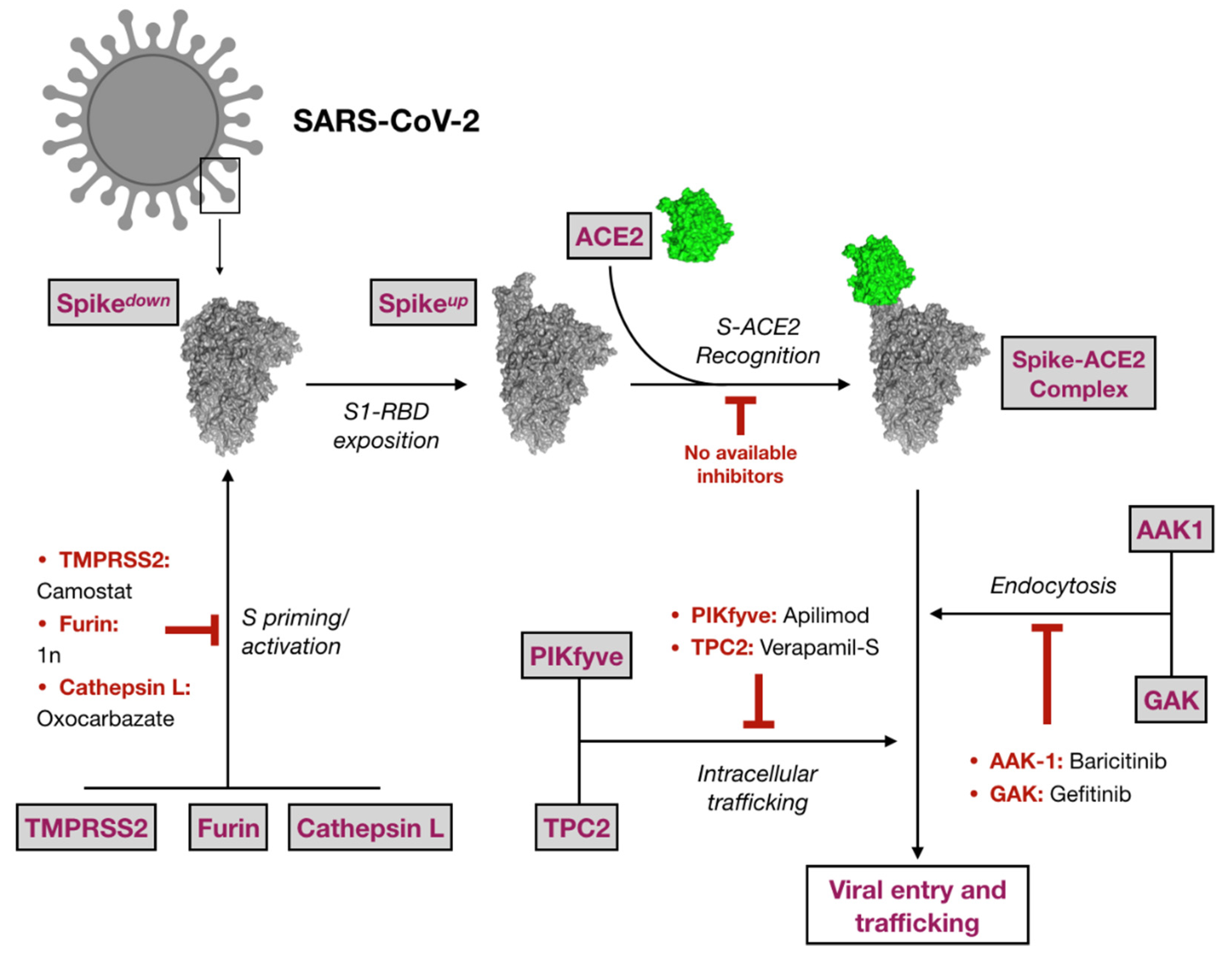
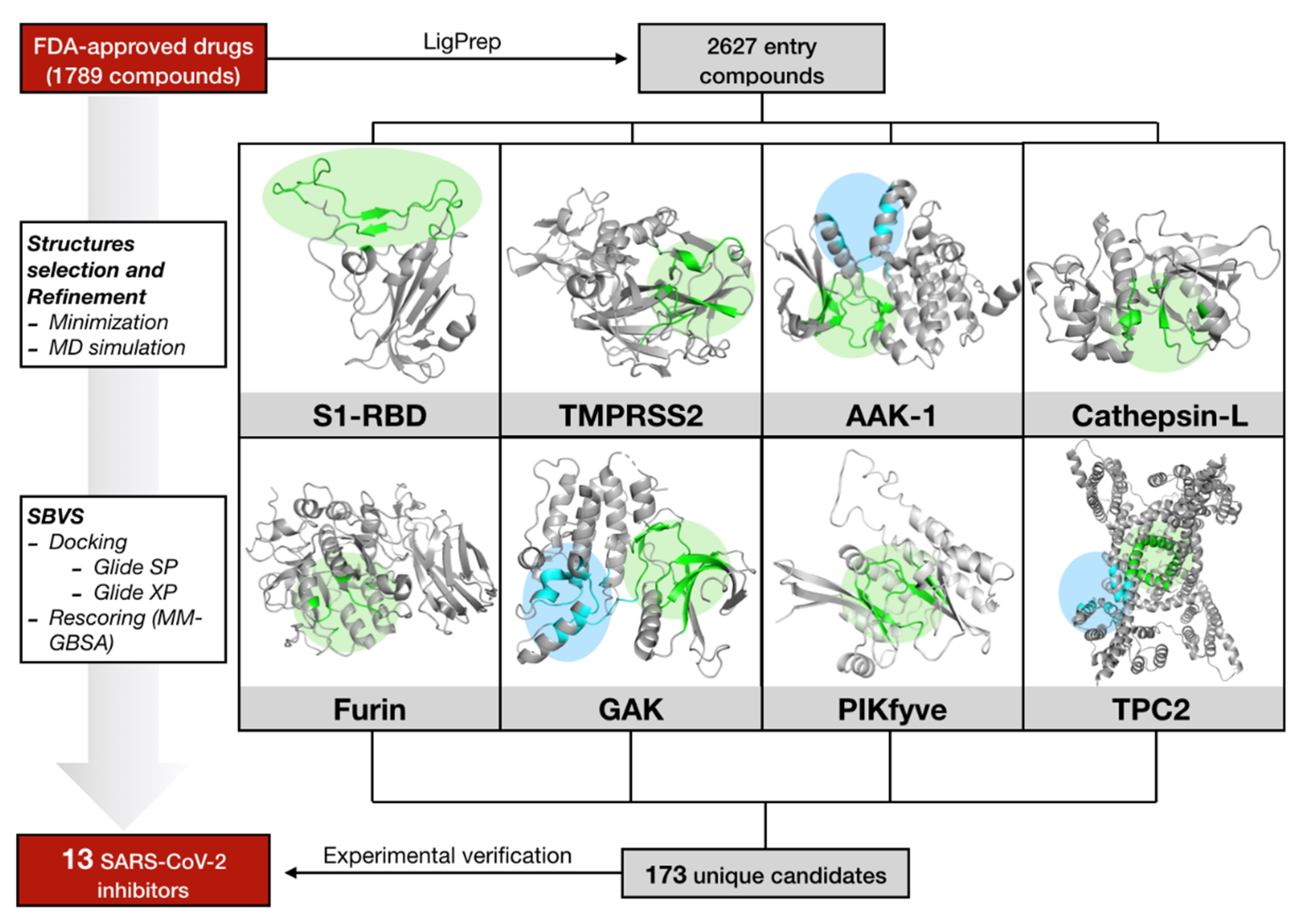
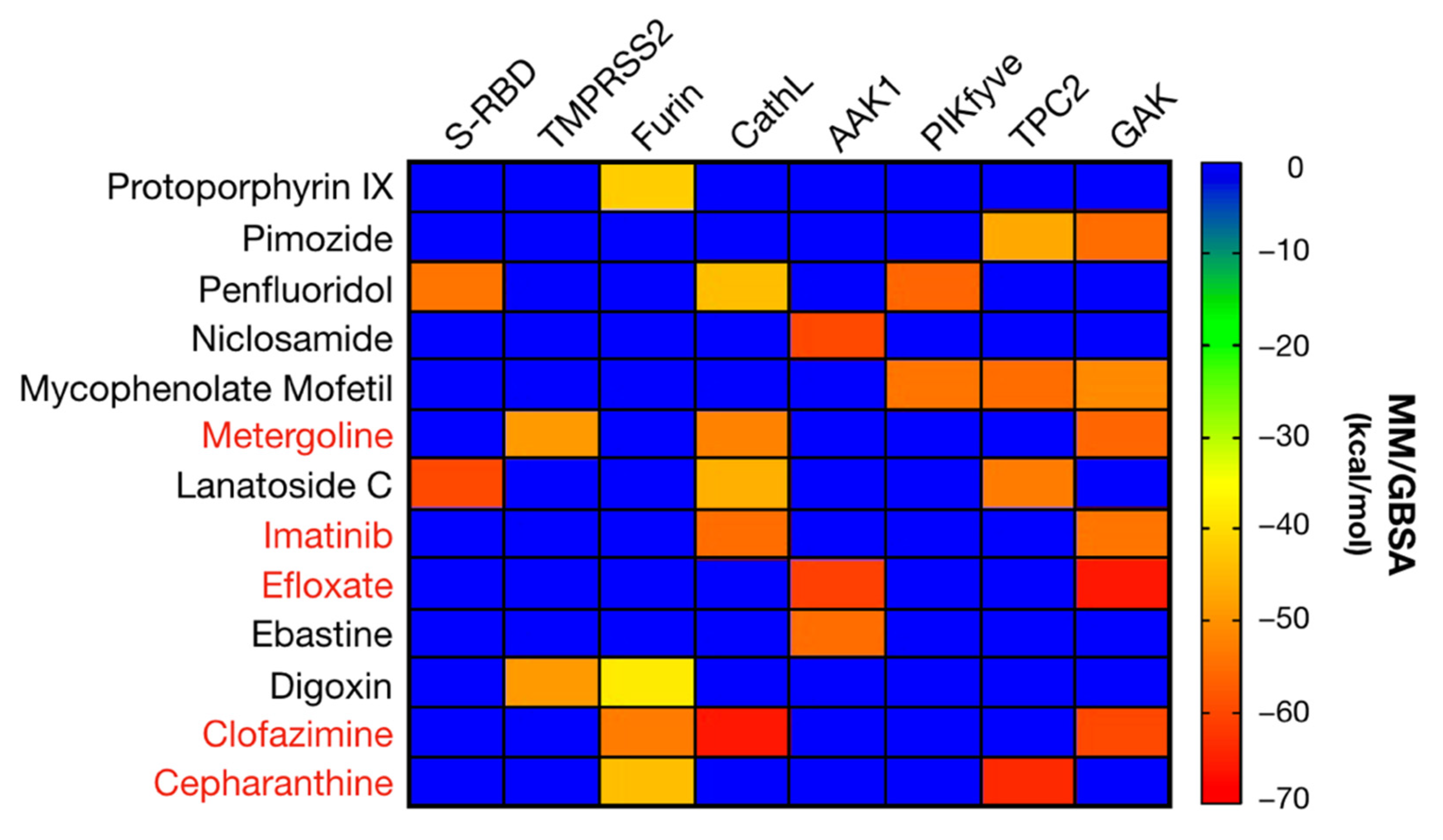
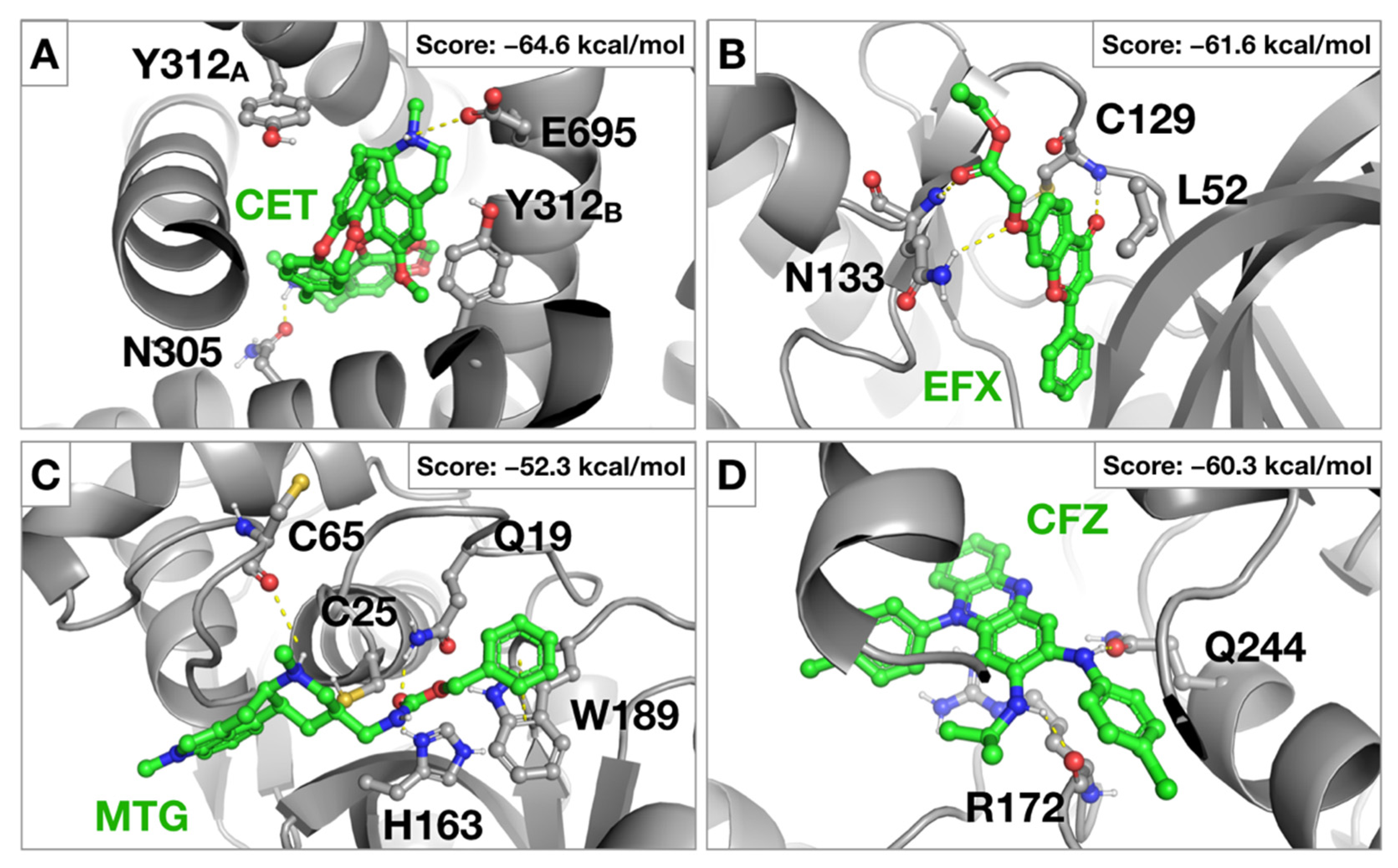
2.1. Virtual Screening against Selected Targets
2.2. SARS-CoV-2 Antiviral Candidate Biological Evaluation: Experimental Screening and Prioritization
2.3. Antiviral Activity of Selected Candidates
2.4. Evaluation of Anti-SARS-CoV-2 Drugs as Entry Inhibitors
3. Discussion
4. Materials and Methods
4.1. Computational Studies
4.1.1. The Drug-Dataset
4.1.2. Protein Targets and MD Simulations
4.1.3. Structure-Based Virtual Screening (SBVS) and MM-GBSA Rescoring
4.2. SARS-CoV-2 Infection Assays
4.2.1. Cell Monolayer Protection Assays
4.2.2. Evaluation of the Antiviral Activity Immunofluorescence Microscopy
4.2.3. Cytotoxicity Measurement by MTT Assays
4.3. SARS-CoV-2 Spike Protein-Pseudotyped Retroviral Vectors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Saul, S.; Einav, S. Old drugs for a new virus: Repurposed approaches for combating COVID-19. ACS Infect. Dis. 2020, 6, 2304–2318. [Google Scholar] [CrossRef]
- Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.P.; Malekzadeh, R.; et al. Repurposed antiviral drugs for COVID-19—Interim WHO Solidarity trial results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment (accessed on 24 March 2021).
- Mulangu, S.; Dodd, L.E.; Davey, R.T., Jr.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharmacol. 2018, 175, 181–191. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palu, G.; Loregian, A. Drug repurposing for viral infectious diseases: How far are we? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramirez, D.; Alonso, C.; Campillo, N.E. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819–e00820. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292e6. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.W.; Okumura, Y.; Kido, H.; Ng, L.F.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Dahms, S.O.; Jiao, G.S.; Than, M.E. Structural studies revealed active site distortions of human Furin by a small molecule inhibitor. ACS Chem. Biol. 2017, 12, 1211–1216. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef]
- Hardes, K.; Becker, G.L.; Lu, Y.; Dahms, S.O.; Kohler, S.; Beyer, W.; Sandvig, K.; Yamamoto, H.; Lindberg, I.; Walz, L.; et al. Novel Furin inhibitors with potent anti-infectious activity. Chem. Med. Chem. 2015, 10, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B., 3rd; Bates, P.; et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Kang, Y.L.; Chou, Y.Y.; Rothlauf, P.W.; Liu, Z.; Soh, T.K.; Cureton, D.; Case, J.B.; Chen, R.E.; Diamond, M.S.; Whelan, S.P. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 20803–20813. [Google Scholar] [CrossRef]
- Filippini, A.; D’Amore, A.; Palombi, F.; Carpaneto, A. Could the inhibition of endo-lysosomal two-pore channels (TPCs) by the natural flavonoid naringenin represent an option to fight SARS-CoV-2 infection? Front. Microbiol. 2020, 11, 970. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-dependent Ca(2+) signaling regulates Middle East respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium. 2018, 75, 30–41. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Weinbach, E.C.; Garbus, J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature 1969, 221, 1016–1018. [Google Scholar] [CrossRef]
- Zhao, J.; He, Q.; Gong, Z.; Chen, S.; Cui, L. Niclosamide suppresses renal cell carcinoma by inhibiting Wnt/β-catenin and inducing mitochondrial dysfunctions. Springerplus 2016, 5, 1436. [Google Scholar] [CrossRef]
- Agathokleous, E.; Calabrese, E.J. Hormesis: The dose response for the 21st century: The future has arrived. Toxicology 2019, 425, 152249. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Roth, S.L.; Bialecki, M.A.; Whittaker, G.R. Internalization and fusion mechanism of vesicular stomatitis virus and related rhabdoviruses. Future Virol. 2010, 5, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Rasko, J.E.; Battini, J.L.; Gottschalk, R.J.; Mazo, I.; Miller, A.D. The RD114/simian type D retrovirus receptor is a neutral amino acid transporter. Proc. Natl. Acad. Sci. USA 1999, 96, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Paolo Rizzardi, G.; D’Agostino, C.; Attinger, A.; Knabenhans, C.; Fleury, S.; Acha-Orbea, H.; Pantaleo, G. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nat. Med. 2000, 6, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Pan, X.; Chen, D.; Xie, X.; Wu, Y.; Shang, W.; Jiang, X.; Sun, Y.; Fan, S.; He, J. Broad-spectrum antivirals of protoporphyrins inhibit the entry of highly pathogenic emerging viruses. Bioorg. Chem. 2021, 107, 104619. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Debing, Y.; Neyts, J.; Delang, L. The future of antivirals: Broad-spectrum inhibitors. Curr. Opin. Infect. Dis. 2015, 28, 596–602. [Google Scholar] [CrossRef]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-directed antiviral therapy. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Rogosnitzky, M.; Okediji, P.; Koman, I. Cepharanthine: A review of the antiviral potential of a Japanese-approved alopecia drug in COVID-19. Pharmacol. Rep. 2020, 72, 1509–1516. [Google Scholar] [CrossRef]
- Chen, C.Z.; Xu, M.; Pradhan, M.; Gorshkov, K.; Petersen, J.D.; Straus, M.R.; Zhu, W.; Shinn, P.; Guo, H.; Shen, M. Identifying SARS-CoV-2 entry inhibitors through drug repurposing screens of SARS-S and MERS-S pseudotyped particles. ACS Pharmacol. Transl. Sci. 2020, 3, 1165–1175. [Google Scholar] [CrossRef]
- Gerndt, S. Novel Chemical Tools for the Modulation of Two Pore Channel 2. Ph.D. Thesis, Ludwig Maximilians University of Munich, Munich, Germany, 2020. [Google Scholar]
- Morales-Ortega, A.; Bernal-Bello, D.; Llarena-Barroso, C.; Frutos-Pérez, B.; Duarte-Millán, M.; García de Viedma-García, V.; Farfán-Sedano, A.I.; Canalejo-Castrillero, E.; Ruiz-Giardín, J.M.; Ruiz-Ruiz, J. Imatinib for COVID-19: A case report. Clin. Immunol. 2020, 218, 108518. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.; Coleman, C.M.; Haupt, R.; Logue, J.; Matthews, K.; Li, Y.; Reyes, H.M.; Weiss, S.R.; Frieman, M.B. Broad anti-coronavirus activity of food and drug administration-approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo. J. Virol. 2020, 94, e01218–e01220. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, Y.J.; Kim, J.H.; Kim, S.I.; Kim, S.S.; Choi, B.S.; Choi, J.H. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci. Rep. 2020, 10, 16200. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.Y.; Chen, K.C.; Chen, H.; Seng, E.K.; Chu, J.J. Antiviral activity of lanatoside C against dengue virus infection. Antiviral Res. 2014, 111, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Jan, J.T.; Chen, C.M.; Hsieh, H.P.; Hwang, D.R.; Liu, H.W.; Liu, C.Y.; Huang, H.W.; Chen, S.C.; Hong, C.F. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob. Agents Chemother. 2004, 48, 2693–2696. [Google Scholar] [CrossRef]
- Cline, J.C.; Nelson, J.D.; Gerzon, K.; Williams, R.H.; Delong, D.C. In vitro antiviral activity of mycophenolic acid and its reversal by guanine-type compounds. Appl. Microbiol. 1969, 18, 14–20. [Google Scholar] [CrossRef]
- Schrödinger LLC. Schrödinger Release 2020-1; Schrödinger LLC: New York, NY, USA, 2020. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–11092. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Zeng, X.; Uyar, A.; Sui, D.; Donyapour, N.; Wu, D.; Dickson, A.; Hu, J. Structural insights into lethal contractural syndrome type 3 (LCCS3) caused by a missense mutation of PIP5Kgamma. Biochem. J. 2018, 475, 2257–2269. [Google Scholar] [CrossRef]
- Available online: http://research.bmh.manchester.ac.uk/bryce/amber/ (accessed on 24 March 2021).
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Baik, J.; Rosania, G.R. Molecular imaging of intracellular drug-membrane aggregate formation. Mol. Pharm. 2011, 8, 1742–1749. [Google Scholar] [CrossRef]
- Gastaminza, P.; Whitten-Bauer, C.; Chisari, F.V. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. USA 2010, 107, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Mingorance, L.; Friesland, M.; Coto-Llerena, M.; Perez-del-Pulgar, S.; Boix, L.; Lopez-Oliva, J.M.; Bruix, J.; Forns, X.; Gastaminza, P. Selective inhibition of hepatitis C virus infection by hydroxyzine and benztropine. Antimicrob. Agents Chemother. 2014, 58, 3451–3460. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ginex, T.; Garaigorta, U.; Ramírez, D.; Castro, V.; Nozal, V.; Maestro, I.; García-Cárceles, J.; Campillo, N.E.; Martinez, A.; Gastaminza, P.; et al. Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods. Pharmaceuticals 2021, 14, 332. https://doi.org/10.3390/ph14040332
Ginex T, Garaigorta U, Ramírez D, Castro V, Nozal V, Maestro I, García-Cárceles J, Campillo NE, Martinez A, Gastaminza P, et al. Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods. Pharmaceuticals. 2021; 14(4):332. https://doi.org/10.3390/ph14040332
Chicago/Turabian StyleGinex, Tiziana, Urtzi Garaigorta, David Ramírez, Victoria Castro, Vanesa Nozal, Inés Maestro, Javier García-Cárceles, Nuria E. Campillo, Ana Martinez, Pablo Gastaminza, and et al. 2021. "Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods" Pharmaceuticals 14, no. 4: 332. https://doi.org/10.3390/ph14040332
APA StyleGinex, T., Garaigorta, U., Ramírez, D., Castro, V., Nozal, V., Maestro, I., García-Cárceles, J., Campillo, N. E., Martinez, A., Gastaminza, P., & Gil, C. (2021). Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods. Pharmaceuticals, 14(4), 332. https://doi.org/10.3390/ph14040332