
Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development




{kind=link}
{kind=link}
Abstract
:1. Brief Introduction to Human Pluripotent Stem Cells (hPSCs) and Their Applications
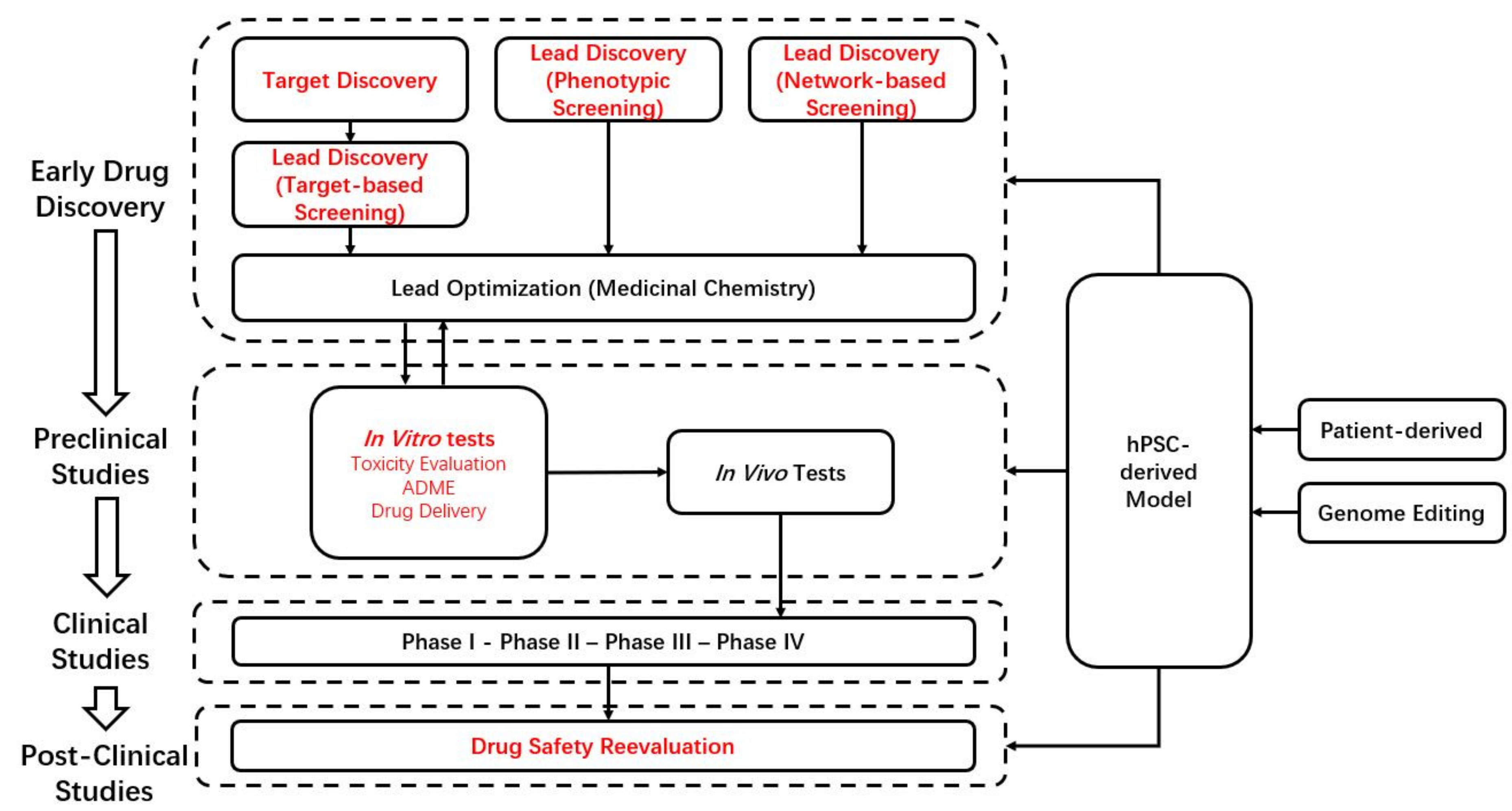
2. Overview of Current Drug Discovery and Development
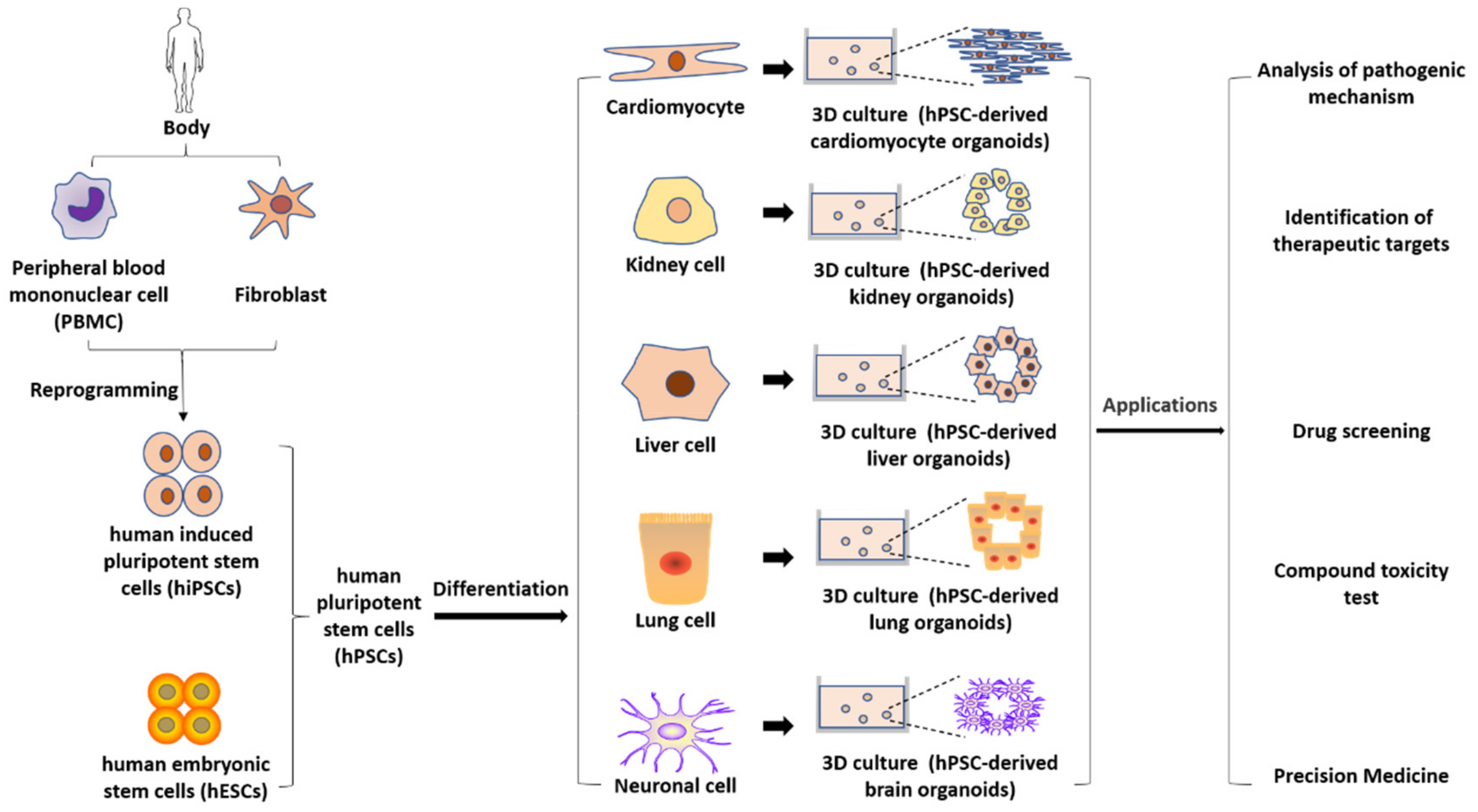
3. hPSC-Derived Disease Models
3.1. Models for the Study of Genetic Disorders
3.1.1. Patient-Derived hPSC Models
3.1.2. Genome-Edited hPSC Models
3.2. Models for the Study of Acquired Diseases
4. hPSC-Derived Models in Drug Discovery and Development
4.1. Target Discovery
4.2. Phenotypic Screening
4.3. Network-Based Screening
4.4. Models for the Study of Disease Mechanisms
4.5. Models for Toxicology
4.6. Models for Precision Medicine
4.7. Clinical Trial in a Dish
4.8. Post-Clinical Studies
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, Y.; Schaffer, D.V. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, E5039–E5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Dai, X.; Zhang, Q.; Dai, Z. Gene expression of OCT4, SOX2, KLF4 and MYC (OSKM) induced pluripotent stem cells: Identification for potential mechanisms. Diagn. Pathol. 2015, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell-Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef]
- Cyranoski, D. How human embryonic stem cells sparked a revolution. Nature 2018, 555, 428–430. [Google Scholar] [CrossRef]
- Augustyniak, J.; Bertero, A.; Coccini, T.; Baderna, D.; Buzanska, L.; Caloni, F. Organoids are promising tools for species-specific in vitro toxicological studies. J. Appl. Toxicol. 2019, 39, 1610–1622. [Google Scholar] [CrossRef]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Silva, M.C.; Haggarty, S.J. Human pluripotent stem cell-derived models and drug screening in CNS precision medicine. Ann. N. Y. Acad. Sci. 2020, 1471, 18–56. [Google Scholar] [CrossRef]
- Garcia-Leon, J.A.; Caceres-Palomo, L.; Sanchez-Mejias, E.; Mejias-Ortega, M.; Nuñez-Diaz, C.; Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Davila, J.C.; Vitorica, J.; Gutierrez, A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6867. [Google Scholar] [CrossRef]
- Han, C.; Chaineau, M.; Chen, C.X.Q.; Beitel, L.K.; Durcan, T.M. Open Science Meets Stem Cells: A New Drug Discovery Approach for Neurodegenerative Disorders. Front. Neurosci. 2018, 12, 47. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, J.L.; Inglese, J.; Walters, M.A. Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discov. 2015, 14, 279–294. [Google Scholar] [CrossRef]
- Lal, S.; Li, A.; Dos Remedios, C. Limitations in Translating Animal Studies to Humans in Cardiovascular Disease. J. Cardiovasc. Transl. Res. 2016, 9, 165–166. [Google Scholar] [CrossRef]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and Disease-Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef]
- Craveiro, N.S.; Lopes, B.S.; Tomás, L.; Almeida, S.F. Drug Withdrawal Due to Safety: A Review of the Data Supporting Withdrawal Decision. Curr. Drug Saf. 2020, 15, 4–12. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- McNeish, J.; Gardner, J.P.; Wainger, B.J.; Woolf, C.J.; Eggan, K. From Dish to Bedside: Lessons Learned While Translating Findings from a Stem Cell Model of Disease to a Clinical Trial. Cell Stem Cell 2015, 17, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.K.; Wu, J.C. Clinical Trial in a Dish: Using Patient-Derived Induced Pluripotent Stem Cells to Identify Risks of Drug-Induced Cardiotoxicity. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1019–1031. [Google Scholar] [CrossRef]
- Pravenec, M.; Křen, V.; Landa, V.; Mlejnek, P.; Musilová, A.; Šilhavý, J.; Šimáková, M.; Zídek, V. Recent progress in the genetics of spontaneously hypertensive rats. Physiol. Res. 2014, 63, S1–S8. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Herault, Y.; Delabar, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Yu, E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Models Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Z.; Hu, F.; Su, L. Cell Culture Models and Animal Models for HBV Study. Adv. Exp. Med. Biol. 2020, 1179, 109–135. [Google Scholar] [CrossRef]
- Chin, R.M.; Panavas, T.; Brown, J.M.; Johnson, K.K. Patient-derived lymphoblastoid cell lines harboring mitochondrial DNA mutations as tool for small molecule drug discovery. BMC Res. Notes 2018, 11, 205. [Google Scholar] [CrossRef] [Green Version]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, Y. A transcriptomic study suggesting human iPSC-derived hepatocytes potentially offer a better in vitro model of hepatotoxicity than most hepatoma cell lines. Cell Biol. Toxicol. 2017, 33, 407–421. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef]
- Chen, L.; Morris, D.L.; Vyse, T.J. Genetic advances in systemic lupus erythematosus: An update. Curr. Opin. Rheumatol. 2017, 29, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, D.; Elefante, E.; Calabresi, E.; Signorini, V.; Bortoluzzi, A.; Tani, C. One year in review 2019: Systemic lupus erythematosus. Clin. Exp. Rheumatol. 2019, 37, 715–722. [Google Scholar] [PubMed]
- Zhu, T.Y.; Tam, L.S.; Li, E.K. Cost-of-illness studies in systemic lupus erythematosus: A systematic review. Arthritis Care Res. 2011, 63, 751–760. [Google Scholar] [CrossRef]
- Rivas-Larrauri, F.; Yamazaki-Nakashimada, M.A. Systemic lupus erythematosus: Is it one disease? Reumatol. Clin. 2016, 12, 274–281. [Google Scholar] [CrossRef]
- Dörner, T.; Furie, R. Novel paradigms in systemic lupus erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef]
- Li, W.; Liu, D.; Zheng, F.; Zeng, Z.; Cai, W.; Luan, S.; Hong, X.; Tang, D.; Yin, L.H.; Dai, Y. Generation of Systemic Lupus Erythematosus Patient-Derived Induced Pluripotent Stem Cells from Blood. Stem Cells Dev. 2021, 30, 227–233. [Google Scholar] [CrossRef]
- Fick, G.M.; Gabow, P.A. Natural history of autosomal dominant polycystic kidney disease. Annu. Rev. Med. 1994, 45, 23–29. [Google Scholar] [CrossRef]
- Benedetti, V.; Brizi, V.; Guida, P.; Tomasoni, S.; Ciampi, O.; Angeli, E.; Valbusa, U.; Benigni, A.; Remuzzi, G.; Xinaris, C. Engineered Kidney Tubules for Modeling Patient-Specific Diseases and Drug Discovery. EBioMedicine 2018, 33, 253–268. [Google Scholar] [CrossRef]
- Mo, J.; Anastasaki, C.; Chen, Z.; Shipman, T.; Papke, J.; Yin, K.; Gutmann, D.H.; Le, L.Q. Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. J. Clin. Investg. 2021, 131, e139807. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Voytas, D.F. TAL effectors: Customizable proteins for DNA targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.W.; Broton, C.; Bogacheva, M.S.; Xiao, A.Z.; Garcia-Castro, M.I.; Lou, Y.R. RNA-based CRISPR-Mediated Loss-of-Function Mutagenesis in Human Pluripotent Stem Cells. J. Mol. Biol. 2020, 432, 3956–3964. [Google Scholar] [CrossRef]
- Christidi, E.; Huang, H.M.; Brunham, L.R. CRISPR/Cas9-mediated genome editing in human stem cell-derived cardiomyocytes: Applications for cardiovascular disease modelling and cardiotoxicity screening. Drug Discov. Today Technol. 2018, 28, 13–21. [Google Scholar] [CrossRef]
- Suh, W. A new era of disease modeling and drug discovery using induced pluripotent stem cells. Arch. Pharm. Res. 2017, 40, 1–12. [Google Scholar] [CrossRef]
- Artero Castro, A.; Lukovic, D.; Jendelova, P.; Erceg, S. Concise Review: Human Induced Pluripotent Stem Cell Models of Retinitis Pigmentosa. Stem Cells 2018, 36, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Le Verche, V.; Przedborski, S. Is amyotrophic lateral sclerosis a mitochondrial channelopathy? Neuron 2010, 67, 523–524. [Google Scholar] [CrossRef] [Green Version]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human Motor Neurons With SOD1-G93A Mutation Generated From CRISPR/Cas9 Gene-Edited iPSCs Develop Pathological Features of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2020, 14, 604171. [Google Scholar] [CrossRef]
- Zhang, T.; Moss, A.; Cong, P.; Pan, M.; Chang, B.; Zheng, L.; Fang, Q.; Zareba, W.; Robinson, J.; Lin, C.; et al. LQTS gene LOVD database. Hum. Mutat. 2010, 31, E1801–E1810. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, P.; Lan, F.; Wu, H.; Lisowski, L.; Gu, M.; Hu, S.; Kay, M.A.; Urnov, F.D.; Shinnawi, R.; et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J. Am. Coll. Cardiol. 2014, 64, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Reith, W. Neurodegenerative diseases. Radiologe 2018, 58, 241–258. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Leon, J.A.; Vitorica, J.; Gutierrez, A. Use of human pluripotent stem cell-derived cells for neurodegenerative disease modeling and drug screening platform. Future Med. Chem. 2019, 11, 1305–1322. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- García-León, J.A.; Cabrera-Socorro, A.; Eggermont, K.; Swijsen, A.; Terryn, J.; Fazal, R.; Nami, F.; Ordovás, L.; Quiles, A.; Lluis, F.; et al. Generation of a human induced pluripotent stem cell-based model for tauopathies combining three microtubule-associated protein TAU mutations which displays several phenotypes linked to neurodegeneration. Alzheimer Dement. 2018, 14, 1261–1280. [Google Scholar] [CrossRef]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.L.; et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.A.; Parmar, M.; Kirkeby, A.; Björklund, A.; Thompson, L.; Brundin, P. Are Stem Cell-Based Therapies for Parkinson’s Disease Ready for the Clinic in 2016? J. Parkinson Dis. 2016, 6, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-Cell Transcriptomic Comparison of Human Fetal Retina, hPSC-Derived Retinal Organoids, and Long-Term Retinal Cultures. Cell Rep. 2020, 30, 1644–1659. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Fogo, M.S.; Pinto, B.G.G.; Alves, A.Y.; Suzuki, A.M.; Morales, A.G.; Ezquina, S.; Sosa, O.J.; Sutton, G.J.; Sunaga-Franze, D.Y.; et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 2021, 26, 1589–1605. [Google Scholar] [CrossRef] [Green Version]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.-K.; Micali, N.; Wang, Y.; Colantuoni, C.; Straub, R.E.; Hoeppner, D.J.; et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11, 462. [Google Scholar] [CrossRef] [Green Version]
- Kolanowski, T.J.; Busek, M.; Schubert, M.; Dmitrieva, A.; Binnewerg, B.; Pöche, J.; Fisher, K.; Schmieder, F.; Grünzner, S.; Hansen, S.; et al. Enhanced structural maturation of human induced pluripotent stem cell-derived cardiomyocytes under a controlled microenvironment in a microfluidic system. Acta Biomater. 2020, 102, 273–286. [Google Scholar] [CrossRef]
- Amaratunga, D.; Göhlmann, H.; Peeters, P.J. 3.05—Microarrays. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 87–106. [Google Scholar] [CrossRef]
- Kobayashi, H.; Hatakeyama, H.; Nishimura, H.; Yokota, M.; Suzuki, S.; Tomabechi, Y.; Shirouzu, M.; Osada, H.; Mimaki, M.; Goto, Y.I.; et al. Chemical reversal of abnormalities in cells carrying mitochondrial DNA mutations. Nat. Chem. Biol. 2021, 17, 335–343. [Google Scholar] [CrossRef]
- Zhang, W.B.; Ross, P.J.; Tu, Y.; Wang, Y.; Beggs, S.; Sengar, A.S.; Ellis, J.; Salter, M.W. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci. Rep. 2016, 6, 23837. [Google Scholar] [CrossRef] [Green Version]
- Warchal, S.J.; Unciti-Broceta, A.; Carragher, N.O. Next-generation phenotypic screening. Future Med. Chem. 2016, 8, 1331–1347. [Google Scholar] [CrossRef]
- Li, Z.; Cvijic, M.E.; Zhang, L. 2.15—Cellular Imaging in Drug Discovery: Imaging and Informatics for Complex Cell Biology. In Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 362–387. [Google Scholar] [CrossRef]
- Eggert, U.S. The why and how of phenotypic small-molecule screens. Nat. Chem. Biol. 2013, 9, 206–209. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Han, H.W.; Seo, H.H.; Jo, H.Y.; Han, H.J.; Falcão, V.C.A.; Delorme, V.; Heo, J.; Shum, D.; Choi, J.H.; Lee, J.M.; et al. Drug Discovery Platform Targeting M. tuberculosis with Human Embryonic Stem Cell-Derived Macrophages. Stem Cell Rep. 2019, 13, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L. Network pharmacology. Nat. Biotechnol. 2007, 25, 1110–1111. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Nichols, E.; Szoeke, C.E.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- James, B.D.; Bennett, D.A. Causes and Patterns of Dementia: An Update in the Era of Redefining Alzheimer’s Disease. Annu. Rev. Public Health 2019, 40, 65–84. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Vergallo, A.; Aguilar, L.F.; Benda, N.; Broich, K.; Cuello, A.C.; Cummings, J.; Dubois, B.; Federoff, H.J.; Fiandaca, M.; et al. Precision pharmacology for Alzheimer’s disease. Pharmacol. Res. 2018, 130, 331–365. [Google Scholar] [CrossRef]
- Park, J.C.; Jang, S.Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef]
- Theodoris, C.V.; Zhou, P.; Liu, L.; Zhang, Y.; Nishino, T.; Huang, Y.; Kostina, A.; Ranade, S.S.; Gifford, C.A.; Uspenskiy, V.; et al. Network-based screen in iPSC-derived cells reveals therapeutic candidate for heart valve disease. Science 2021, 371. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Terada, N.; Ashizawa, T. Human iPSC Models to Study Orphan Diseases: Muscular Dystrophies. Curr. Stem Cell Rep. 2018, 4, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, M.C.; Petersen, J.A.; Stucka, R.; Fischer, D.; Schröder, R.; Vorgerd, M.; Schroers, A.; Schreiber, H.; Hanemann, C.O.; Knirsch, U.; et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J. Med. Genet. 2004, 41, e50. [Google Scholar] [CrossRef] [Green Version]
- Poppe, M.; Cree, L.; Bourke, J.; Eagle, M.; Anderson, L.V.; Birchall, D.; Brockington, M.; Buddles, M.; Busby, M.; Muntoni, F.; et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003, 60, 1246–1251. [Google Scholar] [CrossRef]
- Poppe, M.; Bourke, J.; Eagle, M.; Frosk, P.; Wrogemann, K.; Greenberg, C.; Muntoni, F.; Voit, T.; Straub, V.; Hilton-Jones, D.; et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 2004, 56, 738–741. [Google Scholar] [CrossRef]
- Krag, T.O.; Vissing, J. A New Mouse Model of Limb-Girdle Muscular Dystrophy Type 2I Homozygous for the Common L276I Mutation Mimicking the Mild Phenotype in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.D.; Zimmermann, W.H.; Cyganek, L.; et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [Green Version]
- Bunce, C.; Xing, W.; Wormald, R. Causes of blind and partial sight certifications in England and Wales: April 2007–March 2008. Eye 2010, 24, 1692–1699. [Google Scholar] [CrossRef] [Green Version]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar]
- Gonzales, C.R. Enhanced efficacy associated with early treatment of neovascular age-related macular degeneration with pegaptanib sodium: An exploratory analysis. Retina 2005, 25, 815–827. [Google Scholar] [CrossRef]
- Colquitt, J.L.; Jones, J.; Tan, S.C.; Takeda, A.; Clegg, A.J.; Price, A. Ranibizumab and pegaptanib for the treatment of age-related macular degeneration: A systematic review and economic evaluation. Health Technol. Assess. 2008, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- El-Asrag, M.E.; Sergouniotis, P.I.; McKibbin, M.; Plagnol, V.; Sheridan, E.; Waseem, N.; Abdelhamed, Z.; McKeefry, D.; Van Schil, K.; Poulter, J.A.; et al. Biallelic mutations in the autophagy regulator DRAM2 cause retinal dystrophy with early macular involvement. Am. J. Hum. Genet. 2015, 96, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Cerniauskas, E.; Kurzawa-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 9, 1585–1603. [Google Scholar] [CrossRef]
- Paget, E. The LD50 test. Acta Pharmacol. Toxicol. 1983, 52 (Suppl. S2), 6–19. [Google Scholar] [CrossRef]
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef]
- Rusyn, I.; Gatti, D.M.; Wiltshire, T.; Kleeberger, S.R.; Threadgill, D.W. Toxicogenetics: Population-based testing of drug and chemical safety in mouse models. Pharmacogenomics 2010, 11, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.; Steger-Hartmann, T. A big data approach to the concordance of the toxicity of pharmaceuticals in animals and humans. Regul. Toxicol. Pharmacol. 2018, 96, 94–105. [Google Scholar] [CrossRef]
- Stricker-Krongrad, A.; Shoemake, C.R.; Pereira, M.E.; Gad, S.C.; Brocksmith, D.; Bouchard, G.F. Miniature Swine Breeds in Toxicology and Drug Safety Assessments: What to Expect during Clinical and Pathology Evaluations. Toxicol. Pathol. 2016, 44, 421–427. [Google Scholar] [CrossRef]
- Foote, R.H.; Carney, E.W. The rabbit as a model for reproductive and developmental toxicity studies. Reprod. Toxicol. 2000, 14, 477–493. [Google Scholar] [CrossRef]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Hunt, P.R. The C. elegans model in toxicity testing. J. Appl. Toxicol. 2017, 37, 50–59. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef] [Green Version]
- Li, R.A.; Keung, W.; Cashman, T.J.; Backeris, P.C.; Johnson, B.V.; Bardot, E.S.; Wong, A.O.T.; Chan, P.K.W.; Chan, C.W.Y.; Costa, K.D. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials 2018, 163, 116–127. [Google Scholar] [CrossRef]
- Kulkeaw, K.; Tubsuwan, A.; Tongkrajang, N.; Whangviboonkij, N. Generation of human liver organoids from pluripotent stem cell-derived hepatic endoderms. PeerJ 2020, 8, e9968. [Google Scholar] [CrossRef]
- Steichen, C.; Giraud, S.; Hauet, T. Kidney organoids. Med. Sci. 2019, 35, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V. Kidney organoids-a new tool for kidney therapeutic development. Kidney Int. 2018, 94, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Hesley, J.; Rusyn, I.; Cromwell, E.F. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev. Technol. 2014, 12, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Grimm, F.A.; Ryan, K.R.; Iwata, Y.; Chiu, W.A.; Parham, F.; Wignall, J.A.; Anson, B.; Cromwell, E.F.; Behl, M.; et al. In vitro cardiotoxicity assessment of environmental chemicals using an organotypic human induced pluripotent stem cell-derived model. Toxicol. Appl. Pharmacol. 2017, 322, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Pointon, A.; Pilling, J.; Dorval, T.; Wang, Y.; Archer, C.; Pollard, C. From the Cover: High-Throughput Imaging of Cardiac Microtissues for the Assessment of Cardiac Contraction during Drug Discovery. Toxicol. Sci. 2017, 155, 444–457. [Google Scholar] [CrossRef]
- McKeithan, W.L.; Savchenko, A.; Yu, M.S.; Cerignoli, F.; Bruyneel, A.A.N.; Price, J.H.; Colas, A.R.; Miller, E.W.; Cashman, J.R.; Mercola, M. An Automated Platform for Assessment of Congenital and Drug-Induced Arrhythmia with hiPSC-Derived Cardiomyocytes. Front. Physiol. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Grimm, F.A.; Iwata, Y.; Sirenko, O.; Bittner, M.; Rusyn, I. High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug Dev. Technol. 2015, 13, 529–546. [Google Scholar] [CrossRef]
- Ni, X.; Yang, Z.Z.; Ye, L.Q.; Han, X.L.; Zhao, D.D.; Ding, F.Y.; Ding, N.; Wu, H.C.; Yu, M.; Xu, G.Y.; et al. Establishment of an in vitro safety assessment model for lipid-lowering drugs using same-origin human pluripotent stem cell-derived cardiomyocytes and endothelial cells. Acta Pharmacol. Sin. 2021, 1–11. [Google Scholar] [CrossRef]
- Kandasamy, K.; Chuah, J.K.; Su, R.; Huang, P.; Eng, K.G.; Xiong, S.; Li, Y.; Chia, C.S.; Loo, L.H.; Zink, D. Prediction of drug-induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell-derived cells and machine learning methods. Sci. Rep. 2015, 5, 12337. [Google Scholar] [CrossRef] [Green Version]
- Sherman, S.P.; Bang, A.G. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis. Models. Mech. 2018, 11, dmm031906. [Google Scholar] [CrossRef] [Green Version]
- Klaren, W.D.; Rusyn, I. High-Content Assay Multiplexing for Muscle Toxicity Screening in Human-Induced Pluripotent Stem Cell-Derived Skeletal Myoblasts. Assay Drug Dev. Technol. 2018, 16, 333–342. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Karumbayaram, S.; Novitch, B.G.; Patterson, M.; Umbach, J.A.; Richter, L.; Lindgren, A.; Conway, A.E.; Clark, A.T.; Goldman, S.A.; Plath, K.; et al. Directed differentiation of human-induced pluripotent stem cells generates active motor neurons. Stem Cells 2009, 27, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.X.; Nekanti, U.; Haus, D.L.; Funes, G.; Moreno, D.; Kamei, N.; Cummings, B.J.; Anderson, A.J. Induction of early neural precursors and derivation of tripotent neural stem cells from human pluripotent stem cells under xeno-free conditions. J. Comp. Neurol. 2014, 522, 2767–2783. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef] [Green Version]
- Klima, S.; Brüll, M.; Spreng, A.S.; Suciu, I.; Falt, T.; Schwamborn, J.C.; Waldmann, T.; Karreman, C.; Leist, M. A human stem cell-derived test system for agents modifying neuronal N-methyl-d-aspartate-type glutamate receptor Ca2+-signalling. Arch. Toxicol. 2021, 95, 1703–1722. [Google Scholar] [CrossRef]
- PsychENCODE Consortium. Revealing the brain’s molecular architecture. Science 2018, 362, 1262–1263. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Duong, A.; Evstratova, A.; Sivitilli, A.; Hernandez, J.J.; Gosio, J.; Wahedi, A.; Sondheimer, N.; Wrana, J.L.; Beaulieu, J.M.; Attisano, L.; et al. Characterization of mitochondrial health from human peripheral blood mononuclear cells to cerebral organoids derived from induced pluripotent stem cells. Sci. Rep. 2021, 11, 4523. [Google Scholar] [CrossRef]
- Jameson, J.L.; Longo, D.L. Precision medicine—Personalized, problematic, and promising. N. Engl. J. Med. 2015, 372, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- König, I.R.; Fuchs, O.; Hansen, G.; von Mutius, E.; Kopp, M.V. What is precision medicine? Eur. Respir. J. 2017, 50, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastrinos, F.; Samadder, N.J.; Burt, R.W. Use of Family History and Genetic Testing to Determine Risk of Colorectal Cancer. Gastroenterology 2020, 158, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Knabben, L.; Imboden, S.; Mueller, M.D. Genetic testing in ovarian cancer—Clinical impact and current practices. Horm. Mol. Biol. Clin. Investig. 2019, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Teoh, H.K.; Cheong, S.K. Reprogramming human dermal fibroblast into induced pluripotent stem cells using nonintegrative Sendai virus for transduction. Malays. J. Pathol. 2018, 40, 325–329. [Google Scholar] [PubMed]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strikoudis, A.; Cieślak, A.; Loffredo, L.; Chen, Y.W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.; Rudnick, D.A. Induced Pluripotent Stem Cell-Derived Hepatocytes and Precision Medicine in Human Liver Disease. J. Pediatric. Gastroenterol. Nutr. 2018, 66, 716–719. [Google Scholar] [CrossRef]
- Fermini, B.; Coyne, S.T.; Coyne, K.P. Clinical Trials in a Dish: A Perspective on the Coming Revolution in Drug Development. SLAS Discov. 2018, 23, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Fatkin, D. Familial dilated cardiomyopathy: Current challenges and future directions. Glob. Cardiol. Sci. Pract. 2012, 2012, 8. [Google Scholar] [CrossRef]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Bidault, G.; Garcia, M.; Vantyghem, M.C.; Ducluzeau, P.H.; Morichon, R.; Thiyagarajah, K.; Moritz, S.; Capeau, J.; Vigouroux, C.; Béréziat, V. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Sayed, N.; Liu, C.; Ameen, M.; Himmati, F.; Zhang, J.Z.; Khanamiri, S.; Moonen, J.R.; Wnorowski, A.; Cheng, L.; Rhee, J.W.; et al. Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci. Transl. Med. 2020, 12, eaax9276. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef] [Green Version]
- Burnett, S.D.; Blanchette, A.D.; Grimm, F.A.; House, J.S.; Reif, D.M.; Wright, F.A.; Chiu, W.A.; Rusyn, I. Population-based toxicity screening in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2019, 381, 114711. [Google Scholar] [CrossRef]
- Stillitano, F.; Hansen, J.; Kong, C.W.; Karakikes, I.; Funck-Brentano, C.; Geng, L.; Scott, S.; Reynier, S.; Wu, M.; Valogne, Y.; et al. Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. Elife 2017, 6, e19406. [Google Scholar] [CrossRef]
- Shinozawa, T.; Nakamura, K.; Shoji, M.; Morita, M.; Kimura, M.; Furukawa, H.; Ueda, H.; Shiramoto, M.; Matsuguma, K.; Kaji, Y.; et al. Recapitulation of Clinical Individual Susceptibility to Drug-Induced QT Prolongation in Healthy Subjects Using iPSC-Derived Cardiomyocytes. Stem Cell Rep. 2017, 8, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Blinova, K.; Schocken, D.; Patel, D.; Daluwatte, C.; Vicente, J.; Wu, J.C.; Strauss, D.G. Clinical Trial in a Dish: Personalized Stem Cell-Derived Cardiomyocyte Assay Compared With Clinical Trial Results for Two QT-Prolonging Drugs. Clin. Transl. Sci. 2019, 12, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Sun, P.; Wang, Y.; Chen, J.; Lv, L.; Wei, W.; Jin, C.; Li, W. Generation of Self-Renewing Hepatoblasts From Human Embryonic Stem Cells by Chemical Approaches. Stem Cells Transl. Med. 2015, 4, 1275–1282. [Google Scholar] [CrossRef]
- Raju, R.; Chau, D.; Cho, D.S.; Park, Y.; Verfaillie, C.M.; Hu, W.S. Cell Expansion During Directed Differentiation of Stem Cells Toward the Hepatic Lineage. Stem Cells Dev. 2017, 26, 274–284. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Tan, Z.; Su, Y.; Liu, J.; Chang, M.; Yan, F.; Chen, J.; Chen, T.; Li, C.; et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019, 29, 1009–1026. [Google Scholar] [CrossRef]
- Akbari, S.; Sevinç, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinç, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Mun, S.J.; Hong, Y.H.; Ahn, H.S.; Ryu, J.S.; Chung, K.S.; Son, M.J. Long-Term Expansion of Functional Human Pluripotent Stem Cell-Derived Hepatic Organoids. Int. J. Stem Cells 2020, 13, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Models Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [Green Version]
- Committee on Ethical, Legal, and Regulatory Issues Associated with Neural Chimeras and Organoids; Committee on Science, Technology, and Law; Policy and Global Affairs; National Academies of Sciences, Engineering, and Medicine. The National Academies Collection: Reports funded by National Institutes of Health. In The Emerging Field of Human Neural Organoids, Transplants, and Chimeras: Science, Ethics, and Governance; National Academies Press: Washington, DC, USA, 2021. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Tang, J.; Lou, Y.-R. Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development. Pharmaceuticals 2021, 14, 525. https://doi.org/10.3390/ph14060525
Lin X, Tang J, Lou Y-R. Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development. Pharmaceuticals. 2021; 14(6):525. https://doi.org/10.3390/ph14060525
Chicago/Turabian StyleLin, Xiying, Jiayu Tang, and Yan-Ru Lou. 2021. "Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development" Pharmaceuticals 14, no. 6: 525. https://doi.org/10.3390/ph14060525
APA StyleLin, X., Tang, J., & Lou, Y.-R. (2021). Human Pluripotent Stem-Cell-Derived Models as a Missing Link in Drug Discovery and Development. Pharmaceuticals, 14(6), 525. https://doi.org/10.3390/ph14060525