
The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug–Drug Interactions: Drug–Gene, Drug–Gene–Gene and Drug–Drug–Gene Interactions


Abstract
:1. Intoduction
2. Pharmacokinetic Drug–Drug Interactions
3. Drug–Gene Interactions
- (a)
- a change in the codon, which might change the amino acid that is transcribed;
- (b)
- a premature stop codon (no functional protein is formed);
- (c)
- different intron and exon splice junctions (no functional protein is formed);
- (d)
- an alteration in the stability of the mRNA (no proteins are formed);
- (e)
- a change in enhancer activity (gain of function);
- (f)
- or even no discernible consequence.
4. Genetic Polymorphisms of DME of Phase I Metabolism
5. DMEs of Phase 2 Metabolism
6. Drug Transporters (Phase 3)
7. Drug–Gene–Gene Interactions (DGGIs)
8. Drug–Drug–Gene Interactions (DDGIs) and Phenoconversion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lazarou, J.; Pomeranz, B.H.; Corey, P.N. Incidence of Adverse Drug Reactions in Hospitalized Patients: A Meta-Analysis of Prospective Studies. Surv. Anesthesiol. 1999, 43, 53–54. [Google Scholar] [CrossRef]
- Gurwitz, J.H.; Field, T.S.; Avorn, J.; McCormick, D.; Jain, S.; Eckler, M.; Benser, M.; Edmondson, A.C.; Bates, D.W. Incidence and preventability of adverse drug events in nursing homes. Am. J. Med. 2000, 109, 87–94. [Google Scholar] [CrossRef]
- Leone, R.; Magro, L.; Moretti, U.; Cutroneo, P.; Moschini, M.; Motola, D.; Tuccori, M.; Conforti, A. Identifying Adverse Drug Reactions Associated with Drug-Drug Interactions Data Mining of a Spontaneous Reporting Database in Italy. Drug Saf. 2010, 33, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Roll, S.C. Validierung von Interaktionsdatenbanken in der Psychopharmakotherapie. Der Nervenarzt 2017, 89, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Hahn, M.; Roll, S.C.; Stämpfli, D. Psychiatrische Patienten fragen ihren Arzt und Apotheker- wie kann die ambulante Versrgung optimiert werden? Psychopharmakotherapie 2020, 27, 270–277. [Google Scholar]
- Ramsey, L.B.; Ong, H.H.; Schildcrout, J.S.; Shi, Y.; Tang, L.A.; Hicks, J.K.; El Rouby, N.; Cavallari, L.H.; Tuteja, S.; Aquilante, C.L.; et al. Prescribing Prevalence of Medications With Potential Genotype-Guided Dosing in Pediatric Patients. JAMA Netw. Open 2020, 3, e2029411. [Google Scholar] [CrossRef]
- Turner, R.M.; De Koning, E.M.; Fontana, V.; Thompson, A.; Pirmohamed, M. Multimorbidity, polypharmacy, and drug-drug-gene interactions following a non-ST elevation acute coronary syndrome: Analysis of a multicentre observational study. BMC Med. 2020, 18, 367. [Google Scholar] [CrossRef]
- Abdullah-Koolmees, H.; van Keulen, A.M.; Nijenhuis, M.; Deneer, V.H.M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front. Pharmacol. 2021, 11. [Google Scholar] [CrossRef]
- De Leon, J. The effects of antiepileptic inducers in neuropsychopharmacology, a neglected issue. Part I: A summary of the current state for clinicians. Rev. Psiquiatr. Salud. Ment. 2015, 8, 97–115. [Google Scholar] [CrossRef]
- Fritz, D.; Ceschi, A.; Curkovic, I.; Huber, M.; Egbring, M.; Kullak-Ublick, G.A.; Russmann, S. Comparative evaluation of three clinical decision support systems: Prospective screening for medication errors in 100 medical inpatients. Eur. J. Clin. Pharmacol. 2012, 68, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Haueis, P.; Greil, W.; Huber, M.; Grohmann, R.; Kullak-Ublick, G.A.; Russmann, S. Evaluation of Drug Interactions in a Large Sample of Psychiatric Inpatients: A Data Interface for Mass Analysis With Clinical Decision Support Software. Clin. Pharmacol. Ther. 2011, 90, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedna, K.; Andersson, M.L.; Gyllensten, H.; Hägg, S.; Böttiger, Y. Clinical relevance of alerts from a decision support system, PHARAO, for drug safety assessment in the older adults. BMC Geriatr. 2019, 19, 164. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Last Updated 3/10/2020. 2019. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 28 April 2021).
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polasek, T.M.; Lin, F.P.Y.; Miners, J.O.; Doogue, M.P. Perpetrators of pharmacokinetic drug-drug interactions arising from altered cytochrome P450 activity: A criteria-based assessment. Br. J. Clin. Pharmacol. 2011, 71, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Guttmacher, A.E.; Collins, F.S. Genomic Medicine—A Primer. N. Engl. J. Med. 2002, 347, 1512–1520. [Google Scholar] [CrossRef]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP 2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.; Tybring, G.; Dahl, M.J. Impact of Cytochrome P450 2C19 Polymorphisms on Citalopram/Escitalopram Exposure: A Systematic Review an Meta-Analysis. Clin. Pharmakokinet. 2014, 53, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.; Müller, D.J.; Roll, S.C. Frequencies of Genetic Polymorphisms of Clinically Relevant Gene-Drug Pairs in a German Psychiatric Inpatient Population. Pharmacopsychiatry 2021, 54, 81–89. [Google Scholar] [CrossRef]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; Müller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Gronich, N.; Lavi, I.; Lejbkowicz, F.; Pinchev, M.; Zoabi, Y.; Auriel, E.; Saliba, W.; Rennert, G. Ischemic stroke and myocardial ischemia in clopidogrel users and the association with CYP2C19 loss-of-function homozygocity: A real-world study. Pharmacogenom. J. 2021, 1–7. [Google Scholar] [CrossRef]
- He, W.; Grassmann, F.; Eriksson, M.; Eliasson, E.; Margolin, S.; Thorén, L.; Hall, P.; Czene, K. CYP2D6 Genotype Predicts Tamoxifen Discontinuation and Prognosis in Patients With Breast Cancer. J. Clin. Oncol. 2020, 38, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Cihlar, T.; Robinson, K.L.; Tong, L.; Vela, J.E.; Fuller, M.D.; Wieman, L.M.; Eisenberg, E.J.; Rhodes, G.R. Mechanism of Active Renal Tubular Efflux of Tenofovir. Antimicrob. Agents Chemother. 2006, 50, 3297–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, P.; Brodie, M.J. Potential Role of Drug Transporters in the Pathogenesis of Medically Intractable Epilepsy. Epilepsia 2005, 46, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Van Schaik, R.; Edelbroek, P.M.; Redeker, S.; Aronica, E.; Wadman, W.J.; Marchi, N.; Vezzani, A.; Gorter, J.A. Inhibition of the Multidrug Transporter P-Glycoprotein Improves Seizure Control in Phenytoin-treated Chronic Epileptic Rats. Epilepsia 2006, 47, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Belmonte, C.; Román, M.; Ochoa, D.; Jiang-Zheng, C.; Koller, D.; Mejía, G.; Zubiaur, P.; Wojnicz, A.; Abad-Santos, F. Effect of ABCB1 C3435T Polymorphism on Pharmacokinetics of Antipsychotics and Antidepressants. Basic Clin. Pharmacol. Toxicol. 2018, 123, 474–485. [Google Scholar] [CrossRef]
- Krajcsi, P. Drug-transporter interaction testing in drug discovery and development. World J. Pharmacol. 2013, 2, 35. [Google Scholar] [CrossRef]
- Rosenhagen, M.C. The Clinical Impact of ABCB1 Polymorphisms on the Treatment of Psychiatric Diseases. Curr. Pharm. Des. 2011, 17, 2843–2851. [Google Scholar] [CrossRef]
- Samwald, M.; Xu, H.; Blagec, K.; Empey, P.E.; Malone, D.C.; Ahmed, S.M.; Ryan, P.; Hofer, S.; Boyce, R.D. Incidence of exposure of patients in the United States to multiple drugs for which pharmacogenomic guidelines are available. PLoS ONE 2016, 11, e0164972. [Google Scholar] [CrossRef] [Green Version]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT 15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- DeGorter, M.K.; Tirona, R.G.; Schwarz, U.I.; Choi, Y.-H.; Dresser, G.K.; Suskin, N.; Myers, K.; Zou, G.; Iwuchukwu, O.; Wei, W.-Q.; et al. Clinical and Pharmacogenetic Predictors of Circulating Atorvastatin and Rosuvastatin Concentrations in Routine Clinical Care. Circ. Cardiovasc. Genet. 2013, 6, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, S.; Kirkpatrick, C.M.J.; Byron, K.; Sheffield, L. An analysis of allele, genotype and phenotype frequencies, actionable pharmacogenomic (PGx) variants and phenoconversion in 5408 Australian patients genotyped for CYP2D6, CYP2C19, CYP2C9 and VKORC1 genes. J. Neural Transm. 2018, 126, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Drevin, G.; Picard, N.; Jousset, N.; Briet, M.; Abbara, C. Pitfalls and challenges associated with phenoconversion in forensic toxcicology. Forensic Sci. Int. Genet. 2021, 51, 102433. [Google Scholar] [CrossRef] [PubMed]
- Hefner, G.; Shams, M.E.E.; Unterecker, S.; Falter, T.; Hiemke, C. Inflammation and psychotropic drugs: The relationship between C-reactive protein and antipsychotic drug levels. Psychopharmacol. 2016, 233, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Smith, R.L. Addressing phenoconversion: The Achilles’ heel of personalized medicine. Br. J. Clin. Pharmacol. 2015, 79, 222–240. [Google Scholar] [CrossRef] [Green Version]
- Bahar, M.A.; Lanting, P.; Bos, J.H.J.; Sijmons, R.H.; Hak, E.; Wilffert, B. Impact of Drug-Gene-Interaction, Drug-Drug-Interaction, and Drug-Drug-Gene-Interaction on (es)Citalopram Therapy: The PharmLines Initiative. J. Pers. Med. 2020, 10, 256. [Google Scholar] [CrossRef]
- Storelli, F.; Matthey, A.; Lenglet, S.; Thomas, A.; Desmeules, J.; Daali, Y. Impact of CYP2D6 Functional Allelic Variations on Phenoconversion and Drug-Drug Interactions. Clin. Pharmacol. Ther. 2017, 104, 148–157. [Google Scholar] [CrossRef]
- Verbeurgt, P.; Mamiya, T.; Oesterheld, J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics 2014, 15, 655–665. [Google Scholar] [CrossRef]
- Hefner, G.; Wolff, J.; Hahn, M.; Hiemke, C.; Toto, S.; Roll, S.C.; Messer, T.; Klimke, A. Prevalence and sort of pharmacokinetic drug–drug interactions in hospitalized psychiatric patients. J. Neural Transm. 2020, 127, 1185–1198. [Google Scholar] [CrossRef]
- Monte, A.A.; West, K.; McDaniel, K.T.; Flaten, H.K.; Saben, J.; Shelton, S.; Abdelmawla, F.; Bushman, L.R.; Williamson, K.; Abbott, D.; et al. CYP2D6 Genotype Phenotype Discordance Due to Drug-Drug Interaction. Clin. Pharmacol. Ther. 2018, 104, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Klieber, M.; Oberacher, H.; Hofstaetter, S.; Beer, B.; Neururer, M.; Amann, A.; Alber, H.; Modak, A. CYP2C19 Phenoconversion by Routinely Prescribed Proton Pump Inhibitors Omeprazole and Esomeprazole: Clinical Implications for Personalized Medicines. J. Pharmacol. Exp. Ther. 2015, 354, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klomp, S.D.; Manson, M.L.; Guchelaar, H.-J.; Swen, J.J. Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review. J. Clin. Med. 2020, 9, 2890. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wouden, C.H.; Van Rhenen, M.H.; Jama, W.O.; Ingelman-Sundberg, M.; Lauschke, V.M.; Konta, L.; Schwab, M.; Swen, J.J.; Guchelaar, H. Development of the PG x-Passport: A Panel of Actionable Germline Genetic Variants for Pre-Emptive Pharmacogenetic Testing. Clin. Pharmacol. Ther. 2019, 106, 866–873. [Google Scholar] [CrossRef] [PubMed]
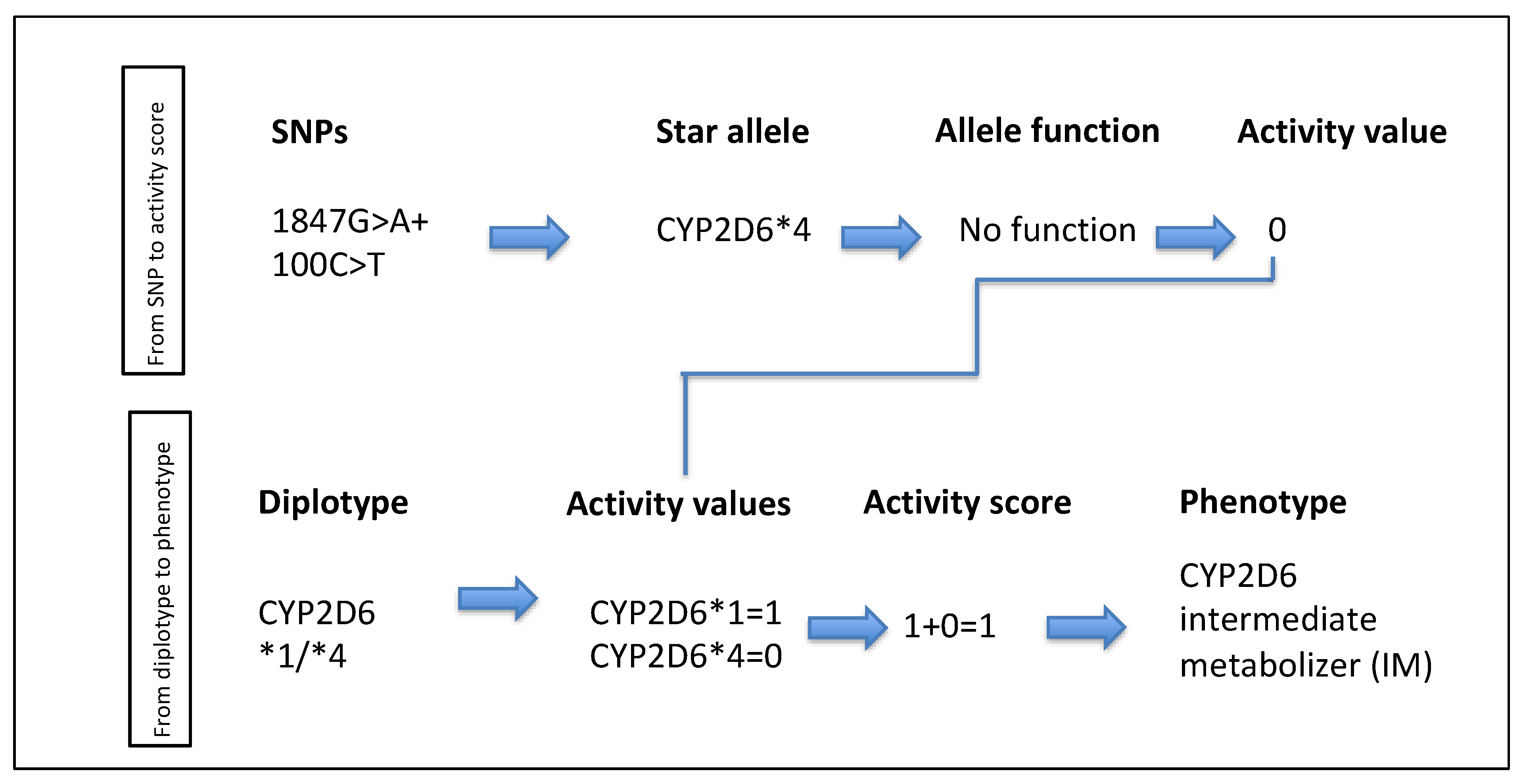


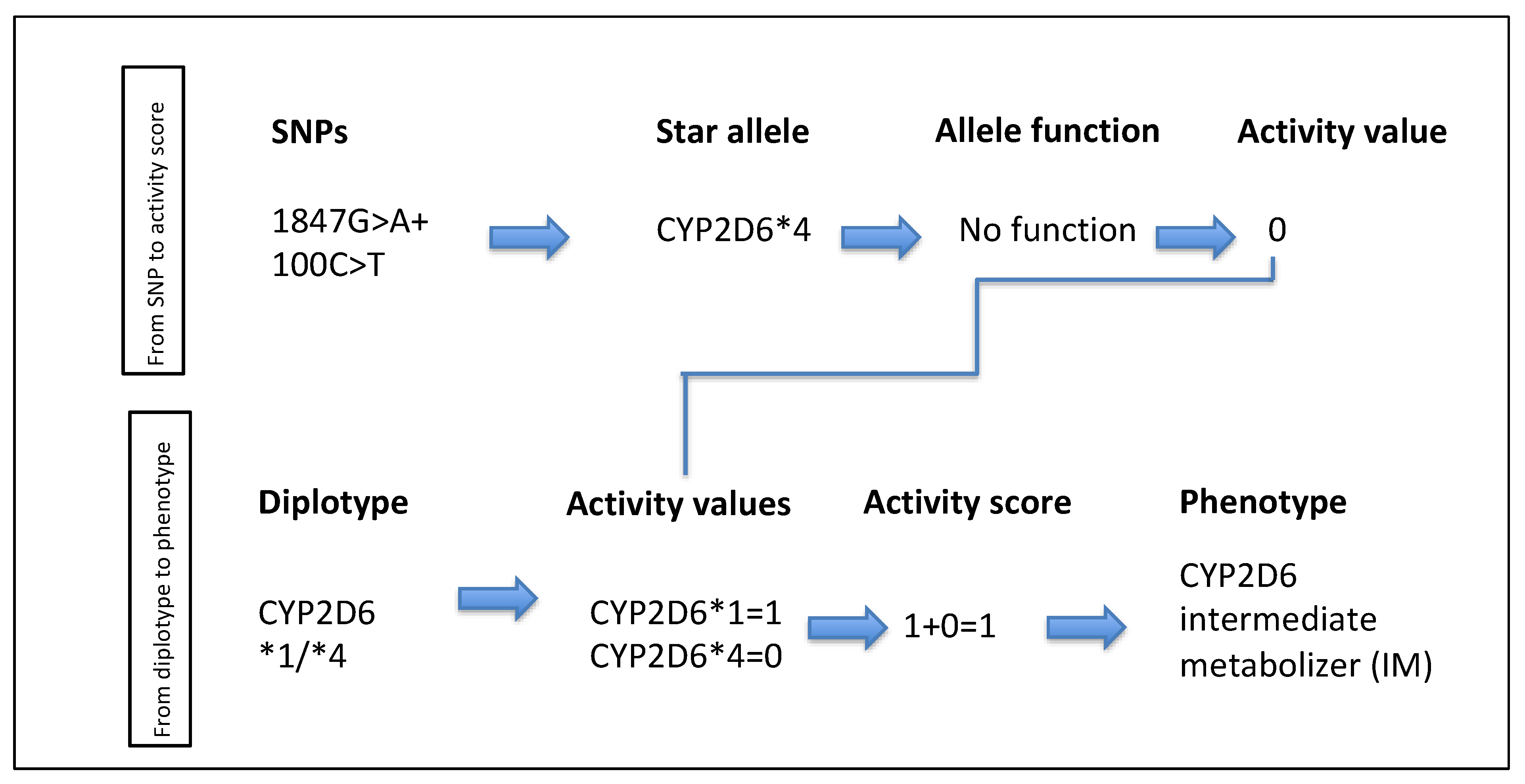{kind=link}
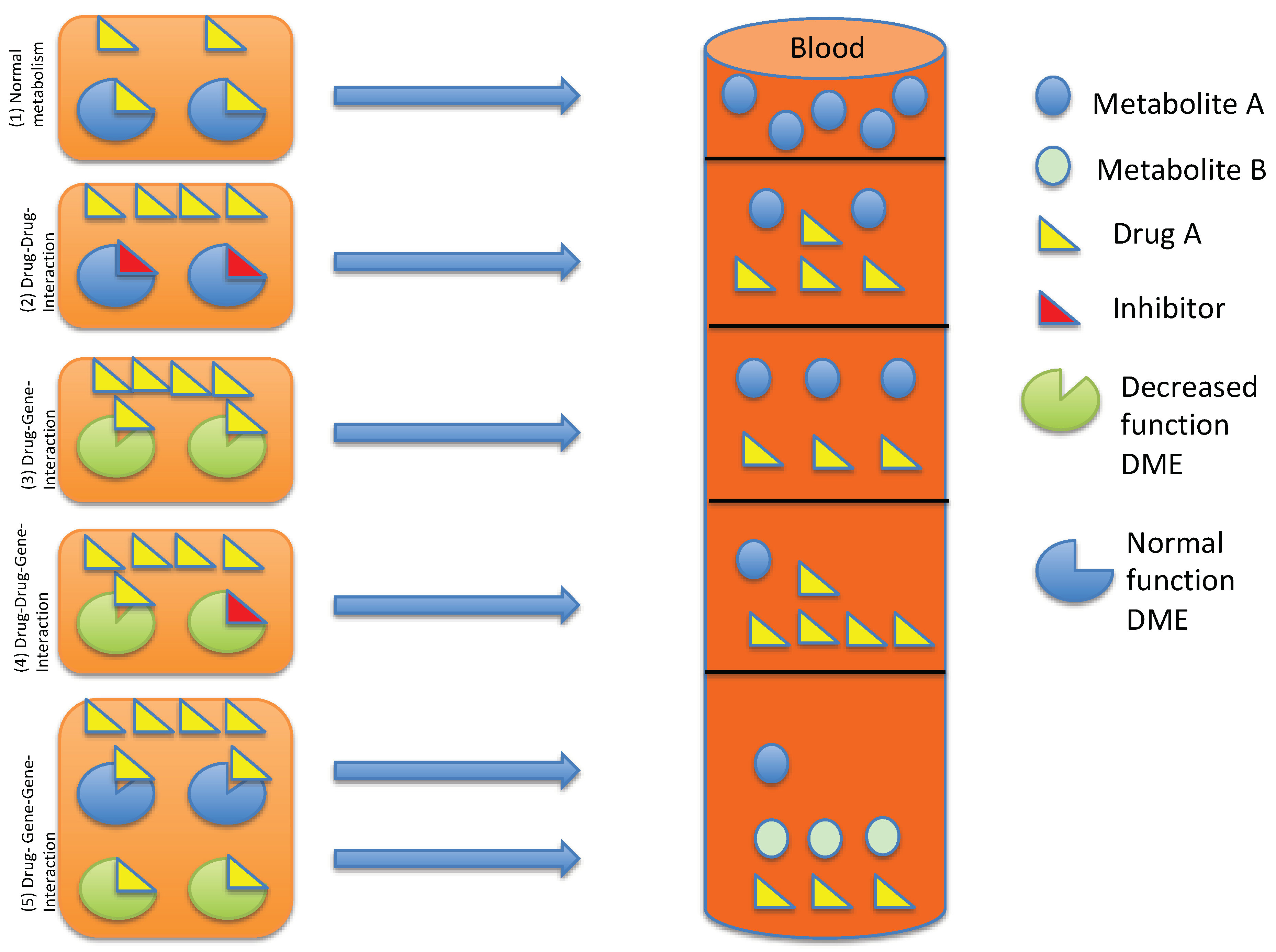{kind=link}
| Term | Definition |
|---|---|
| Drug–Drug Interaction | When a drug in the individual’s regimen affects that individual’s ability to clear another drug. |
| Drug–Gene Interaction | When an individual’s genetic phenotype affects that patient’s ability to clear a drug. |
| Drug–Drug–Gene Interaction | When the individual’s genetic AND another drug in the individual’s regimen affects that individual’s ability to clear a drug. |
| Phenoconversion | Mismatch between the individual’s genotype- based prediction of drug metabolism and true capacity to metabolize drugs due to non-genetic factos (e.g., inflamation, pregnancy, liver failure, GFR, smoking, gender, and comedication). |
| Drug–Gene–Gene Interaction | Mismatch between the expected capacity to metabolize a drug that is caused by a second metabolizing (alternative pathway) enzyme’s genotype. |
| Victim Drug | Substrate of drug-metabolizing enzymes that are induced or inhibited in combination with a perpetrator drug (inhibitor or inducer). The serum levels of the vitim drug changes by this Drug–Drug-Interaction. |
| Perpetrator Drug | Inhibitor or inducer of drug-metabolizing enzymes that increases or decreases the serum levels of the victim drug. The serum level of the perpetrator drug does not change. |
| CYP | Known Phenotypes | Substrates | Phenoconversion |
|---|---|---|---|
| 1A2 | increased funtion normal function unknown function | duloxetine, olanzapin, clozapine, theophyllin, caffeine | fluvoxamine, ciprofloxacine, enoxacine, smoking |
| 2A6 | PM, IM, NM, UM | nicotine | |
| 2B6 | NM, IM, PM, RM, UM | bupropion, cyclophospamide, efavirenz, methadone | clopidogrel, ticlopidine, tenofovir, voriconazole, carbamazepine, efavirenz, rifampin |
| 2C8 | increased function normal function decreased function | glitazones, paclitaxel | gemfibrozil, clopidogrel, teriflunomide, trimethoprim, rifampin, St. John‘s wort |
| 2C9 | NM, IM, PM | losartan, NSAIDs, phenytoin, warfarin, glyburide | amiodarone, fluconazole, miconazole, rifampin |
| 2C19 | NM, IM, PM, RM, UM | clopidogrel, diazepam, proton pump inhibitors (PPI) | fluvoxamine, fluoxetine, fluconazole, omeprazole, ticlopidine, rifampin |
| 2D6 | NM, IM, PM, UM | antidepressants, betablockers, codeine, tramadol, tamoxifen, hydrocodone | bupropion, cimetidine, duloxetine, fluvoxamine, fluoxetine, paroxetine, quinidine, Note: there are no known inducers of CYP2D6. |
| 3A4 | normal function, decreased function, increased function | calcium channel blockers, macrolides, protease inhibitors, statins | azole antimycotics, boceprevir, cobicistat, danoprevir, grapefruit, ritonavir, telaprevir, verapamil, carbamazepine, phenobarbital, phenytoin, rifampin, St. John’s wort |
| 3A5 | NM, IM, PM Note: activity has major influence on CYP3A4 activity, if *1 is present | Tacrolimus, quetiapine | Ciprofloxacin, erythromycin, diltiazem, ketoconazole, verapamil |
| Activity Score | Alleles (Examples) | Type of Allele and Genotype |
|---|---|---|
| >2.25 | *1/*1 × N, *1/*2 × N b*2 a/*2 × N b, *1 × 2/*9 | Increased activity, Ultra rapid metabolizer |
| ≤2.25 to ≥1.25 | *1/*10, *1/*41, *1/*9, *1/*1, *1/*2, *2 × 2/*10 | Wild-type, Normal metabolizer |
| >0 to <1.25 | *4/*10, *4/*41, *10/*10, *10/*41, *41/*41, *1/*5 | Reduced function, Intermediate metabolizer |
| 0 | *3/*4,*4/*4,*5/*5,*5/*6 | Non-functional, Poor metabolizer |
| Enzyme | Known Phenotypes | Substrates | Phenoconversion |
|---|---|---|---|
| UGT1A1 | NM, IM, PM | bilirubin, irinotecan, estradiol | Atazanavir, carbamazepine, phenytoin, phenobarbital, rifampicin, ritonavir, lamotrigin, efavirenz, tyrosine-kinase inhibitors |
| UGT1A4 | Normal function, increased function, decreased function | valproic acid, lamotrigine, allopurinol, febuxostat, tamoxifen, clozapine, anastrozole | methylene blue, ertugliflozin, carbamazepine, phenytoin |
| UGT1A6 | n.a. | allopurinol, febuxostat, methothrexat, valproic acid | troglitazone, fosphenytoin, phenytoin, carbamazepine |
| UGT1A9 | n.a. | allopurinol, febuxostat, methothrexat, valproic acid | vandetanib |
| UGT2B7 | n.a. | zodovudine, oxycodone, efavirenz, methadone, lamotrigine, morphine, codeine, fentanyl. | flunitrazepam, ketoconazole, umifenovir, phenobarbital, mefenamic acid |
| UGT2B15 | normal function decreased funtion | oxazepam, lorazepam | |
| N-acetyltransferase (NAT2) | fast slow | isoniazid, hydralazine, dapsone, caffein, procainamide | |
| Thiopurine Methyl Transferase (TPMT) | NM, IM, possibly intermediate, PM | thiopurines | allopurinol |
| Nudix hydrolase 15 (NUDT 15) | NM, IM, possibly intermediate, PM | thiopurines |
| Gene/Transporter | Known Phenotypes | Substrates | Phenoconversion |
|---|---|---|---|
| OATP1B1/SLCO1B1 gene | normal function, decreased function, poor function | atorvastatin, repaglinide, enalapril, methotrexate, rosuvastatin, simvastatin, eryhtromycin, nateglinide, pitavastatin, pravastatin, lopinavir | astemizole, diazepam, nifedipine |
| BCRP/ABCG2 gene | Normal function, decreased function | allopurinol, asuvastatin, leflunomide, sunitinib, topotecan, pitavastatin, rosuvastatin, sulfasalazine | curcumine, elacridar, cyclosporine A |
| P-glycoprotein/ABCB1/MDR1 gene | normal function, increased function | colchicine, fexofenadine, simvastatin, rifampin, cyclosporine, ondansetron, risperidone, digoxin, fentanyl, methadone, oxycodone, tramadole, phenytoin | amiodarone, carvedilol, clarithromycin, quinidine, verapamil, ritonavir, telaprevir, carbamazepine, St. John’s wort, primidone, rifampin, phenytoin |
| Activity Score CYP2D6 | Genetic Phenotype | Weak Inhibitor and Moderate Inhibitor | Strong Inhibitor |
|---|---|---|---|
| 0 | PM | Activity score × 0.5 = PM | Activity score × 0 = PM |
| > 0 < 1.25 | IM | Activity score × 0.5 = IM | Activity score × 0 = PM |
| > 1.25 < 2.25 | NM | Activity score × 0.5 = IM | Activity score × 0 = PM |
| >2.25 | UM | Activity score × 0.5 = NM | Activity score × 0 = PM |
| Genetic Phenotype CYP2C19 | Comedication of a Moderate or Strong Inhibitor; Predicted Phenotype |
|---|---|
| NM, IM | PM |
| RM, UM | IM |
| PM | PM |
| Comedication of a moderate or strong inducer; Predicted phenotype | |
| NM, RM | UM |
| IM | NM |
| PM | PM |
| UM | UM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, M.; Roll, S.C. The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug–Drug Interactions: Drug–Gene, Drug–Gene–Gene and Drug–Drug–Gene Interactions. Pharmaceuticals 2021, 14, 487. https://doi.org/10.3390/ph14050487
Hahn M, Roll SC. The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug–Drug Interactions: Drug–Gene, Drug–Gene–Gene and Drug–Drug–Gene Interactions. Pharmaceuticals. 2021; 14(5):487. https://doi.org/10.3390/ph14050487
Chicago/Turabian StyleHahn, Martina, and Sibylle C. Roll. 2021. "The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug–Drug Interactions: Drug–Gene, Drug–Gene–Gene and Drug–Drug–Gene Interactions" Pharmaceuticals 14, no. 5: 487. https://doi.org/10.3390/ph14050487
APA StyleHahn, M., & Roll, S. C. (2021). The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug–Drug Interactions: Drug–Gene, Drug–Gene–Gene and Drug–Drug–Gene Interactions. Pharmaceuticals, 14(5), 487. https://doi.org/10.3390/ph14050487