
Insights into an Immunotherapeutic Approach to Combat Multidrug Resistance in Hepatocellular Carcinoma

 , ,
, , 
 and
and
Abstract
:1. Introduction
2. HCC Tumor Resistance: Signaling and Strategies
2.1. Drug Efflux Pump and Drug Metabolism
2.2. Alteration of DNA Repair Pathways and Chemoresistance in HCC
2.3. Tumor Microenvironment
2.4. Micro RNA (miRNA) in HCC
2.5. Chemoresistance due to Epigenetic Regulation
2.6. Topoisomerases in Chemoresistance
2.7. Cancer Stem Cells in Chemoresistance
2.8. Telomerase and Chemo-Resistance
2.9. Impaired Lipid Metabolism
3. Immunotherapy: A Novel Weapon against HCC
3.1. Immune Contexture of HCC
3.1.1. Immunological Organization and Immune Regulation of the Liver
3.1.2. Immune Responses in HCC
Cancer-Immunity Cycle
Mechanism of Immune Evasion in HCC
3.2. HCC as an Ideal Candidate for Immunotherapy
3.2.1. Adoptive Cell Therapy
- ➢
- Cytokine-induced killer-cell therapy
- ➢
- Tumor-infiltrating lymphocyte therapy
- ➢
- Natural killer cell therapy
- ➢ Dendritic cell therapy
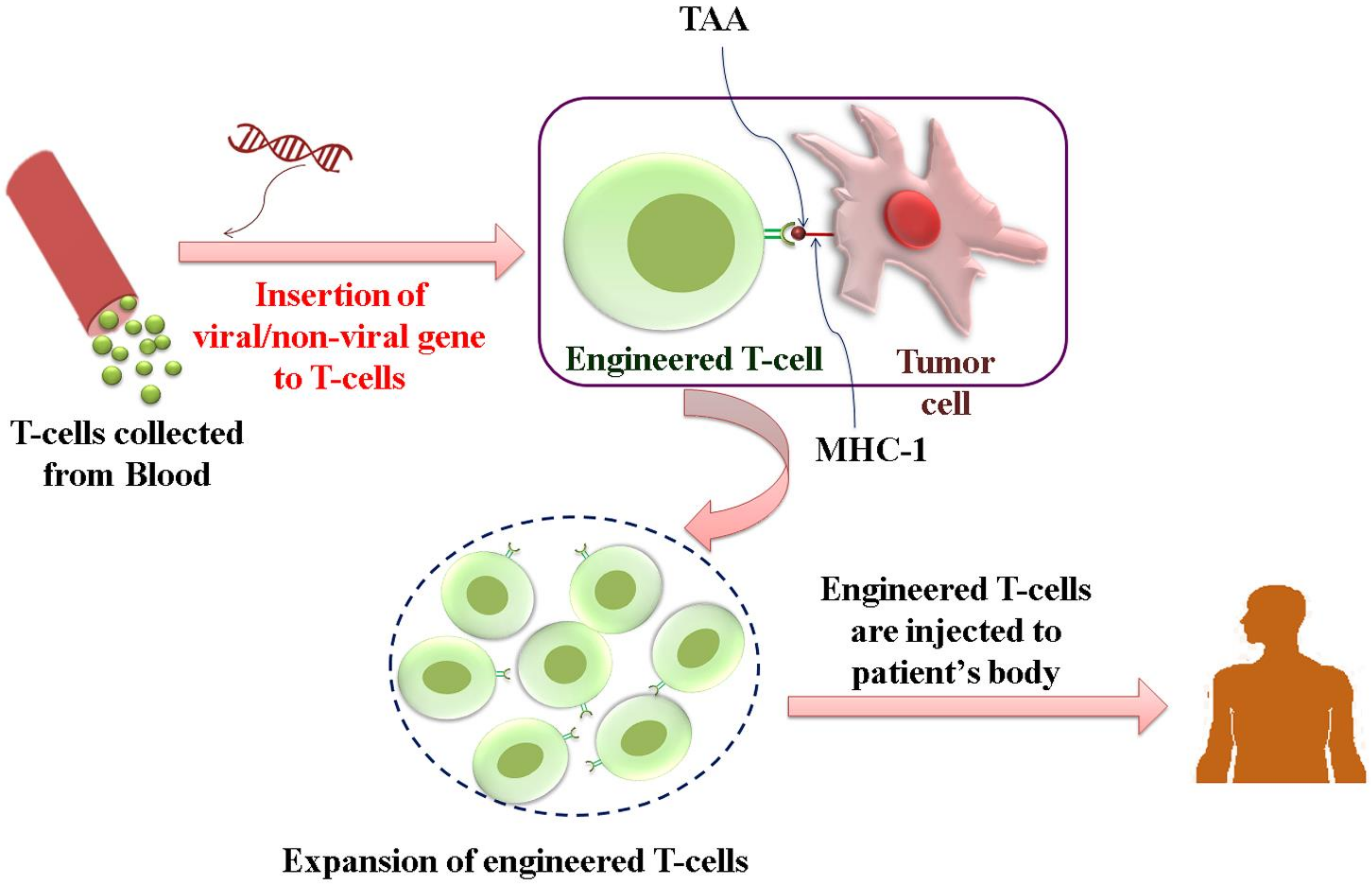
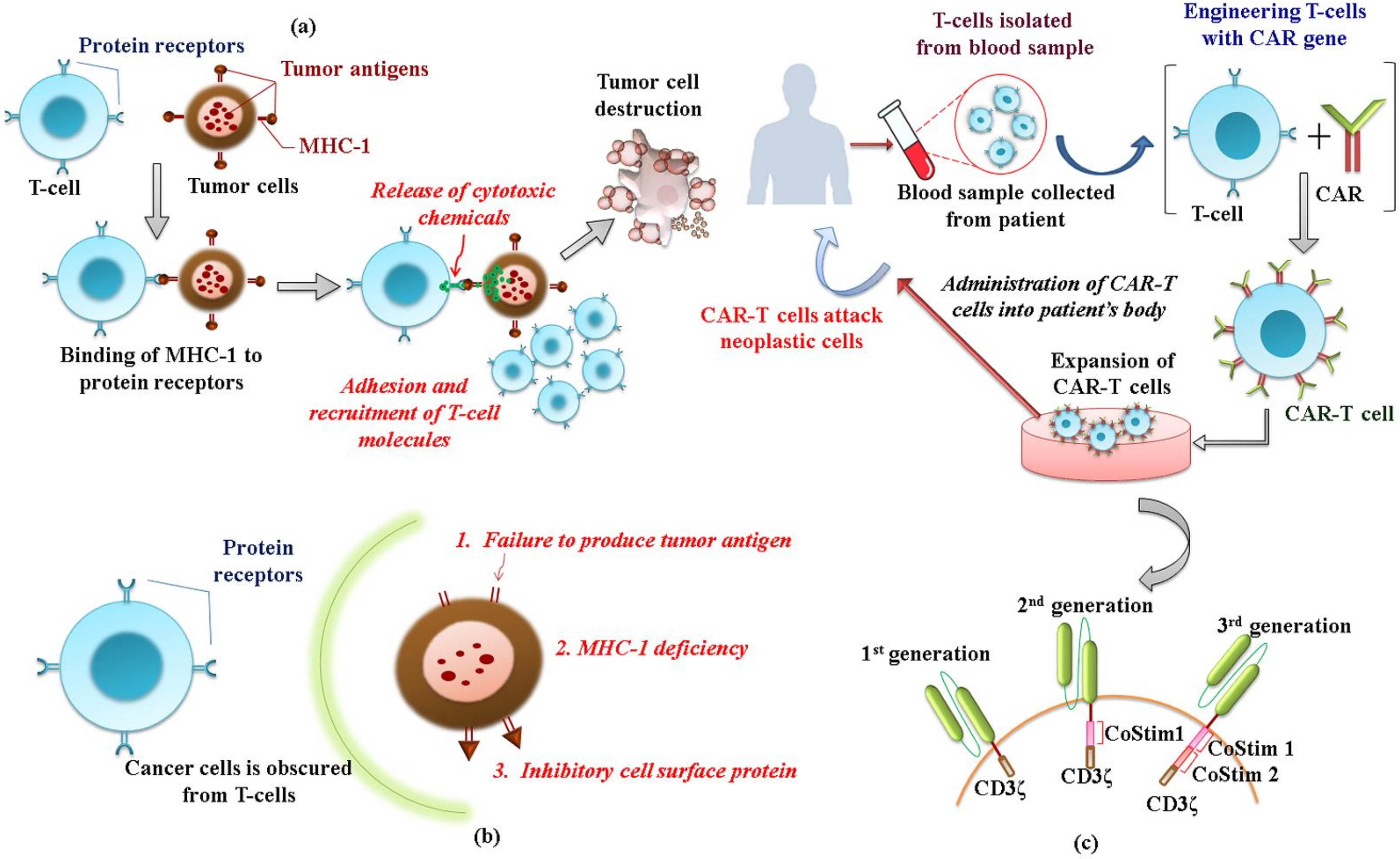
3.2.2. Genetically Modified T-Cell Therapy
- ➢
- TCR-engineered T-cell therapy
- ➢
- CAR-T cell therapy
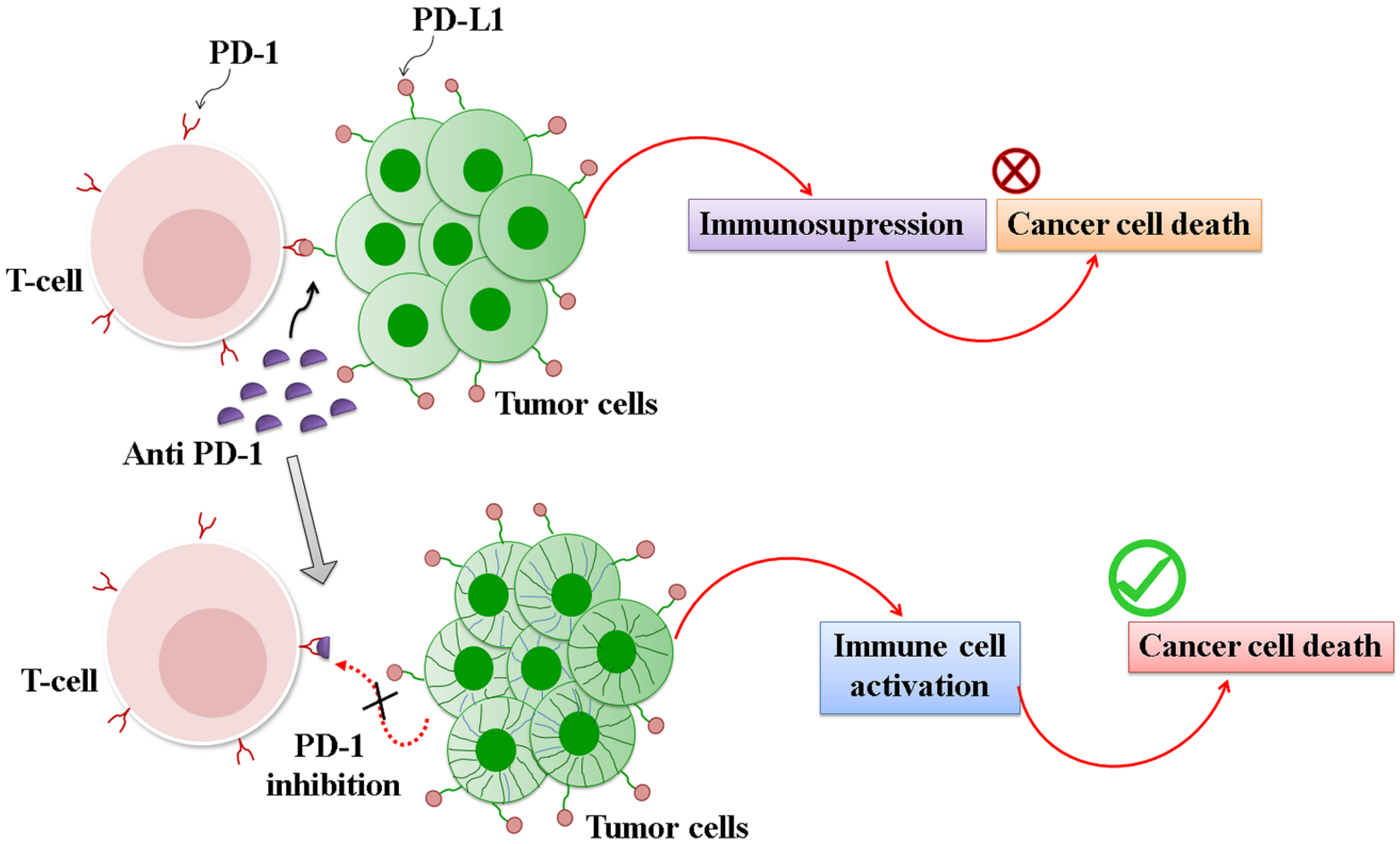
3.2.3. Immune Checkpoint Inhibitors (ICI)
3.2.4. Vaccines
3.2.5. Oncolytic Viruses
3.3. Combination Strategies for Immunotherapy for HCC
4. Challenges and Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Sayiner, M.; Golabi, P.; Younossi, Z.M. Disease burden of hepatocellular carcinoma: A global perspective. Dig. Dis. Sci. 2019, 64, 910–917. [Google Scholar] [CrossRef]
- Nair, B.; Nath, L.R. Inevitable role of TGF-β1 in progression of nonalcoholic fatty liver disease. J. Recept. Signal Transduct 2020, 40, 195–200. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and management of hepatocellular carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieb, B.C.; Goff, L.W.; Goyal, L.; Denlinger, C.S. Evolving landscape of systemic therapy for hepatocellular carcinoma: Breakthroughs, toxicities, and future frontiers. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Baby, J.; Devan, A.R.; Kumar, A.R.; Gorantla, J.N.; Nair, B.; Aishwarya, T.S.; Nath, L.R. Cogent role of flavonoids as key orchestrators of chemoprevention of hepatocellular carcinoma: A review. J. Food Biochem. 2021, 45, e13761. [Google Scholar] [CrossRef]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and future treatment of hepatocellular carcinoma: An updated comprehensive review. J. Clin. Transl. Hepatol. 2018, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Chen, X.; Wang, Q.; Xu, Z.; Zhang, W.; Ye, L. The roles of four multi-drug resistance proteins in hepatocellular carcinoma multidrug resistance. J. Huazhong Univ. Sci. Technol. 2007, 27, 173–175. [Google Scholar] [CrossRef]
- Thakkar, N.; Slizgi, J.R.; Brouwer, K.L. Effect of liver disease on hepatic transporter expression and function. J. Pharm. Sci. 2017, 106, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.J.; Monte, M.J.; Blazquez, A.G.; Macias, R.I.; Serrano, M.A.; Briz, O. The role of reduced intracellular concentrations of active drugs in the lack of response to anticancer chemotherapy. Acta Pharmacol. Sin. 2014, 35, 1–10. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Pérez-Torras, S. Emerging roles of nucleoside transporters. Front. Pharmacol. 2018, 9, 606. [Google Scholar] [CrossRef]
- Perrotton, T.; Trompier, D.; Chang, X.B.; Di Pietro, A.; Baubichon-Cortay, H. (R)-and (S)-verapamil differentially modulate the multidrug-resistant protein MRP1. J. Biol. Chem. 2007, 282, 31542–31548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollt, V.; Kouba, M.; Dietel, M.; Vogt, G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem. Pharmacol. 1992, 43, 2601–2608. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; de Vries, E.G.; Timmer-Bosscha, H.; Meersma, G.J.; Mesander, G.; Vellenga, E.; Mulder, N.H. Effects of amiodarone, cyclosporin A, and PSC 833 on the cytotoxicity of mitoxantrone, doxorubicin, and vincristine in non-P-glycoprotein human small cell lung cancer cell lines. Cancer Res. 1994, 54, 5368–5373. [Google Scholar] [PubMed]
- Wessler, J.D.; Grip, L.; Mendell, J.; Giugliano, R.P. The P-Glycoprotein Transport System and Cardiovascular Drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Furusawa, S.; Nakano, S.; Takahashi, M.; Chiba, H.; Takayanagi, M.; Takayanagi, Y.; Sasaki, K. Reversal of multidrug resistance by tacrolimus hydrate. Methods Find. Exp. Clin. Pharmacol. 1996, 18, 651–658. [Google Scholar] [PubMed]
- Solary, E.; Witz, B.; Caillot, D.; Moreau, P.; Desablens, B.; Cahn, J.Y.; Sadoun, A.; Pignon, B.; Berthou, C.; Maloisel, F.; et al. Combination of quinine as a potential reversing agent with mitoxantrone and cytarabine for the treatment of acute leukemias: A randomized multicenter study. Blood 1996, 88, 1198–1205. [Google Scholar] [CrossRef]
- Deng, T.; Liu, J.C.; Pritchard, K.I.; Eisen, A.; Zacksenhaus, E. Preferential killing of breast tumor initiating cells by N,N-diethyl-2-[4- (phenylmethyl)phenoxy]ethanamine/tesmilifene. Clin. Cancer Res. 2009, 15, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiden, M.V.; Swenerton, K.D.; Matulonis, U.; Campos, S.; Rose, P.; Batist, G.; Ette, E.; Garg, V.; Fuller, A.; Harding, M.W.; et al. A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol. Oncol. 2002, 86, 302–310. [Google Scholar] [CrossRef]
- Abe, T.; Koike, K.; Ohga, T.; Kubo, T.; Wada, M.; Kohno, K.; Mori, T.; Hidaka, K.; Kuwano, M. Chemosensitisation of spontaneous multidrug resistance by a 1,4-dihydropyridine analog and verapamil in human glioma cell lines overexpressing MRP1 or MDR1. Br. J. Cancer 1995, 72, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Vanhoefer, U.; Cao, S.; Mindermann, H.; Tóth, K.; Scheper, R.J.; Slovak, M.L.; Rustum, Y.M. PAK104P, a pyridine analogue, reverses paclitaxel and doxorubicin resistance in cell lines and nude mice bearing xenografts that overexpress the multidrug resistance protein. Clin. Cancer Res. 1996, 2, 369–377. [Google Scholar]
- Hollo, Z.; Homolya, L.; Hegedüs, T.; Sarkadi, B. Transport properties of the multidrug resistance-associated protein (MRP) in human tumor cells. FEBS Lett. 1996, 383, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.G.; Tan, K.W.; Osa-Andrews, B.; Iram, S.H. High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharm. Res. 2017, 119, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Peck, R.A.; Hewett, J.; Harding, M.W.; Wang, Y.-W.; Chaturvedi, P.R.; Ziessman, A.B.; Atkins, F.; Hawkins, M.J. Phase I and pharmacokinetic study of the novel MDR1 and MRP1 inhibitor biricodar administered alone and in combination with doxirubicin. J. Clin. Oncol. 2001, 19, 3130–3141. [Google Scholar] [CrossRef]
- Suzuki, K.; Saito, K.; Tsujimura, S.; Nakayamada, S.; Yamaoka, K.; Sawamukai, N.; Iwata, S.; Nawata, M.; Nakano, K.; Tanaka, Y. Tacrolimus, a calcineurin inhibitor, overcomes treatment unresponsiveness mediated by P-glycoprotein on lymphocytes in refractory rheumatoid arthritis. J. Rheumatol. 2010, 37, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Cullen, K.V.; Davey, R.A.; Davey, M.W. Verapamil-stimulated glutathione transport by the multidrug resistance-associated protein (MRP1) in leukaemia cells. Biochem. Pharm. 2001, 62, 417–424. [Google Scholar] [CrossRef]
- Zhou, X.F.; Yang, X.; Wang, Q.; Coburn, R.A.; Morris, M.E. Effects of dihydropyridines and pyridines on multidrug resistance mediated by breast cancer resistance protein: In Vitro and In Vivo studies. Drug Metab. Dispos. 2005, 33, 1220–1228. [Google Scholar] [CrossRef] [Green Version]
- Pawarode, A.; Shukla, S.; Minderman, H.; Fricke, S.M.; Pinder, E.M.; O’Loughlin, K.L.; Ambudkar, S.V.; Baer, M.R. Differential effects of the immunosuppressive agents cyclosporin A, tacrolimus and sirolimus on drug transport by multidrug resistance proteins. Cancer Chemother Pharm. 2007, 60, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.P.; Scadden, D.T.; D’Aquila, R.T.; De Pasquale, M.P. The protease inhibitor ritonavir inhibits the functional activity of the multidrug resistance related-protein 1 (MRP-1). AIDS 2002, 16, 1743–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letrent, S.P.; Pollack, G.M.; Brouwer, K.R.; Brouwer, K.L. Effect of GF120918, a potent P-glycoprotein inhibitor, on morphine pharmacokinetics and pharmacodynamics in the rat. Pharm. Res. 1998, 15, 599–605. [Google Scholar] [CrossRef]
- Hoffmann, K.; Xiao, Z.; Franz, C.; Mohr, E.; Serba, S.; Büchler, M.W.; Schemmer, P. Involvement of the epidermal growth factor receptor in the modulation of multidrug resistance in human hepatocellular carcinoma cells In Vitro. Cancer cell Int. 2011, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.K.; Lo, R.C. Deregulation of frizzled receptors in hepatocellular carcinoma. Int. J. Mol. Sci. 2018, 19, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, M.A.; Zhang, J.; Ho, C.; Cheung, S.-T.; Fan, S.-T.; Chen, X. Hedgehog signaling in human hepatocellular carcinoma. Cancer Biol. Ther. 2006, 5, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genois, M.M.; Paquet, E.R.; Laffitte, M.C.; Maity, R.; Rodrigue, A.; Ouellette, M.; Masson, J.Y. DNA repair pathways in trypanosomatids: From DNA repair to drug resistance. Microbiol. Mol. Biol. Rev. 2014, 78, 40–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.F.; Chang, C.W.; Wei, R.J.; Shiue, Y.L.; Wang, S.N.; Yeh, Y.T. Involvement of DNA damage response pathways in hepatocellular carcinoma. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Etiévant, C.; Kruczynski, A.; Barret, J.-M.; Perrin, D.; van Hille, B.; Guminski, Y.; Hill, B.T. F 11782, a dual inhibitor of topoisomerases I and II with an original mechanism of action in vitro, and markedly superior in vivo antitumour activity, relative to three other dual topoisomerase inhibitors, intoplicin, aclarubicin and TAS-103. Cancer Chemother. Pharm. 2000, 46, 101–113. [Google Scholar] [CrossRef]
- Schwab, M. (Ed.) Encyclopedia of Cancer; Springer: Berlin/Heidelberg, Germany, 2011; Available online: http://link.springer.com/10.1007/978-3-642-16483-5. (accessed on 30 June 2021).
- Yovine, A.; Riofrio, M.; Blay, J.Y.; Brain, E.; Alexandre, J.; Kahatt, C.; Taamma, A.; Jimeno, J.; Martin, C.; Salhi, Y.; et al. Phase II Study of Ecteinascidin-743 in Advanced Pretreated Soft Tissue Sarcoma Patients. J. Clin. Oncol. 2004, 22, 890–899. [Google Scholar] [CrossRef]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [Green Version]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [Green Version]
- Yanai, M.; Makino, H.; Ping, B.; Takeda, K.; Tanaka, N.; Sakamoto, T.; Yamaguchi, K.; Kodani, M.; Yamasaki, A.; Igishi, T.; et al. DNA-PK Inhibition by NU7441 Enhances Chemosensitivity to Topoisomerase Inhibitor in Non-Small Cell Lung Carcinoma Cells by Blocking DNA Damage Repair. Yonago Acta Med. 2017, 60, 9–15. [Google Scholar]
- Maes, K.; Lemaire, M.; Gauthier, J.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; Wiklund, H.J.; Van der kerken, K.; De Bruyne, E. The DNA Methyltransferase Inhibitor Decitabine Induces DNA Damage, Cell Cycle Arrest and Apoptosis in Multiple Myeloma. Blood 2012, 120, 1833. [Google Scholar] [CrossRef]
- Quinn, J.A.; Jiang, S.X.; Carter, J.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; et al. Phase II trial of Gliadel plus O6-benzylguanine in adults with recurrent glioblastoma multiforme. Clin. Cancer Res. Off, J. Am. Assoc. Cancer Res. 2009, 15, 1064–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, T.C.; Lopitz-Otsoa, F.; Martínez-Chantar, M.L. Post-translational modifiers of liver kinase B1/serine/threonine kinase 11 in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avellini, C.; Orsaria, M.; Baccarani, U.; Adani, G.L.; Lorenzin, D.; Bresadola, V.; Bresadola, F.; Beltrami, C.A. Apurinic apyrimidinic endonuclease/redox effector factor 1 immunoreactivity and grading in hepatocellular carcinoma risk of relapse after liver transplantation. Transplant. Proc. 2010, 42, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.M.; Sheng, H.; Saxena, R.; Skill, N.J.; Bhat-Nakshatri, P.; Yu, M.; Nakshatri, H.; Maluccio, M.A. NF-κB inhibition in human hepatocellular carcinoma and its potential as adjunct to sorafenib based therapy. Cancer Lett. 2009, 278, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Berk, M.; Alkhouri, N.; Partrick, D.A.; Fung, J.J.; Feldstein, A. Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. J. Surg. Res. 2014, 186, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, E.; Lee, S.; Lee, J.; Rashid, M.A.; Kim, Y.W.; Park, J.H.; Park, W.S.; Shokat, K.M.; Ha, J.; Kim, S.S. Human carbonyl reductase 1 upregulated by hypoxia renders resistance to apoptosis in hepatocellular carcinoma cells. J. Hepatol. 2011, 54, 328–339. [Google Scholar] [CrossRef]
- Guo, J.; Li, L.; Guo, B.; Liu, D.; Shi, J.; Wu, C.; Chen, J.; Zhang, X.; Wu, J. Mechanisms of resistance to chemotherapy and radiotherapy in hepatocellular carcinoma. Transl. Cancer Res. 2018, 7, 765–781. [Google Scholar] [CrossRef]
- Drishya, G.; Nambiar, J.; Shaji, S.K.; Vanuopadath, M.; Achuthan, A.; Kumar, A.; Alias, A.; Sherif, A.; Joseph, C.; Divya, P.; et al. RECK and TIMP-2 mediate inhibition of MMP-2 and MMP-9 by Annona muricata. J. Biosci. 2020, 45, 1–11. [Google Scholar] [CrossRef]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 2019, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.S.N.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.C.; Duiker, E.W.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef] [Green Version]
- Orr-Asman, M.A.; Chu, Z.; Jiang, M.; Worley, M.; LaSance, K.; Koch, S.E.; Carreira, V.S.; Dahche, H.M.; Plas, D.R.; Komurov, K.; et al. mTOR Kinase Inhibition Effectively Decreases Progression of a Subset of Neuroendocrine Tumors that Progress on Rapalog Therapy and Delays Cardiac Impairment. Mol. Cancer Ther. 2017, 16, 2432–2441. [Google Scholar] [CrossRef] [Green Version]
- Oudard, S.; Medioni, J.; Aylllon, J.; Barrascourt, E.; Elaidi, R.-T.; Balcaceres, J.; Scotte, F. Everolimus (RAD001): An mTOR inhibitor for the treatment of metastatic renal cell carcinoma. Expert. Rev. Anticancer. Ther. 2009, 9, 705–717. [Google Scholar] [CrossRef]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic Trioxide Inhibits the Hedgehog Pathway Which Is Aberrantly Activated in Acute Promyelocytic Leukemia. Acta Haematol. 2013, 130, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Huang, L.; Liao, Z.; Liu, M.; Li, Q.; Xu, R. Itraconazole inhibits the Hedgehog signaling pathway thereby inducing autophagy-mediated apoptosis of colon cancer cells. Cell Death Dis. 2020, 11, 539. [Google Scholar] [CrossRef]
- Girardi, D.; Barrichello, A.; Fernandes, G.; Pereira, A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells 2019, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A.J.; Wedge, S.R. ZD6474–a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br. J. Cancer 2005, 92, S6–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zhang, W.; Wang, H.; Zhang, Z.; Chu, C.; Liu, X.; Zou, Q. EGFR/HER2 inhibitors effectively reduce the malignant potential of MDR breast cancer evoked by P-gp substrates In Vitro and In Vivo. Oncol. Rep. 2016, 35, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Yahya, S.M.M.; Fathy, S.A.; El-Khayat, Z.A.; El-Toukhy, A.E.; Hamed, A.R.; Hegazy, M.G.A.; Nabih, H.K. Possible Role of microRNA-122 in Modulating Multidrug Resistance of Hepatocellular Carcinoma. Indian J. Clin. Biochem. 2018, 33, 21–30. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Tian, W.; Chen, H.; Deng, Y. MicroRNA-101 sensitizes hepatocellular carcinoma cells to doxorubicin-induced apoptosis via targeting Mcl-1. Mol. Med. Rep. 2016, 13, 1923–1929. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wang, Y.; Li, M.; Zhu, Y.; Liang, H.; Wang, C.; Wang, F.; Zhang, C.-Y.; Zen, K.; Li, L. miR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017, 8, e2540. [Google Scholar] [CrossRef]
- Xue, H.; Yu, Z.; Liu, Y.; Yuan, W.; Yang, T.; You, J.; He, X.; Lee, R.J.; Li, L.; Xu, C. Delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 5271–5287. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zheng, Z.-M.; Li, X.-N.; Li, Z.-F.; Wang, Y.; Geng, Y.-F.; Bai, L.; Zhang, X.-B. miR-223 modulates multidrug resistance via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp. Biol. Med. 2013, 238, 1024–1032. [Google Scholar] [CrossRef]
- Gao, A.M.; Zhang, X.Y.; Hu, J.N.; Ke, Z.-P. Apigenin sensitizes hepatocellular carcinoma cells to doxorubicin through regulating miR-520b/ATG7 axis. Chem. Biol. Interact. 2018, 280, 45–50. [Google Scholar] [CrossRef]
- Xu, N.; Shen, C.; Luo, Y.; Xia, L.; Xue, F.; Xia, Q.; Zhang, J. Upregulated miR-130a increases drug resistance by regulating RUNX3 and Wnt signaling in cisplatin-treated HCC cell. Biochem. Biophys. Res. Commun. 2012, 425, 468–472. [Google Scholar] [CrossRef]
- Qin, J.; Luo, M.; Qian, H.; Chen, W. Upregulated miR-182 increases drug resistance in cisplatin-treated HCC cell by regulating TP53INP1. Gene 2014, 538, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Wang, X.; Shi, K.; Zheng, Y.; Li, J.; Chen, Y.; Jin, L.; Pan, Z. MiR-122 reverses the doxorubicin-resistance in hepatocellular carcinoma cells through regulating the tumor metabolism. PLoS ONE 2016, 11, e0152090. [Google Scholar]
- Xia, H.; Ooi, L.L.P.J.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Pratama, M.Y.; Pascut, D.; Massi, M.N.; Tiribelli, C. The role of microRNA in the resistance to treatment of hepatocellular carcinoma. Ann. Transl. Med. 2019, 7, 577. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.C.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzon, R.; Fabbri, M.; Cimmino, A.; Calin, G.A.; Croce, C.M. MicroRNA expression and function in cancer. Trends. Mol. Med. 2006, 12, 580–587. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dan Dumitru, C.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Gaur, A.; Jewell, D.; Liang, Y.; Ridzon, D.; Moore, J.; Chen, C.; Ambros, V.R.; Israel, M.A. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007, 67, 2456–2468. [Google Scholar] [CrossRef] [Green Version]
- Ohta, K.; Hoshino, H.; Wang, J.; Ono, S.; Iida, Y.; Hata, K.; Huang, S.K.; Colquhoun, S.; Hoon, D.S. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget 2015, 6, 3211. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-F.; Sun, J.-P.; Hou, H.-T.; Li, K.; Liu, X.; Ge, Q.-X. MicroRNA-27b exerts an oncogenic function by targeting Fbxw7 in human hepatocellular carcinoma. Tumor Biol. 2016, 37, 15325–15332. [Google Scholar] [CrossRef]
- Azumi, J.; Tsubota, T.; Sakabe, T.; Shiota, G. miR-181a induces sorafenib resistance of hepatocellular carcinoma cells through downregulation of RASSF 1 expression. Cancer Sci. 2016, 107, 1256–1262. [Google Scholar] [CrossRef]
- Pollutri, D.; Patrizi, C.; Marinelli, S.; Giovannini, C.; Trombetta, E.; Giannone, F.A.; Baldassarre, M.; Quarta, S.; Vandewynckel, Y.P.; Vandierendonck, A.; et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Liu, K.; Liu, S.; Zhang, W.; Jia, B.; Tan, L.; Jin, Z.; Liu, Y. miR-494 promotes cell proliferation migration, invasion; increased sorafenib resistance in hepatocellular carcinoma by targeting, PTEN. Oncol. Rep. 2015, 34, 1003–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Kwon, H.; Han, C.; Zhang, J.; Dash, S.; Lim, K.; Wu, T. Active glycolytic metabolism in CD133 (+) hepatocellular cancer stem cells: Regulation by MIR-122. Oncotarget 2015, 6, 40822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Li, Q.J.; Gong, Z.B.; Zhou, L.; You, N.; Wang, S.; Li, X.L.; Li, J.J.; An, J.Z.; Wang, D.S.; et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol. Cancer Res. Treat. 2014, 13, 77–86. [Google Scholar] [CrossRef]
- Sun, H.; Cui, C.; Xiao, F.; Wang, H.; Xu, J.; Shi, X.; Yang, Y.; Zhang, Q.; Zheng, X.; Yang, X.; et al. miR-486 regulates metastasis chemosensitivity in hepatocellular carcinoma by targeting, CLDN10 and CITRON. Hepatol. Res. 2015, 45, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lin, H.; Li, G.; Sun, Y.; Chen, J.; Shi, L.; Cai, X.; Chang, C. The miR-367-3p increases sorafenib chemotherapy efficacy to suppress hepatocellular carcinoma metastasis through altering the androgen receptor signals. EBioMedicine 2016, 12, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Chen, J.; Zhou, H.; Chen, Y.; Zhi, Y.; Zhang, B.; Chen, L.; Chu, X.; Wang, R.; Zhang, C. PU.1/microRNA-142-3p targets ATG5/ATG16L1 to inactivate autophagy and sensitize hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Sceusi, E.L.; Loose, D.S.; Wray, C.J. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB 2011, 13, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Mansour, L.A.; El Raziky, M.; Mohamed, A.A.; Mahmoud, E.H.; Hamdy, S.; El Sayed, E.H. Circulating hypermethylated RASSF1A as a molecular biomarker for diagnosis of hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2017, 18, 1637. [Google Scholar]
- Panvichian, R.; Tantiwetrueangdet, A.; Angkathunyakul, N.; Leelaudomlipi, S. TOP2A Amplification and Overexpression in Hepatocellular Carcinoma Tissues. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Y.; Liu, X.W.; Zhu, H.; Cao, J.; Zhang, J.; Ding, L.; Lou, J.S.; He, Q.J.; Yang, B. Tirapazamine sensitizes hepatocellular carcinoma cells to topoisomerase I inhibitors via cooperative modulation of hypoxia-inducible factor-1α. Mol. Cancer Ther. 2014, 13, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, S.; Li, M.Y.; Hu, B.G.; Liu, L.P.; Yang, S.L.; Yang, S.; Gong, Z.; Lai, P.B.; Chen, G.G. Cancer stem cells in hepatocellular carcinoma: An overview and promising therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816287. [Google Scholar] [CrossRef]
- Sukowati, C.H.; Rosso, N.; Crocè, L.S.; Tiribelli, C. Hepatic cancer stem cells and drug resistance: Relevance in targeted therapies for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 114. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, Q.; Zhao, J.; Liao, M.; Wen, S.; Yang, M. Phosphorylation of Bcl-2 plays an important role in glycochenodeoxycholate-induced survival and chemoresistance in HCC. Oncol. Rep. 2017, 38, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.K.; Le, Y.; Zhang, Y.F.; Ouyang, H.Y.; Jian, P.E.; Yu, Z.S.; Wang, L.J.; Shi, M. Matrix metalloproteinase 12 expression is associated with tumor FOXP3+ regulatory T cell infiltration and poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2018, 16, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Chang, T.C.; Lin, Y.T.; Yu, Y.L.; Ko, B.S.; Sung, L.Y.; Liou, J.Y. Paracrine regulation of matrix metalloproteinases contributes to cancer cell invasion by hepatocellular carcinoma-secreted 14-3-3σ. Oncotarget 2016, 7, 36988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, Y.; Cao, B.; Yan, A.; Ji, H. Elevated expression levels of androgen receptors and matrix metalloproteinase-2 and-9 in 30 cases of hepatocellular carcinoma compared with adjacent tissues as predictors of cancer invasion and staging. Exp. Ther. Med. 2015, 9, 905–908. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Oda, M.; Yokomori, H.; Han, J.Y. Regulatory mechanisms of hepatic microcirculation. Clin. Hemorheol. Microcirc. 2003, 29, 167–182. [Google Scholar] [PubMed]
- Maslak, E.; Gregorius, A.; Chlopicki, S. Liver sinusoidal endothelial cells (LSECs) function and NAFLD.; NO-based therapy targeted to the liver. Pharmacol. Rep. 2015, 67, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Wake, K.; Decker, K.; Kirn, A.; Knook, D.L.; McCuskey, R.S.; Bouwens, L.; Wisse, E. Cell biology and kinetics of Kupffer cells in the liver. Int. Rev. Cytol. 1989, 118, 173–229. [Google Scholar] [PubMed]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Carambia, A.; Frenzel, C.; Bruns, O.T.; Schwinge, D.; Reimer, R.; Hohenberg, H.; Huber, S.; Tiegs, G.; Schramm, C.; Lohse, A.W.; et al. Inhibition of inflammatory CD4T cell activity by murine liver sinusoidal endothelial cells. J. Hepatol. 2013, 58, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD81T cell tolerance. Hepatology 2008, 47, 296–305. [Google Scholar] [CrossRef]
- Knolle, P.A.; Germann, T.; Treichel, U.; Uhrig, A.; Schmitt, E.; Hegenbarth, S.; Lohse, A.W.; Gerken, G. Endotoxin downregulatesT cell activation by antigen-presenting liver sinusoidal endothelial cells. J. Immunol. 1999, 162, 1401–1407. [Google Scholar] [PubMed]
- Knolle, P.A.; Uhrig, A.; Hegenbarth, S.; Löser, E.; Schmitt, E.; Gerken, G.; Lohse, A.W. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin. Exp. Immunol. 1998, 114, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Hegenbarth, S.I.; Schumak, B.; Scholz, K.; Limmer, A.; Knolle, P.A. Liver sinusoidal endothelial cells veto CD8T cell activationby antigen-presenting dendritic cells. Eur. J. Immunol. 2008, 38, 957–967. [Google Scholar] [CrossRef]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291.12. [Google Scholar] [CrossRef]
- You, Q.; Cheng, L.; Kedl, R.M.; Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008, 48, 978–990. [Google Scholar] [CrossRef]
- Lever, M.; Maini, P.K.; Van Der Merwe, P.A.; Dushek, O. Phenotypic models of T cell activation. Nat. Rev. Immunol. 2014, 14, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Chraa, D.; Naim, A.; Olive, D.; Badou, A. T lymphocyte subsets in cancer immunity: Friends or foes. J. Leukoc. Biol. 2019, 105, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef]
- Weston, C.J.; Zimmermann, H.W.; Adams, D.H. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol 2019, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.Y.; et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013, 58, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Qiu, S.J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-cell therapy for solid tumors. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 193–204. [Google Scholar] [CrossRef]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Kim, Y.; Shuang, Z.Y.; Zhang, Y.J.; Lao, X.M.; Li, Y.Q.; Chen, M.S.; Pawlik, T.M.; Xia, J.C.; et al. A randomized controlled trial on patients with or without adjuvant autologous cytokine-induced killer cells after curative resection for hepatocellular carcinoma. Oncoimmunology 2016, 5, e1083671. [Google Scholar] [CrossRef] [Green Version]
- Toh, U.; Fujii, T.; Seki, N.; Niiya, F.; Shirouzu, K.; Yamana, H. Characterization of IL-2-activated TILs and their use in intrapericardial immunotherapy in malignant pericardial effusion. Cancer Immunol. Immunother. 2006, 55, 1219–1227. [Google Scholar] [CrossRef]
- Jiang, S.S.; Tang, Y.; Zhang, Y.J.; Weng, D.S.; Zhou, Z.G.; Pan, K.; Pan, Q.Z.; Wang, Q.J.; Liu, Q.; He, J.; et al. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget 2015, 6, 41339. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Chang, Y.H.; Campana, D. Expanded and activated natural killer cells for immunotherapy of hepatocellular carcinoma. Cancer Immunol. Res. 2016, 4, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Ohira, M.; Nishida, S.; Levi, D.; Tekin, A.; Selvaggi, G.; Ruiz, P.; Ricordi, C.; Ohdan, H.; Tzakis, A. Adoptive immunotherapy using liver natural killer cells for preventing recurrence of hepatocellular carcinoma in cadaveric donor liver transplantation. J. Immunol. 2012, 188, 53.20. [Google Scholar]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD 19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017, 7, 2091. [Google Scholar] [PubMed]
- Nakamoto, Y.; Mizukoshi, E.; Kitahara, M.; Arihara, F.; Sakai, Y.; Kakinoki, K.; Fujita, Y.; Marukawa, Y.; Arai, K.; Yamashita, T.; et al. Prolonged recurrence-free survival following OK432-stimulated dendritic cell transfer into hepatocellular carcinoma during transarterial embolization. Clin. Exp. Immunol. 2011, 163, 165–177. [Google Scholar] [CrossRef]
- Rizell, M.; SternbyEilard, M.; Andersson, M.; Andersson, B.; Karlsson-Parra, A.; Suenaert, P. Phase 1 trial with the cell-based immune primer ilixadencel, alone, and combined with sorafenib, in advanced hepatocellular carcinoma. Front. Oncol. 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Mizukoshi, E.; Kobayashi, E.; Tamai, T.; Hamana, H.; Ozawa, T.; Kishi, H.; Kitahara, M.; Yamashita, T.; Arai, K.; et al. Association Between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein− Derived Peptides, and Anti-Tumor Effects in Patients with Hepatocellular Carcinoma. Gastroenterology 2017, 152, 1395–1406. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukoshi, E.; Nakagawa, H.; Kitahara, M.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Fushimi, K.; Kobayashi, E.; Kishi, H.; Muraguchi, A.; et al. Immunological features of T cells induced by human telomerase reverse transcriptase-derived peptides in patients with hepatocellular carcinoma. Cancer Lett. 2015, 364, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Dargel, C.; Bassani-Sternberg, M.; Hasreiter, J.; Zani, F.; Bockmann, J.H.; Thiele, F.; Bohne, F.; Wisskirchen, K.; Wilde, S.; Sprinzl, M.F.; et al. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology 2015, 149, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Jiang, X.; Li, Q.; Liu, H.; Huang, L.; Wu, J.; Celis, E.; et al. Identification of α-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018, 68, 574–589. [Google Scholar] [CrossRef] [Green Version]
- Thibodeaux, S.R.; Milone, M.C. Immunotherapy using chimeric antigen receptor-engineered T cells: A novel cellular therapy with important implications for the clinical laboratory. Clin. Chem. 2019, 65, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, K.; Jiang, H.; Song, F.; Gao, H.; Pan, X.; Shi, B.; Bi, Y.; Wang, H.; Wang, H.; et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol. Immunother. 2017, 66, 475–489. [Google Scholar] [CrossRef] [PubMed]
- El Dika, I.; Khalil, D.N.; Abou-Alfa, G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer 2019, 125, 3312–3319. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, Y.; Zhang, J.; Zhang, Y.; Li, Y.; Liu, Z.; Li, Q.; Luo, M.; Liang, R.; Ye, J. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 447. [Google Scholar] [CrossRef]
- Lu, L.C.; Hsu, C.; Shao, Y.Y.; Chao, Y.; Yen, C.J.; Shih, I.L.; Hung, Y.P.; Chang, C.J.; Shen, Y.C.; Guo, J.C.; et al. Differential Organ-Specific Tumor Response to Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Liver Cancer 2019, 8, 480–490. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K. The Use of Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Gastroenterol. Hepatol. 2019, 15, 48. [Google Scholar]
- Liu, J.; Liu, Y.; Meng, L.; Liu, K.; Ji, B. Targeting the PD-L1/DNMT1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol. Rep. 2017, 38, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Parayath, N.; Padmakumar, S.; Nair, S.V.; Menon, D.; Amiji, M.M. Strategies for Targeting Cancer Immunotherapy Through Modulation of the Tumor Microenvironment. Regen. Eng. Transl. Med. 2020, 6, 29–49. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Waidmann, O.; Trojan, J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev. Anticancer. Ther. 2018, 18, 1169–1175. [Google Scholar] [CrossRef]
- Tan, K.W.; Chacko, A.M.; Chew, V. PD-1 expression and its significance in tumour microenvironment of hepatocellular carcinoma. Transl. Gastroenterol. Hepatol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- MacekJilkova, Z.; Aspord, C.; Decaens, T. Predictive Factors for Response to PD-1/PD-L1 Checkpoint Inhibition in the Field of Hepatocellular Carcinoma: Current Status and Challenges. Cancers 2019, 11, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M. Pembrolizumab for the treatment of hepatocellular carcinoma. Liver Cancer 2019, 8, 143–154. [Google Scholar] [CrossRef]
- Zhu, X.D.; Sun, H.C. Emerging agents and regimens for hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 110. [Google Scholar] [CrossRef]
- Liu, Z.; Li, X.; He, X.; Xu, Y.; Wang, X. Complete response to the combination of Lenvatinib and Pembrolizumab in an advanced hepatocellular carcinoma patient: A case report. BMC Cancer 2019, 19, 1062. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.L.; Ni, C.F.; Liang, H.S.; Xu, Y.H.; Wang, W.S.; Shen, J.; Li, M.M.; Zhu, X.L. Upregulation of PD-L1 expression promotes epithelial-to-mesenchymal transition in sorafenib-resistant hepatocellular carcinoma cells. Gastroenterol. Rep. 2020, 8, 390–398. [Google Scholar] [CrossRef]
- Cheng, H.; Sun, G.; Chen, H.; Li, Y.; Han, Z.; Li, Y.; Zhang, P.; Yang, L.; Li, Y. Trends in the treatment of advanced hepatocellular carcinoma: Immune checkpoint blockade immunotherapy and related combination therapies. Am. J. Cancer Res. 2019, 9, 1536. [Google Scholar]
- Xu, Y.; Jiang, X.; Huang, Y. T-cell immunoglobulin and mucin-domain containing-3 in malignant cancers. Transl. Cancer Res. 2017, 6, 613–619. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Y.; Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 2018, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 11, 7005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobuoka, D.; Yoshikawa, T.; Sawada, Y.; Fujiwara, T.; Nakatsura, T. Peptide vaccines for hepatocellular carcinoma. Hum. Vaccines Immunother. 2013, 9, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H.; Economou, J.S.; Gamblin, T.C.; Geller, D.A. Alpha fetoprotein DNA prime and adenovirus boost immunization of two hepatocellular cancer patients. J. Transl. Med. 2014, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Bourke, M.G.; Salwa, S.; Harrington, K.J.; Kucharczyk, M.J.; Forde, P.F.; de Kruijf, M.; Soden, D.; Tangney, M.; Collins, J.K.; O’Sullivan, G.C. The emerging role of viruses in the treatment of solid tumours. Cancer Treat. Rev. 2011, 37, 618–632. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Brown, K.T.; Heo, J.; Borad, M.J.; Luca, A.; Pelusio, A.; Agathon, D.; Lusky, M.; et al. PHOCUS: A phase 3 randomized, open-label study comparing the oncolytic immunotherapy Pexa-Vec followed by sorafenib (SOR) vs SOR in patients with advanced hepatocellular carcinoma (HCC) without prior systemic therapy. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329. [Google Scholar] [CrossRef]
- Hilmi, M.; Neuzillet, C.; Calderaro, J.; Lafdil, F.; Pawlotsky, J.M.; Rousseau, B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: Current knowledge and future research directions. J. Immunother. Cancer 2019, 7, 333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Liang, Q.; Liu, B.; Mei, X.; Ma, Y. Combinatorial immunotherapy of sorafenib and blockade of programmed death-ligand 1 induces effective natural killer cell responses against hepatocellular carcinoma. Tumor Biol. 2015, 36, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol. Med. 2019, 25, 1010–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Liu, K.; Chen, M.; Sun, J.Y.; Caughan, G.W.; Lu, X.J.; Ji, J. Immunotherapy for hepatocellular carcinoma: Recent advances and future perspectives. Ther. Adv. Med. Oncol. 2019, 11, 1758835919862692. [Google Scholar] [CrossRef] [Green Version]
- Johnston, M.P.; Khakoo, S.I. Immunotherapy for hepatocellular carcinoma: Current and future. World J. Gastroenterol. 2019, 25, 2977. [Google Scholar] [CrossRef] [PubMed]
- George, A.P.; Kuzel, T.M.; Zhang, Y.; Zhang, B. The Discovery of Biomarkers in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 484. [Google Scholar] [CrossRef]
- Masucci, G.V.; Cesano, A.; Eggermont, A.; Fox, B.A.; Wang, E.; Marincola, F.M.; Ciliberto, G.; Dobbin, K.; Puzanov, I.; Taube, J.; et al. The need for a network to establish and validate predictive biomarkers in cancer immunotherapy. J. Transl. Med. 2017, 15, 223. [Google Scholar] [CrossRef]
- Flemming, A. Tumour heterogeneity determines immune response. Nat. Rev. Immunol. 2019, 19, 662–663. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter Involved in MDR | Effluxed Cytotoxic Drug | Drug Transporter Inhibitors as Chemosensitizers | |
|---|---|---|---|
| Preclinical Reports | Clinical Reports | ||
| P-gp (ABCB1) | Doxorubicin, Daunorubicin, Etoposide, Teniposide, Methotrexate, Sorafenib, Paclitaxel, Vincristine, Vinblastine | Verapamil [13] Nifedipine [14] Nimodipine [15] Amiodarone [16] Cyclosporine A [17] Tacrolimus [18] | Quinine [19] Tesmilifene [20] Biricodar [21] |
| MRP1 (ABCC1) | Doxorubicin, Daunorubicin, Methotrexate, Irinotecan, Etoposide, Teniposide, Imatinib, Gefitinib, Vincristine, Vinblastine | Disulfiram [22] Pak-104P [23] Cyclosporin [24] | Biricodar [25] Quinine [19] Tacrolimus [26] |
| BCRP (ABCG2) | Doxorubicin, Daunorubicin, Epirubicin, Methotrexate, Irinotecan, Etoposide, Teniposide, Imatinib, Gefitinib. | Nifedipine [27] Dihydropyridine [28] Cyclosporine A [29] Ritonavir [30] | Elacridar [31] |
| DNA Damage Response Mechanism | Affected Cytotoxic Drugs | Drugs Targeting DDR as Chemosensitizers | |
|---|---|---|---|
| Preclinical Reports | Clinical Reports | ||
| Nucleotide Excision Repair | Cisplatin, Alkylating agents | F117 82 [37] UCN01 [38] | Ecteinascidin 743 [39] |
| Homologous Recombination | Doxorubicin | 17-AAG [40] ImatinibErlotinib [41] PCI-24781 [42] B02 [43] | |
| Non-homologous end joining | Topoisomerase inhibitors | NU7441 [44] | |
| DNA mismatch repair | Cisplatin, Carboplatin | Decitabine [45] | |
| O6-methyl guanine DNA methyl transferase | Alkylating agents | O6-benzyl guanine [46] | |
| Tumor Microenvironment Associated Targets Involved in MDR | Drugs Modulating Tumor Microenvironment as Chemosensitizers | |
|---|---|---|
| Preclinical Report | Clinical Report | |
| COLI A2 | Let-7 g [54,55] | |
| Extracellular matrix protein | PI 88 [56] | |
| VEGFR2, FGFR1 | Brivatinib (NCT00858871) | |
| VEGFR2, FGFR, PDGFR | TSU-68 (NCT00784290) | |
| PDGFR | MEDI-575 (NCT01102400) | |
| GPC3 | GC33 (NCT01102400) | |
| Over Expressed Cell Survival Pathway | Cancer Type | Inhibitors of Cell Survival Pathways as Chemosensitizers | |
|---|---|---|---|
| Preclinical reports | Clinical Reports | ||
| PI3k/AKT/mTOR | Ovarian, cervical, gastric, breast, colorectal, hepatocellular, thyroid, endometroid, glioblastoma, acute leukaemia | AZ D8055 [57] CC-2223 [58] | Rapamycin, RAD001 [59] |
| Hedgehog | Gastro oesophageal, pancreatic, hepatocellular, brain, non-small cell lung cancer, glioblastoma | arsenic trioxide [60] Itraconazole [61] | Sonidigib, buparlisib, vismodigib, saridigib, taladigib [62] |
| EGFR | Head and neck, breast, renal, cervix, esophageal, pancreatic, non-small cell lung, colon, liver, bladder, gastric | Ramucirumab [63,64] Erlotinib Lapatinib Cetuximab Vandetanib | |
| miRNA | Resistance Conferring Targets Modified by MiRNA |
|---|---|
| MiR-122 | IGF-1R [81] SRF ADAM10 PDK4 SLC7A1 GALNT |
| miR-34a | BCL-2 [82] |
| MiR-27b | P53 [83] |
| let-7 | Bcl-XL [84] |
| miR-193b | MCL1 [85] |
| miR-486 | CITRON CLDN10 [86] AR |
| miR-367-3p | CITRON CLDN10 [87] AR |
| miR-338-3p | HIF-1α [88] |
| miR-142-3p | ATG5 ATG16L1 [89] |
| miR-7 | TYRO3 TYRO3-AXL-MER [90] |
| Inhibitor | Target | Reference/Trial ID |
|---|---|---|
| Nivolumab | PD L1 | NCT01658878 |
| Pembrolizumab | PD 1 | NCT02702414 |
| Pidilizumab | DLL 1, PD 1 | NCT00966251 |
| Ipilimumab | CTLA 4 | NCT03510871 |
| Tremelimumab | CTLA 4 | NCT03638141 |
| Dual blockade antibody | TIM 3 | NCT03680508 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devan, A.R.; Kumar, A.R.; Nair, B.; Anto, N.P.; Muraleedharan, A.; Mathew, B.; Kim, H.; Nath, L.R. Insights into an Immunotherapeutic Approach to Combat Multidrug Resistance in Hepatocellular Carcinoma. Pharmaceuticals 2021, 14, 656. https://doi.org/10.3390/ph14070656
Devan AR, Kumar AR, Nair B, Anto NP, Muraleedharan A, Mathew B, Kim H, Nath LR. Insights into an Immunotherapeutic Approach to Combat Multidrug Resistance in Hepatocellular Carcinoma. Pharmaceuticals. 2021; 14(7):656. https://doi.org/10.3390/ph14070656
Chicago/Turabian StyleDevan, Aswathy R., Ayana R. Kumar, Bhagyalakshmi Nair, Nikhil Ponnoor Anto, Amitha Muraleedharan, Bijo Mathew, Hoon Kim, and Lekshmi R. Nath. 2021. "Insights into an Immunotherapeutic Approach to Combat Multidrug Resistance in Hepatocellular Carcinoma" Pharmaceuticals 14, no. 7: 656. https://doi.org/10.3390/ph14070656
APA StyleDevan, A. R., Kumar, A. R., Nair, B., Anto, N. P., Muraleedharan, A., Mathew, B., Kim, H., & Nath, L. R. (2021). Insights into an Immunotherapeutic Approach to Combat Multidrug Resistance in Hepatocellular Carcinoma. Pharmaceuticals, 14(7), 656. https://doi.org/10.3390/ph14070656