
Myeloperoxidase Inhibitory and Antioxidant Activities of (E)-2-Hydroxy-α-aminocinnamic Acids Obtained through Microwave-Assisted Synthesis

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
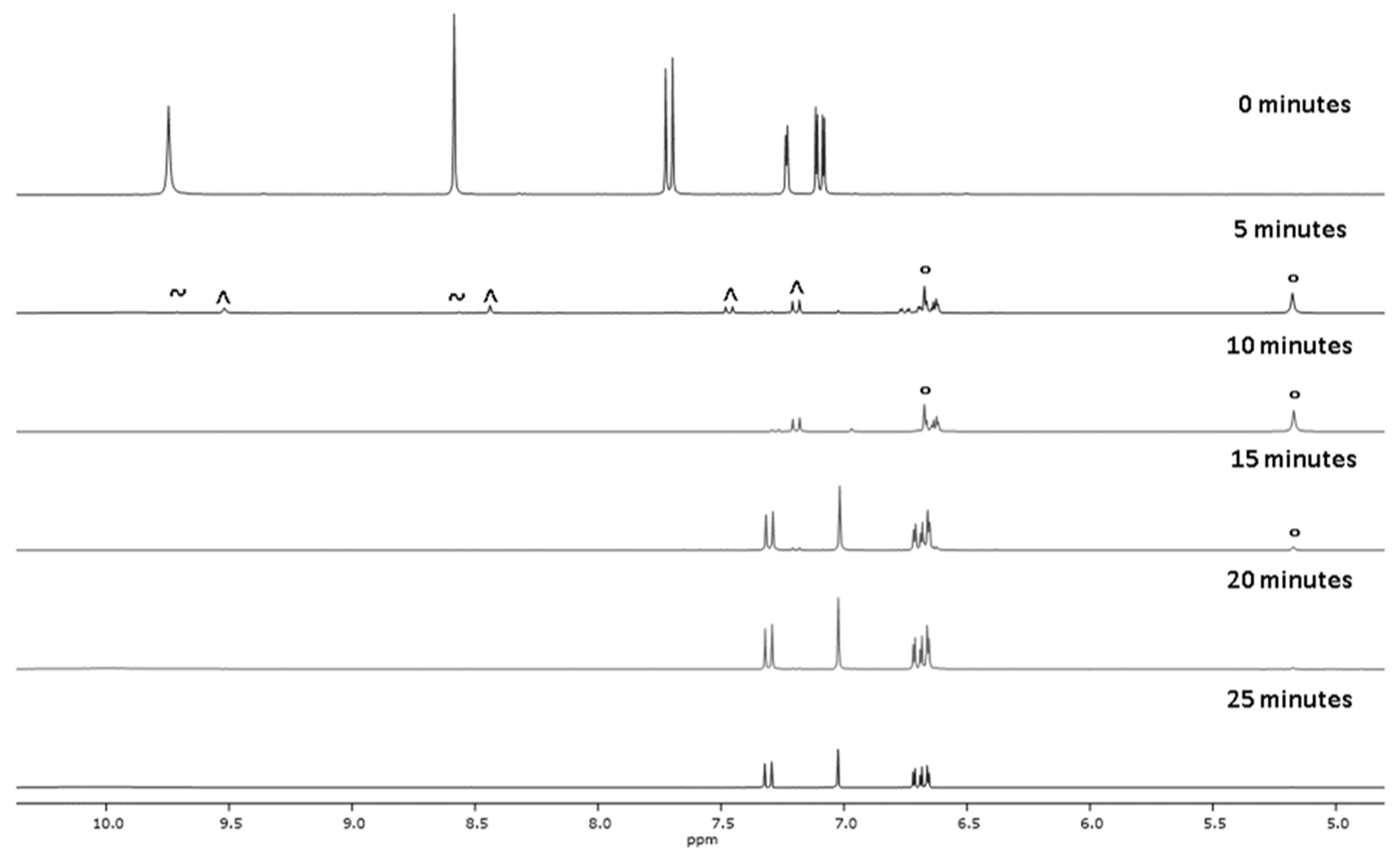
2.1. Chemistry
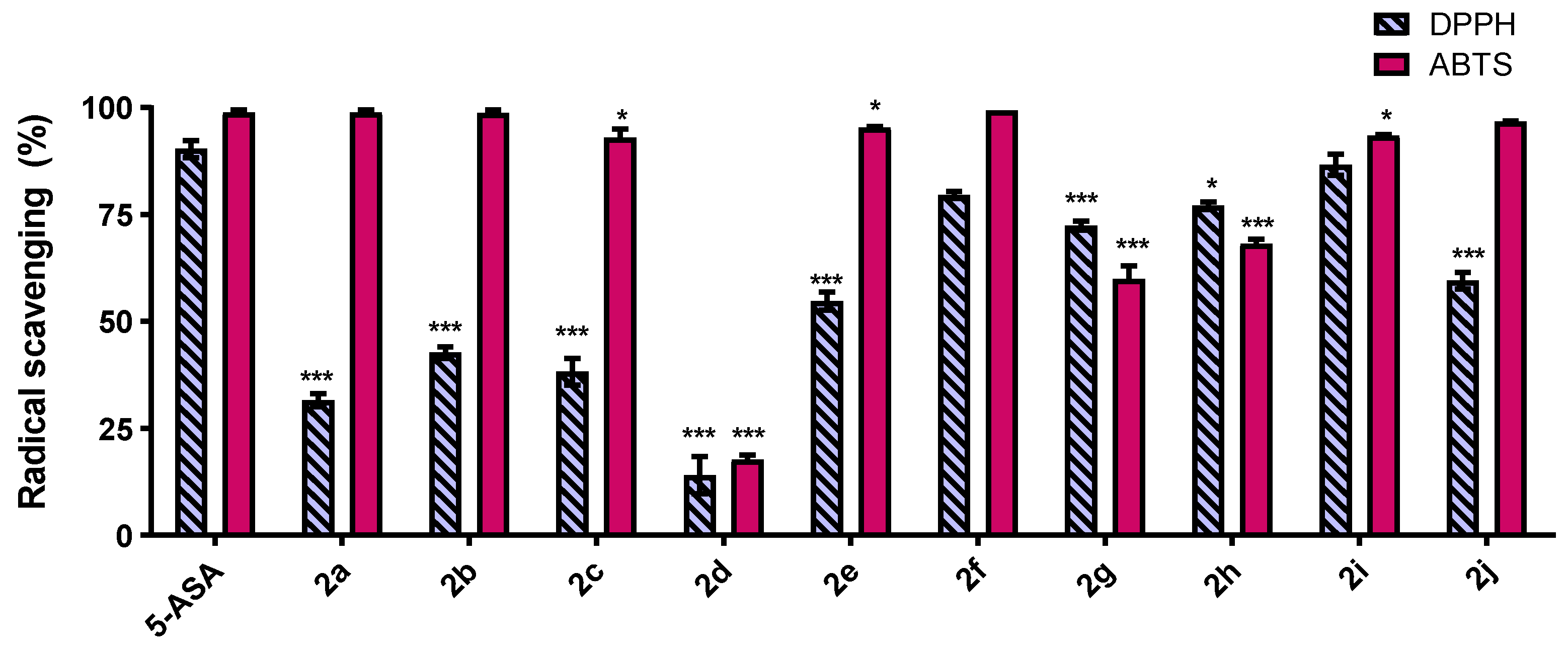
2.2. DPPH and ABTS Free Radical-Scavenging Activity
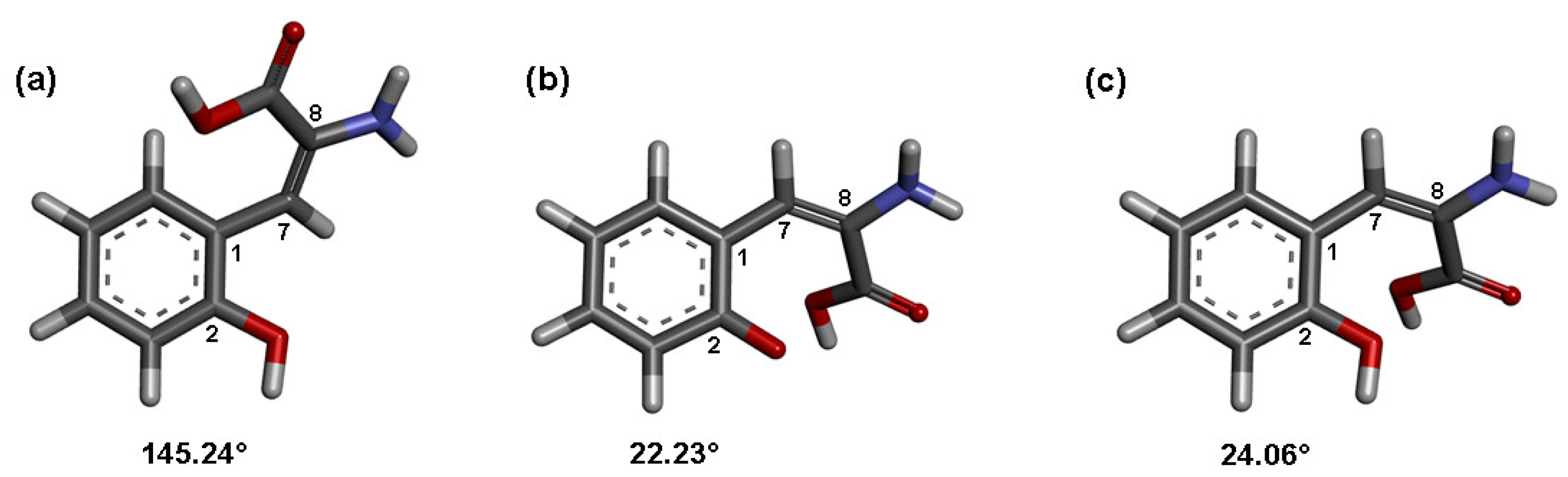
2.3. Molecular Orbital Calculations and Antioxidant Mechanism
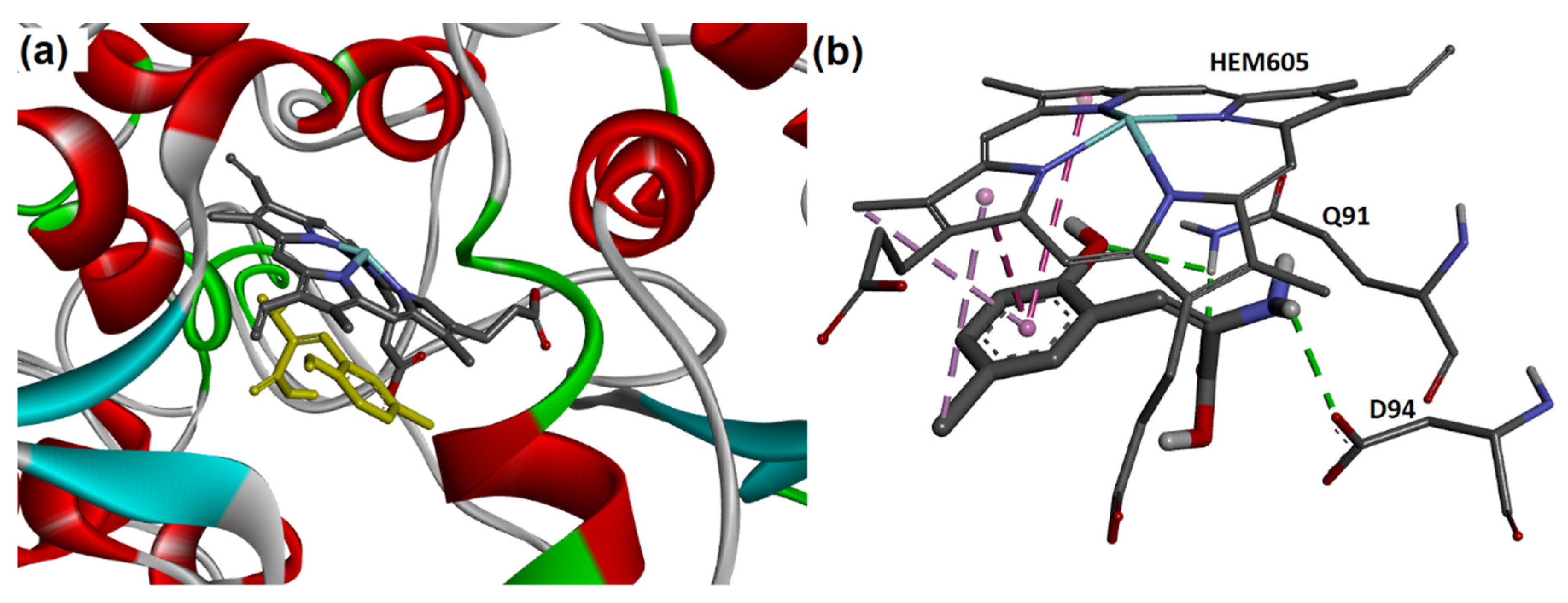
2.4. Docking of 2a–j with MPO
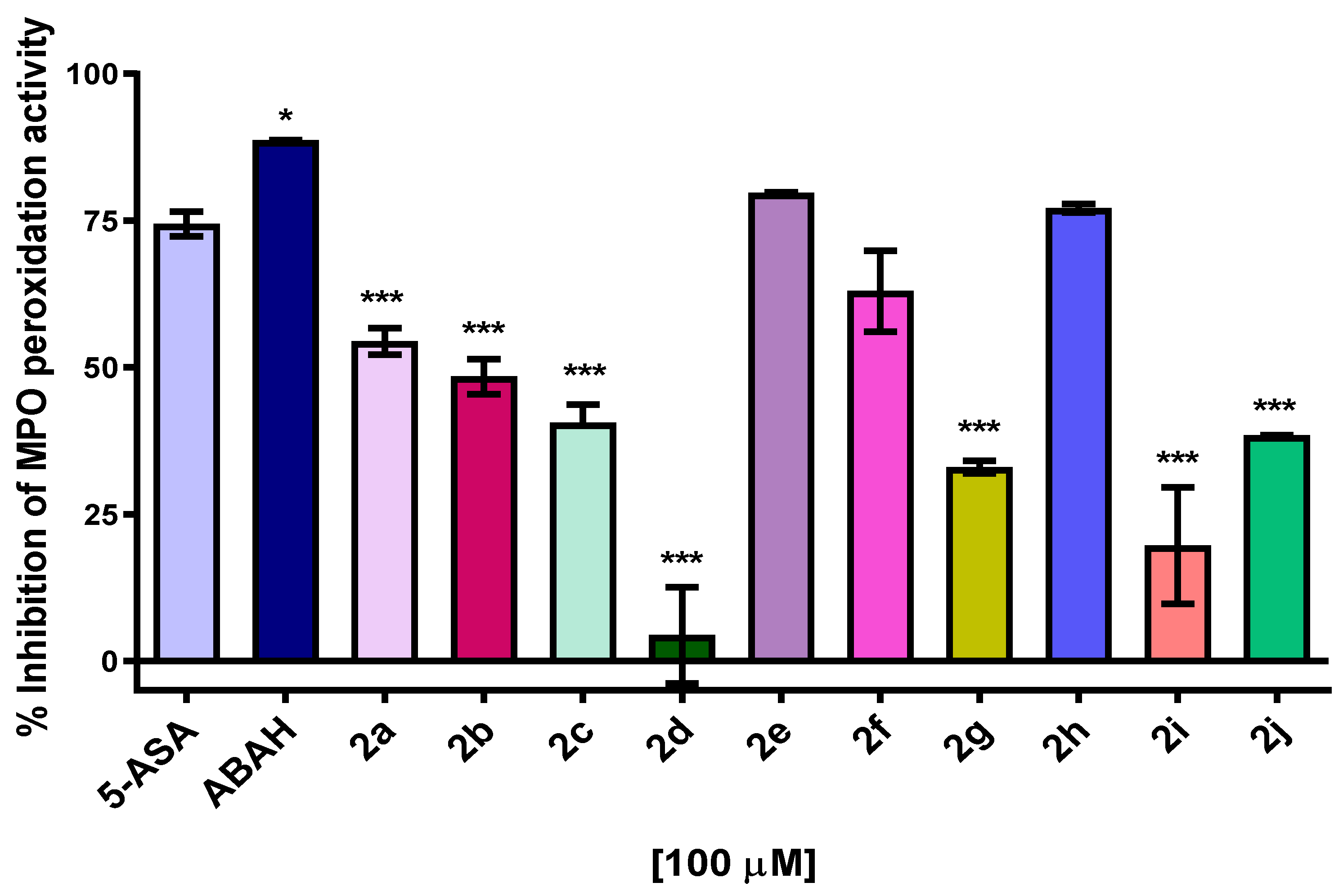
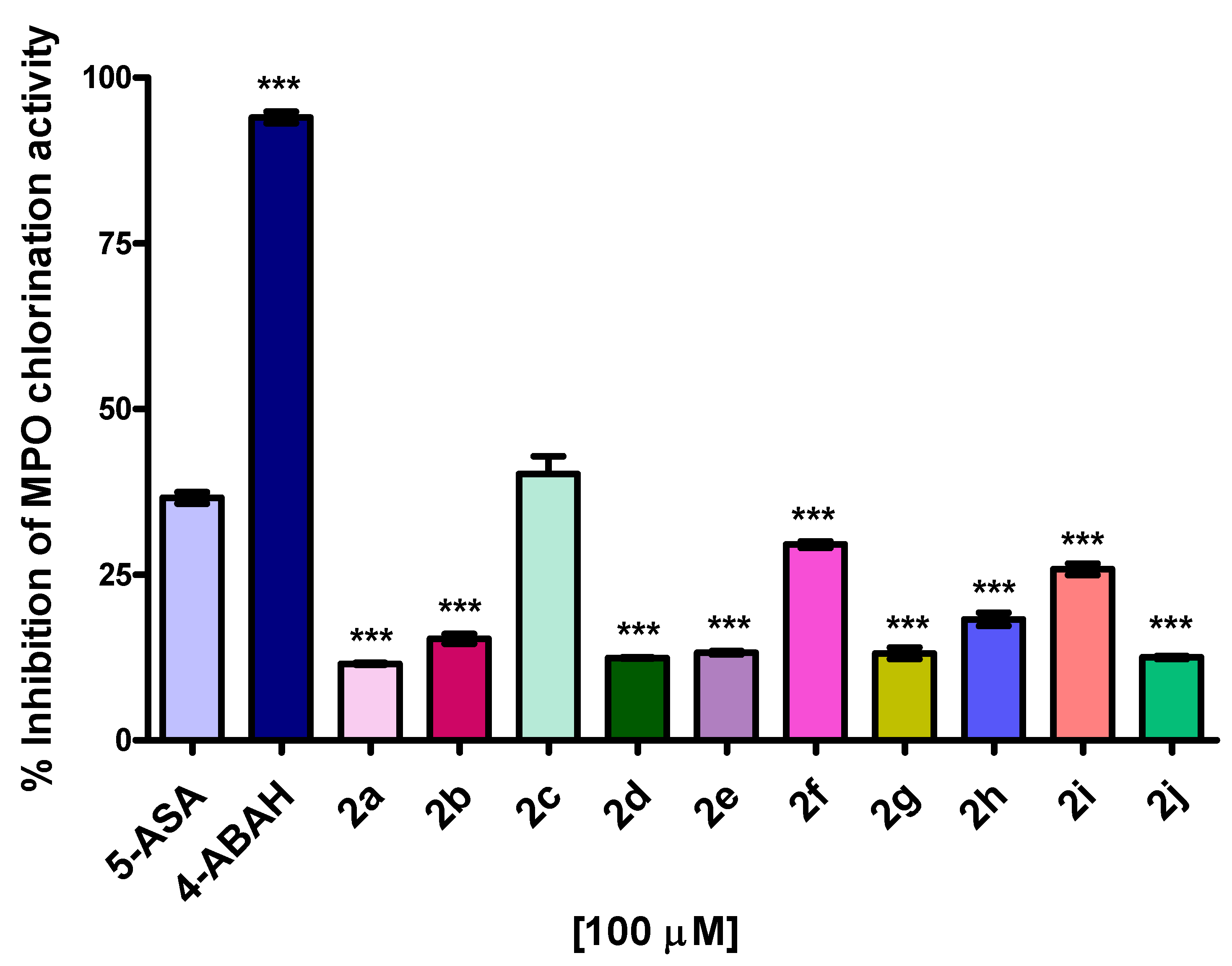
2.5. MPO Enzymatic Activity of Peroxidation and Chlorination
2.6. Cell Viability
3. Materials and Methods
3.1. Synthetic Procedures and Characterization of Compounds 2a–j
3.2. DPPH Assay (2,2-Diphenyl-1-picrylhydrazyl)
3.3. ABTS Test (2,2-Azino-bis(3-ethylbenzothiazolin)-6-sulfonic Acid)
3.4. MPO Enzymatic Activity of Peroxidation
3.5. MPO Enzymatic Activity of Chlorination
3.6. Cell Viability
3.7. Docking MPO
3.8. Molecular Orbital Calculations
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Nybo, T.; Cai, H.; Chuang, C.Y.; Gamon, L.F.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of human plasma fibronectin by myeloperoxidase-derived oxidants, and its consequences for smooth muscle cell function. Redox Biol. 2018, 19, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Gamon, L.F.; Dieterich, S.; Ignasiak, M.T.; Schrameyer, V.; Davies, M.J. Iodide modulates protein damage induced by the inflammation-associated heme enzyme myeloperoxidase. Redox Biol. 2020, 28, 101331. [Google Scholar] [CrossRef] [PubMed]
- Khalilova, I.S.; Dickerhof, N.; Mocatta, T.J.; Bhagra, C.J.; McClean, D.R.; Obinger, C.; Kettle, A.J. A myeloperoxidase precursor, pro-myeloperoxidase, is present in human plasma and elevated in cardiovascular disease patients. PLoS ONE 2018, 13, e0192952. [Google Scholar] [CrossRef] [PubMed]
- Mariani, F.; Roncucci, L. Role of the Vanins–Myeloperoxidase Axis in Colorectal Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellhaar, S.; Sunnemark, D.; Eriksson, H.; Olson, L.; Galter, D. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson’s and Alzheimer’s disease. Cell Tissue Res. 2017, 369, 445–454. [Google Scholar] [CrossRef]
- Yan, Y.; Ren, F.; Wang, P.; Sun, Y.; Xing, J. Synthesis and evaluation of a prodrug of 5-aminosalicylic acid for the treatment of ulcerative colitis. Iran. J. Basic Med. Sci. 2019, 22, 1452–1461. [Google Scholar]
- Forbes, L.V.; Sjögren, T.; Auchère, F.; Jenkins, D.W.; Thong, B.; Laughton, D.; Hemsley, P.; Pairaudeau, G.; Turner, R.; Eriksson, H.; et al. Potent Reversible Inhibition of Myeloperoxidase by Aromatic Hydroxamates. J. Biol. Chem. 2013, 288, 36636–36647. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Pérez, L.C.; Mendieta-Wejebe, J.E.; Hernández, R.M.; Fragoso, M.J.; Salazar, J.R.; Correa-Basurto, J.; Pa-dilla-Martínez, I.I.; Rosales-Hernández, M.C. Novel 5-aminosalicylic derivatives as anti-inflammatories and myeloperoxidase inhibitors evaluated in silico, in vitro and ex vivo. Arab. J. Chem. 2019, 12, 5278–5291. [Google Scholar] [CrossRef]
- Van der Veen, B.S.; de Winther, M.P.J.; Heeringa, P. Myeloperoxidase: Molecular Mechanisms of Action and Their Relevance to Human Health and Disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef]
- Huang, J.; Smith, F.; Panizzi, J.R.; Goodwin, D.C.; Panizzi, P. Inactivation of myeloperoxidase by benzoic acid hydrazide. Arch. Biochem. Biophys. 2015, 570, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poljšak, B.; Fink, R. The Protective Role of Antioxidants in the Defence against ROS/RNS-Mediated Environmental Pollution. Oxid. Med. Cell Longev. 2014, 2014, 671539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peperidou, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Voulgari, E.; Avgoustakis, K. Multifunctional Cinnamic Acid Derivatives. Molecules 2017, 22, 1247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, J.; Luan, G.; Zhang, S.; Zhuoma, Y.; Xie, J.; Zhou, W. Quantitative Analyses of Nine Phenolic Compounds and Their Antioxidant Activities from Thirty-Seven Varieties of Raspberry Grown in the Qinghai-Tibetan Plateau Region. Molecules 2019, 24, 3932. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.T.; Bento, C.M.; Pena, A.C.; Figueiredo, L.M.; Prudêncio, C.; Aguiar, L.; Silva, T.; Ferraz, R.; Gomes, M.S.; Teixeira, C.; et al. Cinnamic Acid Conjugates in the Rescuing and Repurposing of Classical Antimalarial Drugs. Molecules 2019, 25, 66. [Google Scholar] [CrossRef] [Green Version]
- Leite-Diniz, L.R.; De Santana-Souza, M.T.; Nascimento-Barboza, J.; Nóbrega-de Almeida, R.; Pergentino de Sousa, D. An-tidepressant Potential of Cinnamic Acids: Mechanisms of Action and Perspectives in Drug Development. Molecules 2019, 24, 4469. [Google Scholar] [CrossRef] [Green Version]
- Adisakwattana, S. Cinnamic Acid and Its Derivatives: Mechanisms for Prevention and Management of Diabetes and Its Complications. Nutrients 2017, 9, 163. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Gaspar, A.; Garrido, E.M.; Garrido, J.; Borges, F. Hydroxycinnamic Acid Antioxidants: An Electrochemical Overview. BioMed Res. Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Vega, M.R.G.; De Carvalho, M.G.; Vieira, I.J.C.; Filho, R.B. Chemical constituents from the Paraguayan medicinal plant, Eupatorium macrocephalum Less. J. Nat. Med. 2007, 62, 122–123. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, S.; Long, L. Two (2)-4-hydroxycinnamic acid glycosides and their application. China’s Patent CN 1515580 A 20040728, 28 July 2004. [Google Scholar]
- Yen, G.-C.; Chen, Y.-L.; Sun, F.-M.; Chiang, Y.-L.; Lu, S.-H.; Weng, C.-J. A comparative study on the effectiveness of cis- and transform of cinnamic acid treatments for inhibiting invasive activity of human lung adenocarcinoma cells. Eur. J. Pharm. Sci. 2011, 44, 281–287. [Google Scholar] [CrossRef]
- Salum, M.L.; Balsells, R.E. High Purity cis-Cinnamic Acid Preparation for Studying Physiological Role of trans-Cinnamic and cis-Cinnamic Acids in Higher Plants. Environ. Control. Biol. 2013, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Horaguchi, T.; Nobuyuki, H.; Tanemura, K.; Suzuki, T. Photocyclization Reactions. Part 8. Synthesis of 2-quinolone, quinoline and coumarin derivatives using trans-cis isomerization by photoreaction. J. Heterocyclic Chem. 2002, 39, 61–67. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2009, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Ondruschka, B.; Bonrath, W.; Stuerga, D. Development and Design of Reactors in Microwave-Assisted Chemistry. In Microwaves in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2013; Volume 1, pp. 57–103. [Google Scholar]
- Bogart, J.W.; Bowers, A.A. Dehydroamino acids: Chemical multi-tools for late-stage diversification. Org. Biomol. Chem. 2019, 17, 3653–3669. [Google Scholar] [CrossRef] [PubMed]
- Veloso, S.R.; Jervis, P.J.; Silva, J.F.; Hilliou, L.; Moura, C.; Pereira, D.M.; Coutinho, P.J.; Martins, J.; Castanheira, E.M.; Ferreira, P.M. Supramolecular ultra-short carboxybenzyl-protected dehydropeptide-based hydrogels for drug delivery. Mater. Sci. Eng. C 2021, 122, 111869. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Sarkar, S.; Khan, M.; Belal, M.; Khan, A.T. A mild and efficient method for large scale synthesis of 3-aminocoumarins and its further application for the preparation of 4-bromo-3-aminocoumarins. Tetrahedron Lett. 2014, 55, 4869–4874. [Google Scholar] [CrossRef]
- Lončarić, M.; Gašo-Sokač, D.; Jokić, S.; Molnar, M. Recent Advances in the Synthesis of Coumarin Derivatives from Dif-ferent Starting Materials. Biomolecules 2020, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Kudale, A.A.; Kendall, J.; Warford, C.C.; Wilkins, N.D.; Bodwell, G.J. Hydrolysis-free synthesis of 3-aminocoumarins. Tetrahedron Lett. 2007, 48, 5077–5080. [Google Scholar] [CrossRef]
- Martínez-Martínez, F.J.; Padilla-Martínez, I.I.; Trujillo-Ferrara, J. 1H and 13C NMR assignments of 2-oxo-2H-1-benzopyran-3-acyl and -3-amide derivatives. Magn. Reson. Chem. 2001, 39, 765–767. [Google Scholar] [CrossRef]
- Borges, R.S.; Castle, S.L. The antioxidant properties of salicylate derivatives: A possible new mechanism of anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2015, 25, 4808–4811. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, F.S.; Aguiar, R.P.; Wiirzler, L.A.M.; Aguayo-Ortiz, R.; Aljuhani, N.; Cuman, R.K.N.; Medina-Franco, J.L.; Siraki, A.G.; Velázquez-Martínez, C.A. Anti-inflammatory and antioxidant properties of a novel resveratrol-salicylate hybrid analogue. Bioorg. Med. Chem. Lett. 2016, 26, 1411–1415. [Google Scholar]
- Gülçin, İ. Antioxidant activity of caffeic acid (3,4-dihydroxycinnamic acid). Toxicology 2006, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, S.; Abraham, T.E.; Zakaria, Z.A. Reactivity of phenolic compounds towards free radicals under in vitro conditions. J. Food Sci. Technol. 2015, 52, 5790–5798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Apak, R.; Özyürek, M.; Güçlü, K.; Çapanoğlu, E. Antioxidant Activity/Capacity Measurement. 1. Classification, Physicochemical Principles, Mechanisms, and Electron Transfer (ET)-Based Assays. J. Agric. Food Chem. 2016, 64, 997–1027. [Google Scholar] [CrossRef]
- Giacomelli, C.; Miranda, F.D.S.; Gonçalves, N.S.; Spinelli, A. Antioxidant activity of phenolic and related compounds: A density functional theory study on the O–H bond dissociation enthalpy. Redox Rep. 2004, 9, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nenadis, N.; Wang, L.-F.; Tsimidou, M.; Zhang, H.-Y. Estimation of Scavenging Activity of Phenolic Compounds Using the ABTS•+Assay. J. Agric. Food Chem. 2004, 52, 4669–4674. [Google Scholar] [CrossRef]
- Woldu, A.S.; Mai, J. Computation of the bond dissociation enthalpies and free energies of hydroxylic antioxidants using the ab initio Hartree–Fock method. Redox Rep. 2012, 17, 252–274. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Carmona, J.R.; Alvarez-Idaboy, J.R.; Galano, A. On the peroxyl scavenging activity of hydroxycinnamic acid derivatives: Mechanisms, kinetics, and importance of the acid–base equilibrium. Phys. Chem. Chem. Phys. 2012, 14, 12534–12543. [Google Scholar] [CrossRef]
- Szeląg, M.; Urbaniak, A.; Bluyssen, H.A. A theoretical antioxidant pharmacophore for natural hydroxycinnamic acids. Open Chem. 2015, 13, 17–31. [Google Scholar] [CrossRef]
- Zouchoune, B. How the ascorbic acid and hesperidin do improve the biological activities of the cinnamon: Theoretical investigation. Struct. Chem. 2020, 31, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Durán, L.A.; Rosales-Hernández, M.C.; Hernández-Rodríguez, M.; Mendieta-Wejebe, J.E.; Trujillo-Ferrara, J.; Correa-Basurto, J. Mapping myeloperoxidase to identify its promiscuity properties using docking and molecular dynamics simulations. Curr. Pharm. Des. 2013, 19, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.D.S.; Ramos, R.D.S.; Costa, K.D.S.L.; Brasil, D.D.S.B.; Silva, C.H.T.D.P.D.; Ferreira, E.F.B.; Borges, R.D.S.; Campos, J.M.; Macêdo, W.J.D.C.; Dos Santos, C.B.R. An in Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods. Molecules 2018, 23, 2801. [Google Scholar] [CrossRef] [Green Version]
- Shacter, E.; Lopez, R.L.; Pati, S. Inhibition of the myeloperoxidase-H2O2-Cl− system of neutrophils by indomethacin and other non-steroidal anti-inflammatory drugs. Biochem. Pharmacol. 1991, 41, 975–984. [Google Scholar] [CrossRef]
- Duclos, F.; Abell, L.M.; Harden, D.G.; Pike, K.; Nowak, K.; Locke, G.A.; Duke, G.J.; Liu, X.; Fernando, G.; Shaw, S.A.; et al. Triazolopyrimidines identified as reversible myeloperoxidase inhibitors. MedChemComm 2017, 8, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Kotora, P.; Šeršeň, F.; Filo, J.; Loos, D.; Gregáň, J.; Gregáň, F. The Scavenging of DPPH, Galvinoxyl and ABTS Radicals by Imine Analogs of Resveratrol. Molecules 2016, 21, 127. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.; Christensen, R.; Rothstein, G. Cellular and extracelular myeloperoxidase in pyogenic inflammation. Blood 1982, 60, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Kettle, A.J.; A Gedye, C.; Hampton, M.B.; Winterbourn, C.C. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem. J. 1995, 308, 559–563. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- DiLabio, G.; Pratt, D.; Wright, J. Theoretical calculation of gas-phase ionization potentials for mono- and polysubstituted benzenes. Chem. Phys. Lett. 1999, 311, 215–220. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, A.E.R.; DiLabio, G.A. Predicting the Activity of Phenolic Antioxidants: Theoretical Method, Analysis of Substituent Effects, and Application to Major Families of Antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Vakarelska-Popovska, M.H.; Velkov, Z. Monohydroxy flavones. Part IV: Ehthalpies of different ways of O–H bond dissociation. Comput. Theor. Chem. 2016, 1077, 87–91. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Reaction Conditions | ||
|---|---|---|---|
| Aq. H2SO4 conc. (% v/v) | Temperature (°C) | Time (min) | |
| 2f | 5 | 120 | 15 |
| 2a, 2e, 2g, 2h, 2j | 15 | 120 | 15 |
| 2b, 2c, 2d, 2i | 15 | 160 | 20 |
| Comp. | BDEOH (kcal mol−1) | IP (kcal mol−1) | EH (eV) | EL (eV) | ΔEH-L (eV) |
|---|---|---|---|---|---|
| 5-ASA [33] | 92.60 | 165.87 | −5.66 | −1.81 | 3.85 |
| 5-ASA | 95.44 | 165.71 | −5.79 | −1.85 | 3.94 |
| 2a | 62.02 | 138.84 | −5.80 | −1.05 | 4.75 |
| 2b | 52.99 | 141.24 | −5.93 | −1.33 | 4.60 |
| 2c | 75.68 | 143.20 | −5.90 | −1.27 | 4.63 |
| 2d | 61.95 | 145.54 | −5.90 | −1.24 | 4.66 |
| 2e | 62.79 | 134.50 | −5.76 | −0.95 | 4.81 |
| 2f | 65.50 55.14 a | 135.67 | −5.61 | −0.96 | 4.65 |
| 2g | 73.01 | 156.95 | −5.55 | −1.02 | 4.53 |
| 2h | 62.85 53.95 b | 136.42 | −5.58 | −1.05 | 4.53 |
| 2i | 79.29 | 152.65 | −6.42 | −2.51 | 3.91 |
| 2j | 81.13 | 150.22 | −5.39 | −0.78 | 4.61 |
| Comp. | ΔG (kcal mol−1) | Interaction Residue |
|---|---|---|
| 4-ABAH | −6.0 | HEM605, Q91, D94, R239, M243 |
| 5-ASA | −6.1 | HEM605, Q91, D94, R239, E242, M243, L246, H336 |
| 2a | −6.3 | HEM605, Q91, D94, R239, M243, L246 |
| 2b | −6.5 | HEM605, Q91, D94, R239, M243, L246 |
| 2c | −6.4 | HEM605, Q91, D94, R239, M243, L246 |
| 2d | −6.3 | HEM605, Q91, D94, R239, S240, M243, L246 |
| 2e | −6.3 | HEM605, Q91, D94, R239, E242, M243, L246, H336, F365 |
| 2f | −6.1 | HEM605, Q91, D94, R239, M243, L246, H336 |
| 2g | −6.2 | HEM605, Q91, D94, R239, M243, L246 |
| 2h | −6.1 | HEM605, Q91, D94, R239, E242, M243, L246, H336, F365 |
| 2i | −6.3 | HEM605, Q91, D94, R239, M243 |
| 2j | −6.0 | HEM605, Q91, D94, R239, M243, L246 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Antonio, A.; Rosales-Hernández, M.C.; Balbuena-Rebolledo, I.; Santiago-Quintana, J.M.; Mendieta-Wejebe, J.E.; Correa-Basurto, J.; García-Vázquez, J.B.; García-Báez, E.V.; Padilla-Martínez, I.I. Myeloperoxidase Inhibitory and Antioxidant Activities of (E)-2-Hydroxy-α-aminocinnamic Acids Obtained through Microwave-Assisted Synthesis. Pharmaceuticals 2021, 14, 513. https://doi.org/10.3390/ph14060513
Rivera-Antonio A, Rosales-Hernández MC, Balbuena-Rebolledo I, Santiago-Quintana JM, Mendieta-Wejebe JE, Correa-Basurto J, García-Vázquez JB, García-Báez EV, Padilla-Martínez II. Myeloperoxidase Inhibitory and Antioxidant Activities of (E)-2-Hydroxy-α-aminocinnamic Acids Obtained through Microwave-Assisted Synthesis. Pharmaceuticals. 2021; 14(6):513. https://doi.org/10.3390/ph14060513
Chicago/Turabian StyleRivera-Antonio, Astrid, Martha Cecilia Rosales-Hernández, Irving Balbuena-Rebolledo, José Martín Santiago-Quintana, Jessica Elena Mendieta-Wejebe, José Correa-Basurto, Juan Benjamín García-Vázquez, Efrén Venancio García-Báez, and Itzia I. Padilla-Martínez. 2021. "Myeloperoxidase Inhibitory and Antioxidant Activities of (E)-2-Hydroxy-α-aminocinnamic Acids Obtained through Microwave-Assisted Synthesis" Pharmaceuticals 14, no. 6: 513. https://doi.org/10.3390/ph14060513