
Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches



Abstract
:1. Introduction
2. ADCs
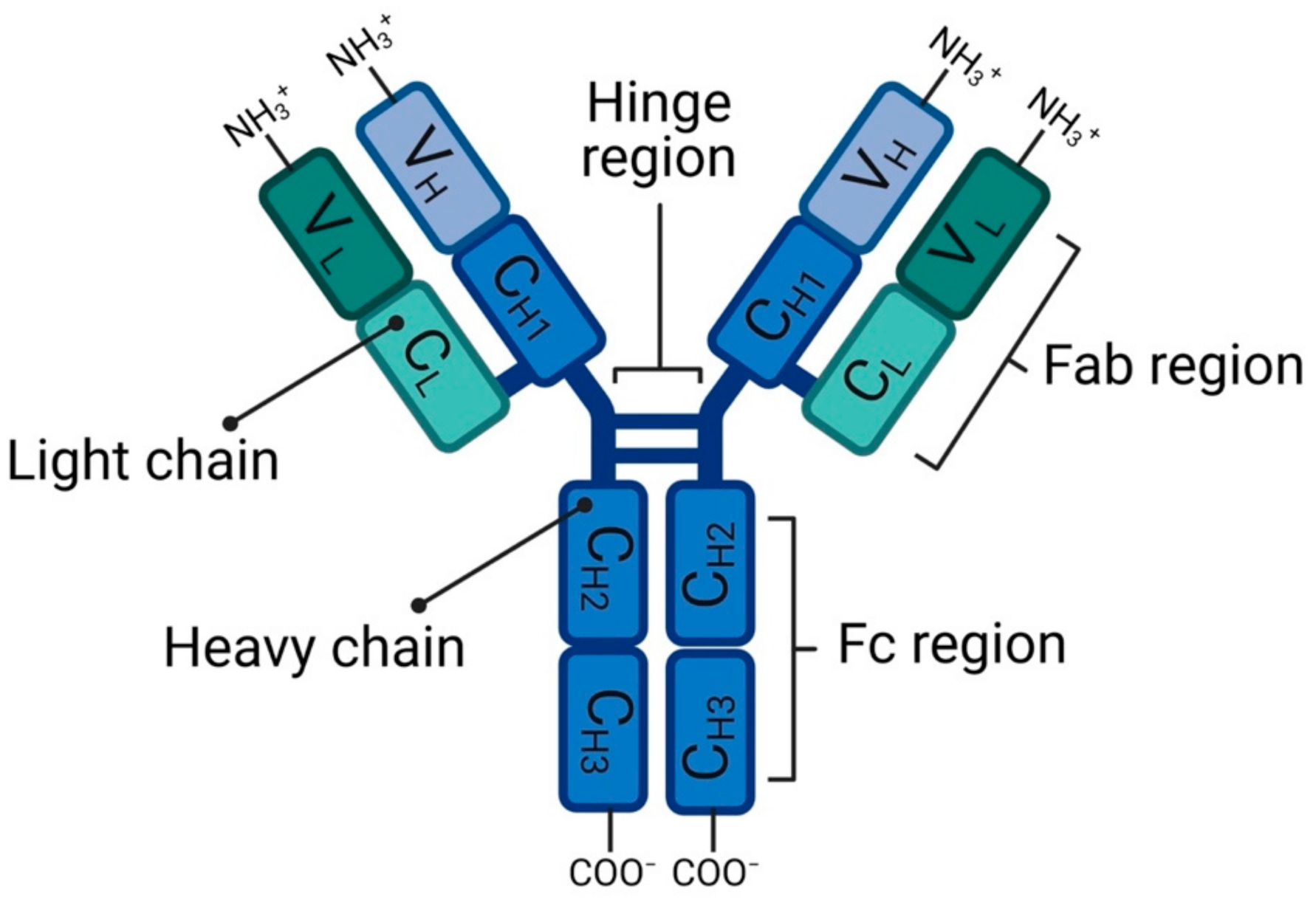
2.1. Antibody
2.2. Target Antigen
2.3. Linker
2.4. Drug
2.5. Conjugation Method
3. Enzyme-Based Conjugation Approaches
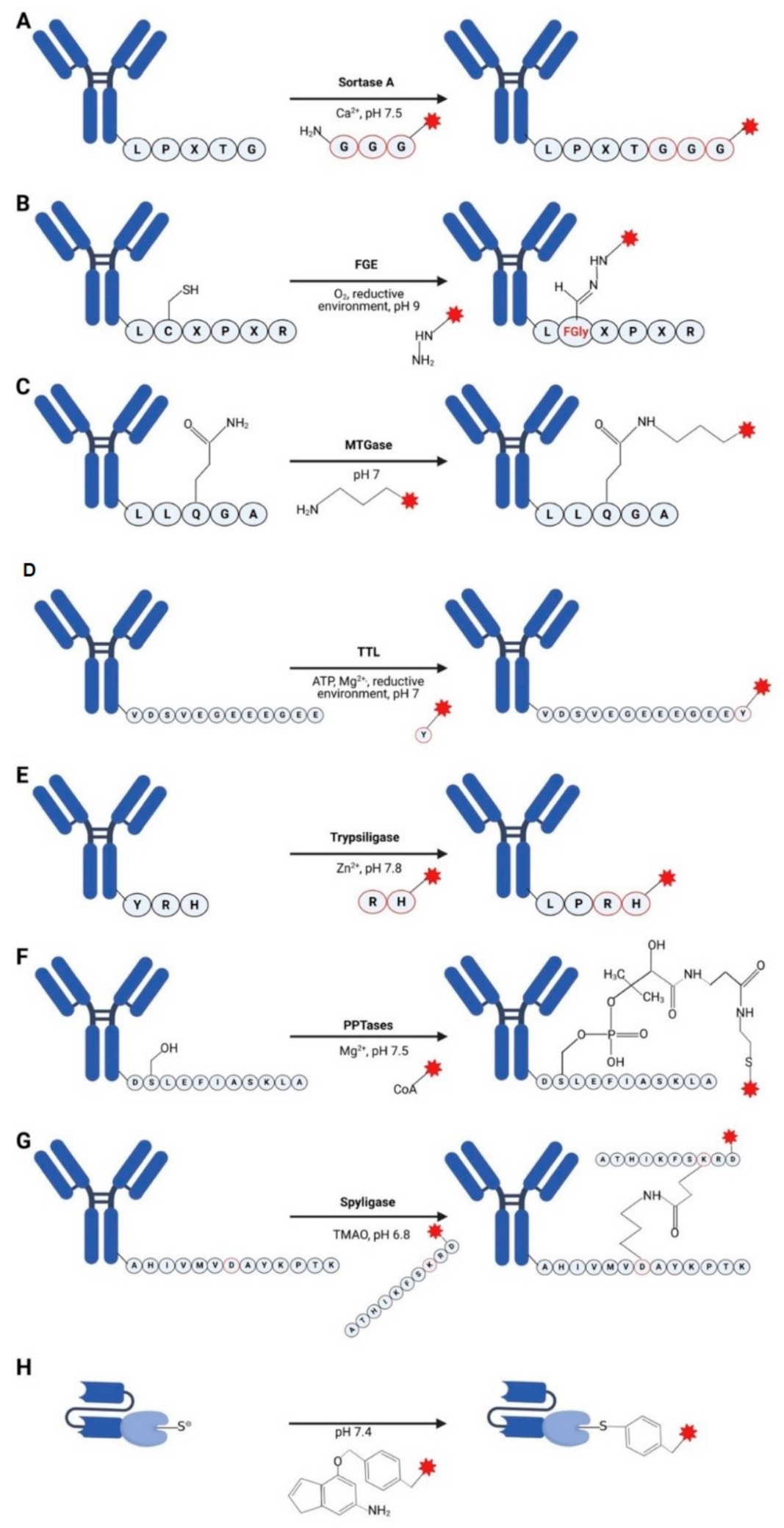
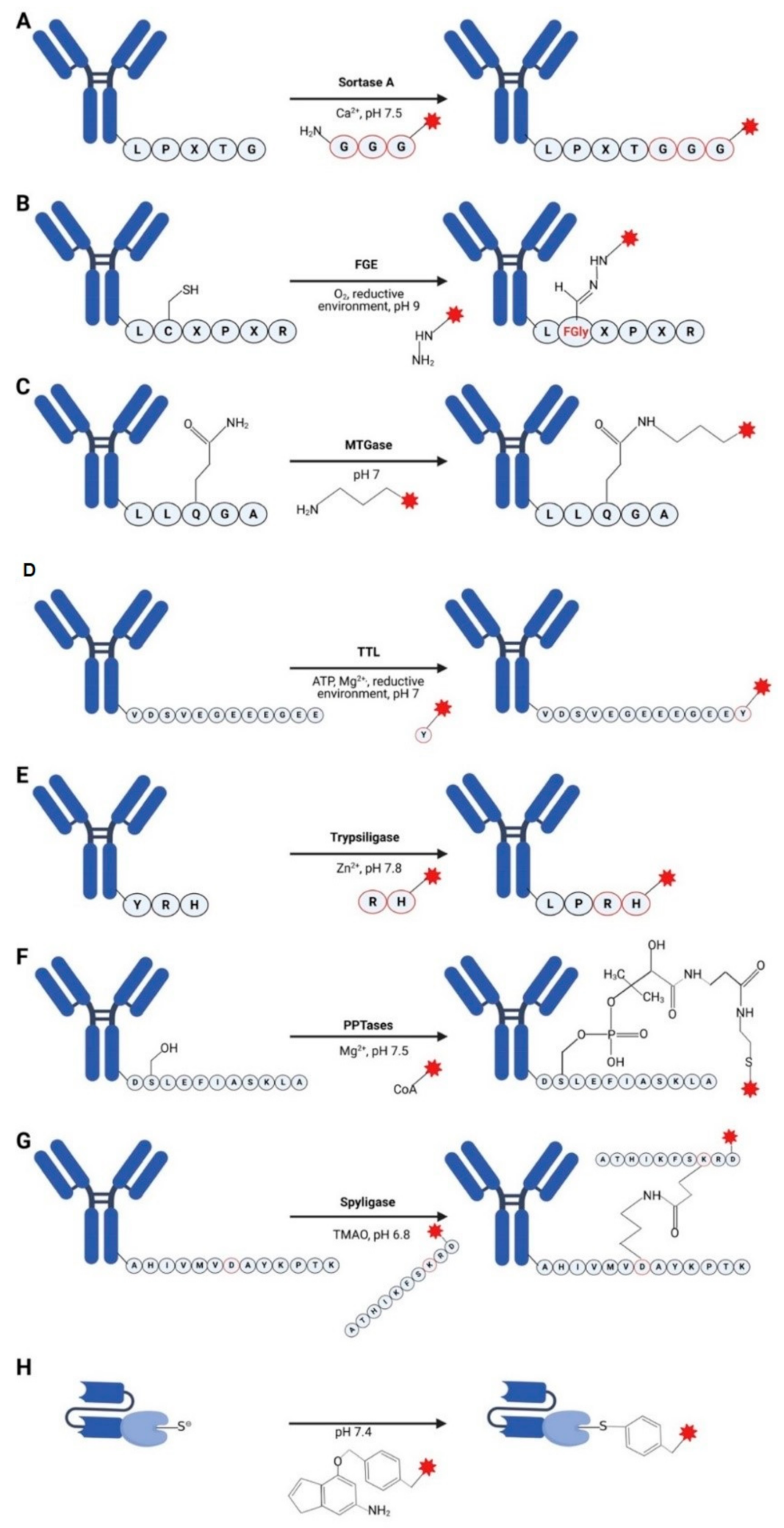
3.1. Sortases
3.2. Formylglycine-Based Methods
3.3. Transglutaminases
3.4. Tubulin Tyrosine Ligase
3.5. Trypsiligase and Subtiligase
3.6. Phosphopantetheinyl Transferase
3.7. Spyligase
3.8. O6-Alkylguanine-DNA Alkyltransferase (AGT) (SNAP-Tag)
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Adaptive Immune System. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Chapter 24; Available online: https://www.ncbi.nlm.nih.gov/books/NBK21070/ (accessed on 1 December 2020).
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.J.; Travers, P.; Walport, M.; Shlomchik, M.J. The structure of a typical antibody molecule. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001; Available online: https://www.ncbi.nlm.nih.gov/books/NBK27144/ (accessed on 1 December 2020).
- Dübel, S.; Breitling, F.; Frenzel, A.; Jostock, T.; Marschall, L.A.; Schirrmann, T.; Hust, M. Rekombinante Antikörper: Lehrbuch und Kompendium für Studium und Praxis; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Heinrich, P.C.; Müller, M.; Graeve, L. Löffler/Petrides Biochemie und Pathobiochemie; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.F.U.H.; Wang, R.; Ling, S.; Wang, S. Antibody Engineering for Pursuing a Healthier Future. Front. Microbiol. 2017, 8, 495. [Google Scholar] [CrossRef] [Green Version]
- Lua, W.H.; Ling, W.L.; Yeo, J.Y.; Poh, J.J.; Lane, D.P.; Gan, S.K. The effects of Antibody Engineering CH and CL in Trastuzumab and Pertuzumab recombinant models: Impact on antibody production and antigen-binding. Sci. Rep. 2018, 8, 718. [Google Scholar] [CrossRef] [Green Version]
- Su, C.T.; Lua, W.H.; Ling, W.L.; Gan, S.K. Allosteric Effects between the Antibody Constant and Variable Regions: A Study of IgA Fc Mutations on Antigen Binding. Antibodies 2018, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Ling, W.-L.; Lua, W.-H.; Gan, S.K.-E. Sagacity in antibody humanization for therapeutics, diagnostics and research purposes: Considerations of antibody elements and their roles. Antib. Ther. 2020, 3, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef] [Green Version]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates-An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar]
- Al-Salama, Z.T. Inotuzumab Ozogamicin: A Review in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukaemia. Target. Oncol. 2018, 13, 525–532. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [Green Version]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H.; Lowman, H.B. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Li, X.; Fang, T.; Boons, G.-J. Preparation of Well-Defined Antibody-Drug Conjugates Through Glycan Remodeling and Strain-Promoted Azide-Alkyne Cycloadditions. Angewandte Chemie 2014, 53, 7179–7182, (International ed. in English). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Trieu-Cuot, P.; Bierne, H. Sorting sortases: A nomenclature proposal for the various sortases of Gram-positive bacteria. Res. Microbiol. 2005, 156, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef]
- Pan, L.; Zhao, W.; Lai, J.; Ding, D.; Zhang, Q.; Yang, X.; Huang, M.; Jin, S.; Xu, Y.; Zeng, S.; et al. Sortase A-Generated Highly Potent Anti-CD20-MMAE Conjugates for Efficient Elimination of B-Lineage Lymphomas. Small 2017, 13. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, S.; Zhao, W.; Liu, W.; Ding, D.; Zhou, J.; Chen, S. A Versatile Chemo-Enzymatic Conjugation Approach Yields Homogeneous and Highly Potent Antibody-Drug Conjugates. Int. J. Mol. Sci. 2017, 18, 2284. [Google Scholar] [CrossRef] [Green Version]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; De Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Krüger, T.; Weiland, S.; Falck, G.; Gerlach, M.; Boschanski, M.; Alam, S.; Müller, K.M.; Dierks, T.; Sewald, N. Two-fold Bioorthogonal Derivatization by Different Formylglycine-Generating Enzymes. Angew. Chem. Int. Ed. 2018, 57, 7245–7249. [Google Scholar] [CrossRef]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Bregeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjugate Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef]
- Schumacher, D.; Hackenberger, C.P.R.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Liebscher, S.; Schöpfel, M.; Aumüller, T.; Sharkhuukhen, A.; Pech, A.; Höss, E.; Parthier, C.; Jahreis, G.; Stubbs, M.T.; Bordusa, F. N-Terminal Protein Modification by Substrate-Activated Reverse Proteolysis. Angew. Chem. Int. Ed. 2014, 53, 3024–3028. [Google Scholar] [CrossRef]
- Meyer, C.; Liebscher, S.; Bordusa, F. Selective Coupling of Click Anchors to Proteins via Trypsiligase. Bioconjugate Chem. 2015, 27, 47–53. [Google Scholar] [CrossRef]
- Grünewald, J.; Brock, A.; Geierstanger, B.H. Site-Specific Antibody Labeling Using Phosphopantetheinyl Transferase-Catalyzed Ligation. Breast Cancer 2019, 2012, 237–278. [Google Scholar]
- Grünewald, J.; Klock, H.E.; Cellitti, S.E.; Bursulaya, B.; McMullan, D.; Jones, D.H.; Chiu, H.-P.; Wang, X.; Patterson, P.; Zhou, H.; et al. Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Phosphopantetheinyl Transferases. Bioconjugate Chem. 2015, 26, 2554–2562. [Google Scholar] [CrossRef]
- Grünewald, J.; Jin, Y.; Vance, J.; Read, J.; Wang, X.; Wan, Y.; Zhou, H.; Ou, W.; Klock, H.E.; Peters, E.C.; et al. Optimization of an Enzymatic Antibody–Drug Conjugation Approach Based on Coenzyme A Analogs. Bioconjugate Chem. 2017, 28, 1906–1915. [Google Scholar] [CrossRef]
- Alam, K.; El-Sayed, A.; Barreto, K.; Bernhard, W.; Fonge, H.; Geyer, C.R. Site-Specific Fluorescent Labeling of Antibodies and Diabodies Using SpyTag/SpyCatcher System for In Vivo Optical Imaging. Mol. Imaging Biol. 2018, 21, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, V.; Piater, B.; Zakeri, B.; Eichhorn, T.; Fischer, F.; Deutsch, C.; Becker, S.; Toleikis, L.; Hock, B.; Betz, U.A.K.; et al. Spontaneous Isopeptide Bond Formation as a Powerful Tool for Engineering Site-Specific Antibody-Drug Conjugates. Sci. Rep. 2016, 6, 39291. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.F.; Kampmeier, F.; Von Felbert, V.; Merk, H.-F.; Tur, M.K.; Barth, S. SNAP-Tag Technology Mediates Site Specific Conjugation of Antibody Fragments with a Photosensitizer and Improves Target Specific Phototoxicity in Tumor Cells. Bioconjugate Chem. 2011, 22, 2487–2495. [Google Scholar] [CrossRef] [PubMed]
- Woitok, M.; Klose, D.; Niesen, J.; Richter, W.; Abbas, M.; Stein, C.; Fendel, R.; Bialon, M.; Puttmann, C.; Fischer, R.; et al. The efficient elimination of solid tumor cells by EGFR-specific and HER2-specific scFv-SNAP fusion proteins conjugated to benzylguanine-modified auristatin F. Cancer Lett. 2016, 381, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.F.; Heppenstall, P.A.; Kampmeier, F.; Meinhold-Heerlein, I.; Barth, S. One-step site-specific antibody fragment auto-conjugation using SNAP-tag technology. Nat. Protoc. 2019, 14, 3101–3125. [Google Scholar] [CrossRef] [PubMed]
- Cosma, M.P.; Pepe, S.; Annunziata, I.; Newbold, R.F.; Grompe, M.; Parenti, G.; Ballabio, A. The Multiple Sulfatase Deficiency Gene Encodes an Essential and Limiting Factor for the Activity of Sulfatases. Cell 2003, 113, 445–456. [Google Scholar] [CrossRef]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321–322. [Google Scholar] [CrossRef]
- Kudirka, R.A.; Barfield, R.M.; McFarland, J.M.; Drake, P.M.; Carlson, A.; Bañas, S.; Zmolek, W.; Garofalo, A.W.; Rabuka, D. Site-Specific Tandem Knoevenagel Condensation–Michael Addition To Generate Antibody–Drug Conjugates. ACS Med. Chem. Lett. 2016, 7, 994–998. [Google Scholar] [CrossRef] [Green Version]
- Strop, P. Versatility of Microbial Transglutaminase. Bioconjugate Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef]
- Zheng, K.; Bantog, C.; Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011, 3, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Ebenig, A.; Juettner, N.E.; Deweid, L.; Avrutina, O.; Fuchsbauer, H.L.; Kolmar, H. Efficient Site-Specific Antibody-Drug Conjugation by Engineering a Nature-Derived Recognition Tag for Microbial Transglutaminase. ChemBiochem 2019, 20, 2411–2419. [Google Scholar] [CrossRef]
- Walker, J.A.; Bohn, J.J.; Ledesma, F.; Sorkin, M.R.; Kabaria, S.R.; Thornlow, D.N.; Alabi, C.A. Substrate Design Enables Heterobifunctional, Dual “Click” Antibody Modification via Microbial Transglutaminase. Bioconjugate Chem. 2019, 30, 2452–2457. [Google Scholar] [CrossRef]
- Gerlach, M.; Stoschek, T.; Leonhardt, H.; Hackenberger, C.P.R.; Schumacher, D.; Helma, J. Tubulin Tyrosine Ligase-Mediated Modification of Proteins. Breast Cancer 2019, 2012, 327–355. [Google Scholar]
- Schumacher, D.; Helma, J.; Mann, F.A.; Pichler, G.; Natale, F.; Krause, E.; Cardoso, M.C.; Hackenberger, C.P.R.; Leonhardt, H. Versatile and Efficient Site-Specific Protein Functionalization by Tubulin Tyrosine Ligase. Angew. Chem. Int. Ed. 2015, 54, 13787–13791. [Google Scholar] [CrossRef]
- Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Abrahmsen, L.; Tom, J.; Burnier, J.; Butcher, K.A.; Kossiakoff, A.; Wells, J.A. Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry 1991, 30, 4151–4159. [Google Scholar] [CrossRef]
- Liebscher, S.; Bordusa, F. Trypsiligase-Catalyzed Peptide and Protein Ligation. Methods Mol. Biol. 2019, 2012, 111–133. [Google Scholar]
- Liebscher, S.; Bordusa, F. Site-Specific Modification of Proteins via Trypsiligase. Methods Mol. Biol. 2019, 2033, 95–115. [Google Scholar]
- Weeks, A.M.; Wells, J.A. Subtiligase-Catalyzed Peptide Ligation. Chem. Rev. 2019, 120, 3127–3160. [Google Scholar] [CrossRef]
- Weeks, A.M.; Wells, J.A. Engineering peptide ligase specificity by proteomic identification of ligation sites. Nat. Chem. Biol. 2017, 14, 50–57. [Google Scholar] [CrossRef]
- Yin, J.; Liu, F.; Li, X.; Walsh, C.T. Labeling Proteins with Small Molecules by Site-Specific Posttranslational Modification. J. Am. Chem. Soc. 2004, 126, 7754–7755. [Google Scholar] [CrossRef]
- George, N.; Pick, H.; Vogel, H.; Johnsson, N.; Johnsson, K. Specific Labeling of Cell Surface Proteins with Chemically Diverse Compounds. J. Am. Chem. Soc. 2004, 126, 8896–8897. [Google Scholar] [CrossRef]
- Yin, J.; Straight, P.D.; McLoughlin, S.M.; Zhou, Z.; Lin, A.J.; Golan, D.E.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA 2005, 102, 15815–15820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Cironi, P.; Lin, A.J.; Xu, Y.; Hrvatin, S.; Golan, D.E.; Silver, P.A.; Walsh, C.T.; Yin, J. Genetically Encoded Short Peptide Tags for Orthogonal Protein Labeling by Sfp and AcpS Phosphopantetheinyl Transferases. ACS Chem. Biol. 2007, 2, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, J.O.; Veggiani, G.; Howarth, M. SpyLigase peptide–peptide ligation polymerizes affibodies to enhance magnetic cancer cell capture. Proc. Natl. Acad. Sci. USA 2014, 111, E1176–E1181. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E.; Byers, T.L. Repair of DNA containing O6-alkylguanine. FASEB J. 1992, 6, 2302–2310. [Google Scholar] [CrossRef]
- Sedgwick, B. Molecular cloning of a gene which regulates the adaptive response to alkylating agents in Escherichia coli. Mol. Genet. Genom. 1983, 191, 466–472. [Google Scholar] [CrossRef]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Demple, B. Self-methylation by suicide DNA repair enzymes. In Protein Methylation; Paik, W.K.a.K.S., Ed.; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, Function, and Inhibition of O6-Alkylguanine-DNA Alkyltransferase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 167–223. [Google Scholar]
- Bojkowska, K.; De Sio, F.S.; Barde, I.; Offner, S.; Verp, S.; Heinis, C.; Johnsson, K.; Trono, D. Measuring In Vivo Protein Half-Life. Chem. Biol. 2011, 18, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Bosch, P.J.; Corrêa, I.R.; Sonntag, M.H.; Ibach, J.; Brunsveld, L.; Kanger, J.S.; Subramaniam, V. Evaluation of Fluorophores to Label SNAP-Tag Fused Proteins for Multicolor Single-Molecule Tracking Microscopy in Live Cells. Biophys. J. 2014, 107, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Foraker, A.B.; Camus, S.M.; Evans, T.M.; Majeed, S.R.; Chen, C.-Y.; Taner, S.B.; Corrêa, I.R.; Doxsey, S.J.; Brodsky, F.M. Clathrin promotes centrosome integrity in early mitosis through stabilization of centrosomal ch-TOG. J. Cell Biol. 2012, 198, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Liss, V.; Barlag, B.; Nietschke, M.; Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep. 2016, 5, 17740. [Google Scholar] [CrossRef]
- Mie, M.; Naoki, T.; Uchida, K.; Kobatake, E. Development of a split SNAP-tag protein complementation assay for visualization of protein–protein interactions in living cells. Analyst 2012, 137, 4760–4765. [Google Scholar] [CrossRef]
- Prifti, E.; Reymond, L.; Umebayashi, M.; Hovius, R.; Riezman, H.; Johnsson, K. A Fluorogenic Probe for SNAP-Tagged Plasma Membrane Proteins Based on the Solvatochromic Molecule Nile Red. ACS Chem. Biol. 2014, 9, 606–612. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Amoury, M.; Bauerschlag, D.; Zeppernick, F.; von Felbert, V.; Berges, N.; di Fiore, S.; Mintert, I.; Bleilevens, A.; Maass, N.; Brautigam, K.; et al. Photoimmunotheranostic agents for triple-negative breast cancer diagnosis and therapy that can be activated on demand. Oncotarget 2016, 7, 54925–54936. [Google Scholar] [CrossRef] [Green Version]
- Maynard, J.; Georgiou, G. Antibody engineering. Annu. Rev. Biomed. Eng. 2000, 2, 339–376. [Google Scholar] [CrossRef]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [Green Version]
- Bauerschlag, D.; Meinhold-Heerlein, I.; Maass, N.; Bleilevens, A.; Bräutigam, K.; Al Rawashdeh, W.; Di Fiore, S.; Haugg, A.M.; Gremse, F.; Steitz, J.; et al. Detection and Specific Elimination of EGFR+ Ovarian Cancer Cells Using a Near Infrared Photoimmunotheranostic Approach. Pharm. Res. 2017, 34, 696–703. [Google Scholar] [CrossRef]
- Chouman, K.; Woitok, M.; Mladenov, R.; Kessler, C.; Weinhold, E.; Hanz, G.; Fischer, R.; Meinhold-Heerlein, I.; Bleilevens, A.; Gresch, G.; et al. Fine tuning antibody conjugation methods using SNAP-tag technology. Anti-Cancer Agents Med. Chem. 2017, 17, 1434–1440. [Google Scholar] [CrossRef]
- Von Felbert, V.; Bauerschlag, D.; Maass, N.; Bräutigam, K.; Meinhold-Heerlein, I.; Woitok, M.; Barth, S.; Hussain, A.F. A specific photoimmunotheranostics agent to detect and eliminate skin cancer cells expressing EGFR. J. Cancer Res. Clin. Oncol. 2016, 142, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Krzyscik, M.A.; Opalinski, L.; Otlewski, J. Novel Method for Preparation of Site-Specific, Stoichiometric-Controlled Dual Warhead Conjugate of FGF2 via Dimerization Employing Sortase A-Mediated Ligation. Mol. Pharm. 2019, 16, 3588–3599. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, L.N.; O’Donnell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjugate Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Conjugation Method | Substrate | Coupling Method | Applications | References |
|---|---|---|---|---|
| Sortases | LPXTG | Labeling peptide | Full length antibody, Fab | [32] |
| Formylglycine-based methods | LCTPSR | Aldehyde coupling chemistry | Full length antibody | [34,35] |
| Transglutaminases | LLQGA | Labeled alkyl- or oligo-amine | Full length antibody | [36] |
| Tubulin tyrosine ligase | VDSVEGEGEEEGEE, Tub-tag | Labeled tyrosine | scFv | [37] |
| Trypsiligase and subtiligase | YRH | Labeled peptide | Fab | [38,39] |
| Phosphopantetheinyl transferase | DSLEFIASKLA | Labeled CoaA | Full length antibody | [40,41,42] |
| Spyligase | AHIVMVDAYKPTK | Peptide-peptide ligation | Full length antibody | [43,44] |
| SNAP-tag | BG-modified molecules | Irreversible transfer of an alkyl group to a cysteine residue | scFv | [45,46,47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, A.F.; Grimm, A.; Sheng, W.; Zhang, C.; Al-Rawe, M.; Bräutigam, K.; Abu Mraheil, M.; Zeppernick, F.; Meinhold-Heerlein, I. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals 2021, 14, 343. https://doi.org/10.3390/ph14040343
Hussain AF, Grimm A, Sheng W, Zhang C, Al-Rawe M, Bräutigam K, Abu Mraheil M, Zeppernick F, Meinhold-Heerlein I. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals. 2021; 14(4):343. https://doi.org/10.3390/ph14040343
Chicago/Turabian StyleHussain, Ahmad Fawzi, Armin Grimm, Wenjie Sheng, Chaoyu Zhang, Marwah Al-Rawe, Karen Bräutigam, Mobarak Abu Mraheil, Felix Zeppernick, and Ivo Meinhold-Heerlein. 2021. "Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches" Pharmaceuticals 14, no. 4: 343. https://doi.org/10.3390/ph14040343
APA StyleHussain, A. F., Grimm, A., Sheng, W., Zhang, C., Al-Rawe, M., Bräutigam, K., Abu Mraheil, M., Zeppernick, F., & Meinhold-Heerlein, I. (2021). Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals, 14(4), 343. https://doi.org/10.3390/ph14040343