
Aminoalkylamides of Eremomycin Exhibit an Improved Antibacterial Activity


 and
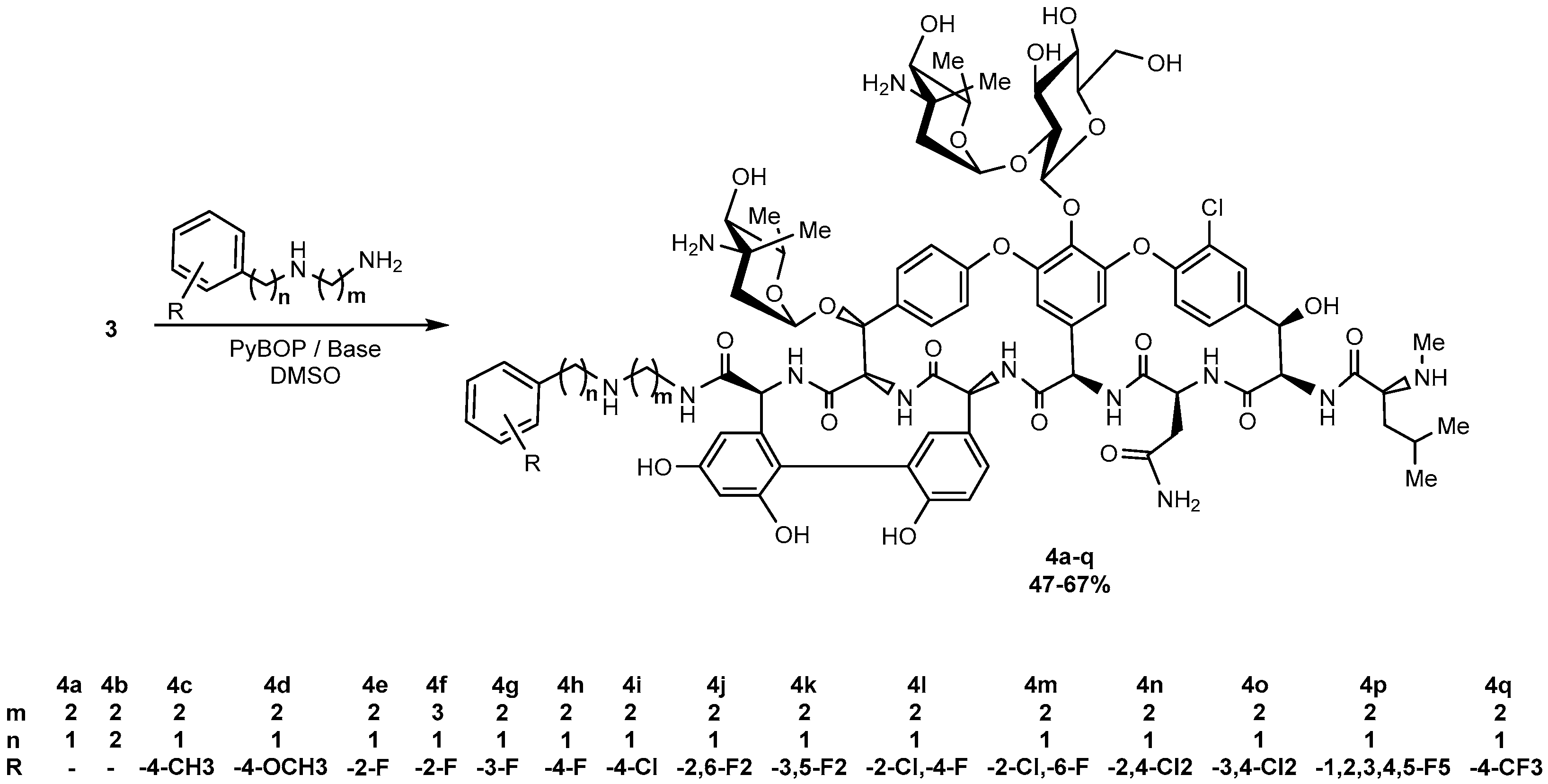
and
Abstract
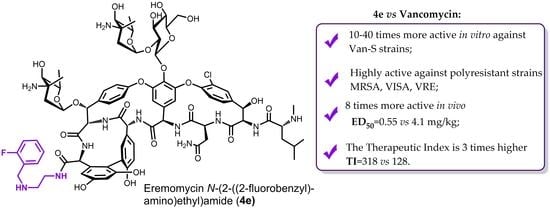
1. Introduction
2. Results
3. Discussion
- Eremomycin N-(2-((2-fluorobenzyl)amino)ethyl)amide (4e) TI = 175/0.55 = 318;
- Vancomycin TI = 525/4.1 = 128.
4. Materials and Methods
4.1. Synthesis, General Procedure
4.2. Compounds Characterization
- Eremomycin N-(2-(benzylamino)ethyl)amide (4a). Yield 47%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 35% (30 min): Rt = 8.8 min (96.9%). HRSM (ESI) calculated for C82H102ClN12O25 [M + H]+: 1689.6762; found 1689.6787.
- Eremomycin N-(2-phenethylamino)ethyl)amide (4b). Yield 48%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 35% (30 min): Rt = 12.7 min (95.4%). HRSM (ESI) calculated for C83H104ClN12O25 [M + H]+: 1703.6919; found 1703.6934.
- Eremomycin N-(2-((4-methylbenzyl)amino)ethyl)amide (4c). Yield 44%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 13.2 min (96.7%). HRSM (ESI) calculated for C83H104ClN12O25 [M + H]+: 1703.6919; found 1703.6905.
- Eremomycin N-(2-((4-methoxybenzyl)amino)ethyl)amide (4d). Yield 60%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 12.3 min (97.2%). HRSM (ESI) calculated for C83H104ClN12O26 [M + H]+: 1719.6868; found 1719.6882.
- Eremomycin N-(2-((2-fluorobenzyl)amino)ethyl)amide (4e). Yield 54%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.6%) pH = 7.8, B—MeCN; gradient B 24–30% (30 min): Rt = 9.4 min (97.6%). HRSM (ESI) calculated for C82H101ClFN12O25 [M + H]+: 1707.6668; found 1707.6677. Found, %: C 50.19, H 5.96, N 8.57; calculated for C82H100ClFN12O25*4HCl*6H2O, %: C 50.34, H 5.80, N 8.59.
- Eremomycin N-(2-((2-fluorobenzyl)amino)propyl)amide (4f). Yield 58%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10–35% (30 min): Rt = 8.8 min (98.1%). HRSM (ESI) calculated for C83H103ClFN12O25 [M + H]+: 1721.6824; found 1721.6798.
- Eremomycin N-(2-((3-fluorobenzyl)amino)ethyl)amide (4g). Yield 52%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 12.2 min (96.4%). HRSM (ESI) calculated for C82H101ClFN12O25 [M + H]+: 1707.6668; found 1707.6645.
- Eremomycin N-(2-((4-fluorobenzyl)amino)ethyl)amide (4h). Yield 57%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 150 mm, LW = 270 nm, eluent: A—HCOONH4 (0.2%) pH = 6.5, B—MeCN; gradient B, 5–60% (30 min): Rt = 20.9 min (96.7%). HRMS (ESI) calculated for C82H101ClFN12O25 [M + H]+: 1707.6668; found 1707.6653.
- Eremomycin N-(2-((4-chlorobenzyl)amino)ethyl)amide (4i). Yield 50%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 14.2 min (95.6%). HRSM (ESI) calculated for C82H101Cl2N12O25 [M + H]+: 1723.6372; found 1723.6364.
- Eremomycin N-(2-((2,6-difluorobenzyl)amino)ethyl)amide (4j). Yield 48%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 14.0 min (95.4%). HRSM (ESI) calculated for C82H100ClF2N12O25 [M + H]+: 1725.6574; found 1725.6534.
- Eremomycin N-(2-((3,5-difluorobenzyl)amino)ethyl)amide (4k). Yield 49%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 35% (30 min): Rt = 11.9 min (95.3%). HRSM (ESI) calculated for C82H100ClF2N12O25 [M + H]+: 1725.6574; found 1725.6546.
- Eremomycin N-(2-((2-chloro-4-fluorobenzyl)amino)ethyl)amide (4l). Yield 45%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 60% (30 min): Rt = 10.4 min (97.3%). HRSM (ESI) calculated for C82H100Cl2FN12O25 [M + H]+: 1741.6278; found 1741.6260.
- Eremomycin N-(2-((2-chloro-6-fluorobenzyl)ethyl)amide (4m). Yield 46%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 35% (30 min): Rt = 10.7 min (96.8%). HRSM (ESI) calculated for C82H100Cl2FN12O25 [M + H]+: 1741.6278, found 1741.6253.
- Eremomycin N-(2-((2,4-dichlorobenzyl)amino)ethyl)amide (4n). Yield 61%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 15.2 min (97.1%). HRSM (ESI) calculated for C82H100Cl3N12O25 [M + H]+: 1757.5983; found 1757.5883.
- Eremomycin N-(2-((3,4-dichlorobenzyl)amino)ethyl)amide (4o). Yield 48%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 5 → 60% (30 min): Rt = 16.1 min (95.7%). HRSM (ESI) calculated for C82H100Cl3N12O25 [M + H]+: 1757.5983; found 1757.6009.
- Eremomycin N-(2-((2,3,4,5,6-pentafluorobenzyl)amino)ethyl)amide (4p). Yield 63%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 35% (30 min): Rt = 16.7 min (96.6%). HRSM (ESI) calculated for C82H97ClF5N12O25 [M + H]+: 1779.6291; found 1779.6276.
- Eremomycin N-(2-((4-trifluoromethylbenzyl)amino)ethyl)amide (4q). Yield 52%. HPLC (column Kromasil-100-5-μm C-18 4.6 × 250 mm, LW = 260 nm, eluent: A—HCOONH4 (0.2%) pH = 4.5, B—MeCN; gradient B 10 → 60% (30 min): Rt = 13.3 min (95.2%). HRSM (ESI) calculated for C83H101ClF3N12O25 [M + H]+: 1757.6636; found 1757.6802.
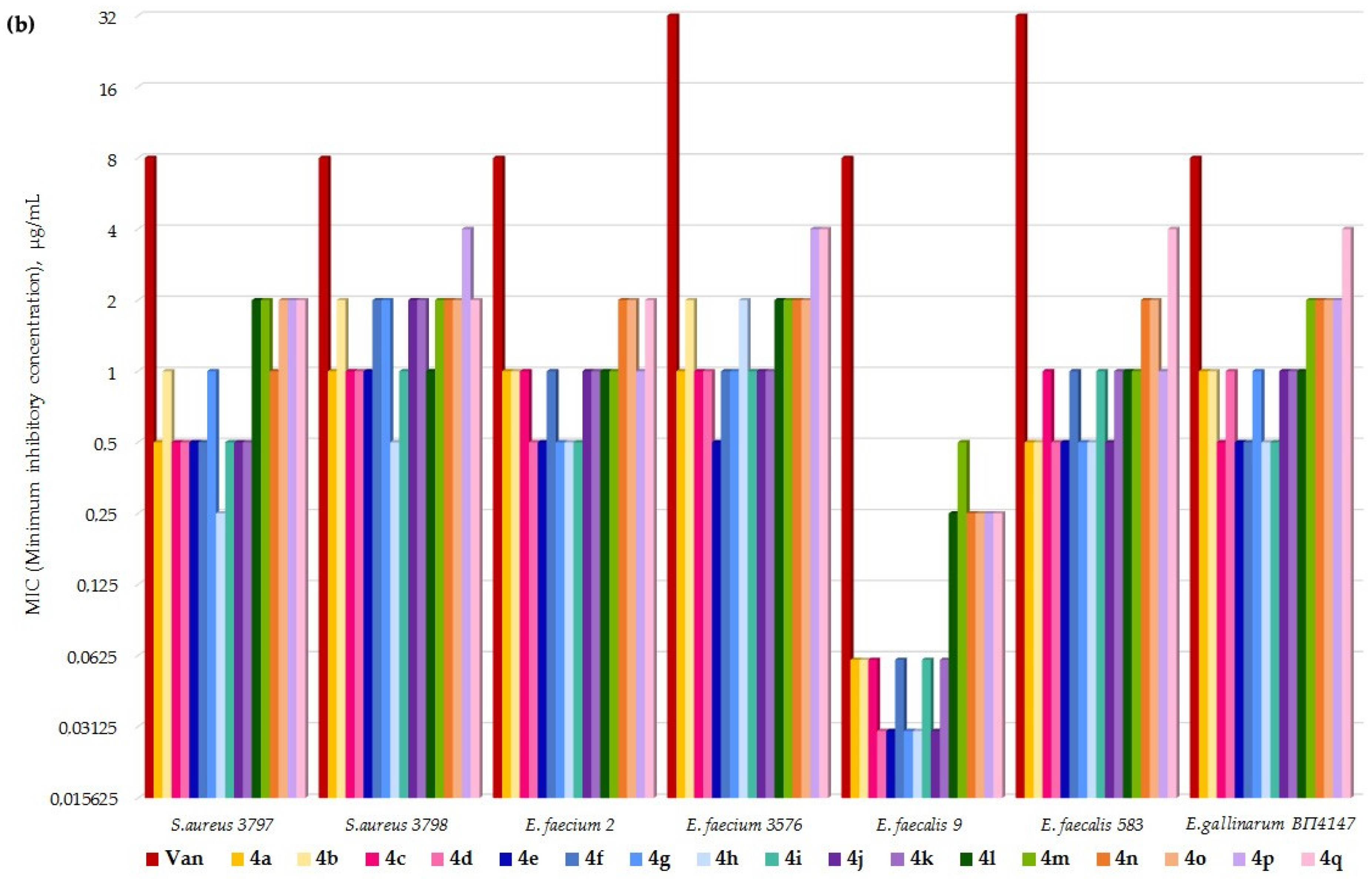
4.3. MIC Values Determination
4.4. In Vivo Efficiency Study
4.5. Acute Toxicity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-PubMed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef]
- Antibiotic Resistance Threats in the United States, 2019 (2019 AR Threats Report). Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 31 December 2019).
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a026989. [Google Scholar] [CrossRef]
- Vehreschild, M.J.G.T.; Haverkamp, M.; Biehl, L.M.; Lemmen, S.; Fätkenheuer, G. Vancomycin-resistant enterococci (VRE): A reason to isolate? Infection 2019, 47, 7–11. [Google Scholar] [CrossRef]
- Dancer, S.J. Glycopeptide resistance in Staphylococcus aureus. J. Antimicrob. Chemother. 2003, 51, 1309–1311. [Google Scholar] [CrossRef]
- Hubert, S.K.; Mohammed, J.M.; Fridkin, S.K.; Gaynes, R.P.; McGowan, J.E.; Tenover, F.C. Glycopeptide-Intermediate Staphylococcus aureus: Evaluation of a Novel Screening Method and Results of a Survey of Selected U.S. Hospitals. J. Clin. Microbiol. 1999, 37, 3590–3593. [Google Scholar] [CrossRef]
- Leadbetter, M.R.; Adams, S.M.; Bazzini, B.; Fatheree, P.R.; Karr, D.E.; Krause, K.M.; Lam, B.M.T.; Linsell, M.S.; Nodwell, M.B.; Pace, J.L.; et al. Hydrophobic vancomycin derivatives with improved ADME properties: Discovery of telavancin (TD-6424). J. Antibiot. 2004, 57, 326–336. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.; Adam, H.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. New Lipoglycopeptides. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef] [PubMed]
- Moiseenko, E.I.; Grammatikova, N.E.; Shchekotikhin, A.E. Eremomycin Picolylamides and Their Cationic Lipoglycopeptides: Synthesis and Antimicrobial Properties. Macroheterocycles 2019, 12, 98–106. [Google Scholar] [CrossRef]
- Allen, N.E.; Nicas, T.I. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol. Rev. 2003, 26, 511–532. [Google Scholar] [CrossRef]
- Allen, N.E.; Letourneau, D.L.; Hobbs, J.N. The Role of Hydrophobic Side Chains as Determinants of Antibacterial Activity of Semisynthetic Glycopeptide Antibiotics. J. Antibiot. 1997, 50, 677–684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ge, M.; Chen, Z.; Russell, H.; Onishi, H.R.; Kohler, J.; Silver, L.L.; Kerns, R.; Fukuzawa, S.; Thompson, C.; Kahne, D. Vancomycin Derivatives That Inhibit Peptidoglycan Biosynthesis Without Binding D-Ala-D-Ala. Science 1999, 284, 507–511. [Google Scholar] [CrossRef]
- Kerns, R.; Dong, S.D.; Fukuzawa, S.; Carbeck, J.; Kohler, J.; Silver, L.L.; Kahne, D. The Role of Hydrophobic Substituents in the Biological Activityof Glycopeptide Antibiotics. J. Am. Chem. Soc. 2000, 122, 12608–12609. [Google Scholar] [CrossRef]
- Printsevskaya, S.S.; Pavlov, A.Y.; Olsufyeva, E.N.; Mirchink, E.P.; Isakova, E.B.; Reznikova, M.I.; Goldman, R.C.; Branstrom, A.A.; Baizman, E.R.; Longley, C.B.; et al. Synthesis and Mode of Action of Hydrophobic Derivatives of the Glycopeptide Antibiotic Eremomycin and Des-(N-methyl-d-leucyl)eremomycin against Glycopeptide-Sensitive and -Resistant Bacteria. J. Med. Chem. 2002, 45, 1340–1347. [Google Scholar] [CrossRef]
- Pavlov, A.Y.; Preobrazhenskaya, M.N.; Malabarba, A.; Ciabatti, R.; Colombo, L. Mono and Double Modified Teicoplanin Aglycon Derivatives on the Amino Acid No. 7; Structure-activity Relationship. J. Antibiot. 1998, 51, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Yasukata, T.; Shindo, H.; Yoshida, O.; Sumino, Y.; Munekage, T.; Narukawa, Y.; Nishitani, Y. An efficient and practical method for solid-phase synthesis of tripeptide-bearing glycopeptide antibiotics: Combinatorial parallel synthesis of carboxamide derivatives of chloroorienticin B. Bioorganic Med. Chem. Lett. 2002, 12, 3033–3036. [Google Scholar] [CrossRef]
- Gauze, G.F.; Brazhnikova, M.G.; Laĭko, A.V.; Sveshnikova, M.A.; Preobrazhenskaia, T.P. Eremomycin—A new antibiotic from the cyclic glycopeptide group. Antibiot. I Meditsinskaia Biotekhnologiia Antibiot. Med. Biotechnol. 1987, 32, 571–576. [Google Scholar]
- Filippos’Iants, S.T.; Malkova, I.V.; Gol’Dberg, L.E. Glycopeptide antibiotics: Eremomycin, vancomycin, and teicoplanin. Comparison of several parameters of pharmacokinetics and antimicrobial activity. Antibiot. I Khimioterapiia Antibiot. Chemoterapy 1989, 34, 523–526. [Google Scholar]
- Maples, K.R.; Wheeler, C.; Ip, E.; Plattner, J.J.; Chu, D.; Zhang, Y.-K.; Preobrazhenskaya, M.N.; Printsevskaya, S.S.; Solovieva, S.E.; Olsufyeva, E.N.; et al. Novel Semisynthetic Derivative of Antibiotic Eremomycin Active against Drug-Resistant Gram-Positive Pathogens IncludingBacillusanthracis. J. Med. Chem. 2007, 50, 3681–3685. [Google Scholar] [CrossRef]
- Pavlov, A.Y.; Miroshnikova, O.V.; Printsevskaya, S.S.; Olsufyeva, E.N.; Preobrazhenskaya, M.N.; Goldman, R.C.; Branstrom, A.A.; Baizman, E.R.; Longley, C.B. Synthesis of Hydrophobic N′-Mono and N′,N″-Double Alkylated Eremomycins Inhibiting the Transglycosylation Stage of Bacterial Cell Wall Biosynthesis. J. Antibiot. 2001, 54, 455–459. [Google Scholar] [CrossRef][Green Version]
- Olsufyeva, E.N.; Shchekotikhin, A.E.; Bychkova, E.N.; Pereverzeva, E.R.; Treshalin, I.D.; Mirchink, E.P.; Isakova, E.B.; Chernobrovkin, M.G.; Kozlov, R.S.; Dekhnich, A.V.; et al. Eremomycin pyrrolidide: A novel semisynthetic glycopeptide with improved chemotherapeutic properties. Drug Des. Dev. Ther. 2018, 12, 2875–2885. [Google Scholar] [CrossRef]
- Moiseenko, E.I.; Grammatikova, N.E.; Shchekotikhin, A.E. Synthesis and Antibacterial Activity of Aminoalkylamides of Eremomycin. Macroheterocycles 2020, 13, 298–304. [Google Scholar] [CrossRef]
- Wang, K.; Qian, X.; Cui, J. One step from nitro to oxime: A convenient preparation of unsaturated oximes by the reduction of the corresponding vinylnitro compounds. Tetrahedron 2009, 65, 10377–10382. [Google Scholar] [CrossRef]
- Izsépi, L.; Erdei, R.; Tevyashova, A.; Grammatikova, N.; Shchekotikhin, A.; Herczegh, P.; Batta, G. Bacterial Cell Wall Analogue Peptides Control the Oligomeric States and Activity of the Glycopeptide Antibiotic Eremomycin: Solution NMR and Antimicrobial Studies. Pharmaceuticals 2021, 14, 83. [Google Scholar] [CrossRef]
- Groves, P.; Searle, M.S.; Mackay, J.P.; Williams, D.H. The structure of an asymmetric dimer relevant to the mode of action of the glycopeptide antibiotics. Structure 1994, 2, 747–754. [Google Scholar] [CrossRef]
- Product Monograph–Vancomycin Hydrochloride for Injection, USP, 2017:21. Available online: https://www.fresenius-kabi.com/en-ca/documents/Vanco-for-Inj.-PM-ENG-v7.0-011218.pdf (accessed on 20 December 2017).
- Vancomycin Hydrochloride for Injection, USP, 2019:02. Available online: https://www.athenexpharma.com/wp-content/uploads/materials/SDS/Vancomycin_SDS.pdf (accessed on 18 March 2020).
- Clinical and Laboratory Standarts Institute M100 Performance Standards for Antimicrobial Susceptibility Testing. 2020. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 23 March 2021).
- Council of Europe European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Purposes. Strasbourg: 1986, 18.III.1986, Council of Europe, ETS No.123. Available online: https://rm.coe.int/168007a67b (accessed on 28 August 2018).
- Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes EN. Official Journal of the European Union, L 276/33-276/79 (20.10.2010). Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:EN:PDF (accessed on 27 April 2020).
- National State Standard GOST 33044-2014 the Russian Federation Standard “The Principles of Good Laboratory Practice” (Approved and Put into Effect by the Order of the Federal Agency for Technical Regulation and Metrology of October 20, 2014), No 1700. Available online: https://docs.cntd.ru/document/1200115791 (accessed on 1 August 2015). (In Russian).
- Behrens, B. Zur Auswertung der Digitalisblätter im Forschversuch. Arch. Exper. Path. Pharm. 1929, 140, 237. [Google Scholar] [CrossRef]
- Binder, M.D.; Hirokawa, N.; Windhorst, U. (Eds.) (CBAxC57BL/6)F1. In Encyclopedia of Neuroscience; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2008; p. 587. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound. | Dose, mg/kg | Survival, % | ED50, mg/kg |
|---|---|---|---|
| Eremomycin N-2-((2-fluorobenzyl)amino)ethyl) amide (4e) | 0.1 | 20 | 0.55 |
| 0.25 | 30 | ||
| 0.5 | 50 | ||
| 1 | 60 | ||
| 1.5 | 80 | ||
| 2.5 | 100 | ||
| Vancomycin (1) | 2.5 | 20 | 4.1 |
| 3.5 | 40 | ||
| 4.5 | 50 | ||
| 5.5 | 80 | ||
| 6.5 | 90 | ||
| 7.5 | 100 | ||
| Eremomycin (3) | 0.5 | 20 | 1.8 |
| 1.5 | 40 | ||
| 2.5 | 80 | ||
| 3.5 | 80 | ||
| 4.5 | 100 | ||
| 5.5 | 100 | ||
| Control dose S. aureus | - | 0 |
| Parameter | 4e |
|---|---|
| LD50 (mg/kg) | 175.5 (161.7 ÷ 189.8) * |
| MTD (LD10) (mg/kg) | 144.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moiseenko, E.I.; Erdei, R.; Grammatikova, N.E.; Mirchink, E.P.; Isakova, E.B.; Pereverzeva, E.R.; Batta, G.; Shchekotikhin, A.E. Aminoalkylamides of Eremomycin Exhibit an Improved Antibacterial Activity. Pharmaceuticals 2021, 14, 379. https://doi.org/10.3390/ph14040379
Moiseenko EI, Erdei R, Grammatikova NE, Mirchink EP, Isakova EB, Pereverzeva ER, Batta G, Shchekotikhin AE. Aminoalkylamides of Eremomycin Exhibit an Improved Antibacterial Activity. Pharmaceuticals. 2021; 14(4):379. https://doi.org/10.3390/ph14040379
Chicago/Turabian StyleMoiseenko, Elena I., Réka Erdei, Natalia E. Grammatikova, Elena P. Mirchink, Elena B. Isakova, Eleonora R. Pereverzeva, Gyula Batta, and Andrey E. Shchekotikhin. 2021. "Aminoalkylamides of Eremomycin Exhibit an Improved Antibacterial Activity" Pharmaceuticals 14, no. 4: 379. https://doi.org/10.3390/ph14040379
APA StyleMoiseenko, E. I., Erdei, R., Grammatikova, N. E., Mirchink, E. P., Isakova, E. B., Pereverzeva, E. R., Batta, G., & Shchekotikhin, A. E. (2021). Aminoalkylamides of Eremomycin Exhibit an Improved Antibacterial Activity. Pharmaceuticals, 14(4), 379. https://doi.org/10.3390/ph14040379