
New (Iso)quinolinyl-pyridine-2,6-dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Pyridine-2,6-dicarboxamide Derivatives
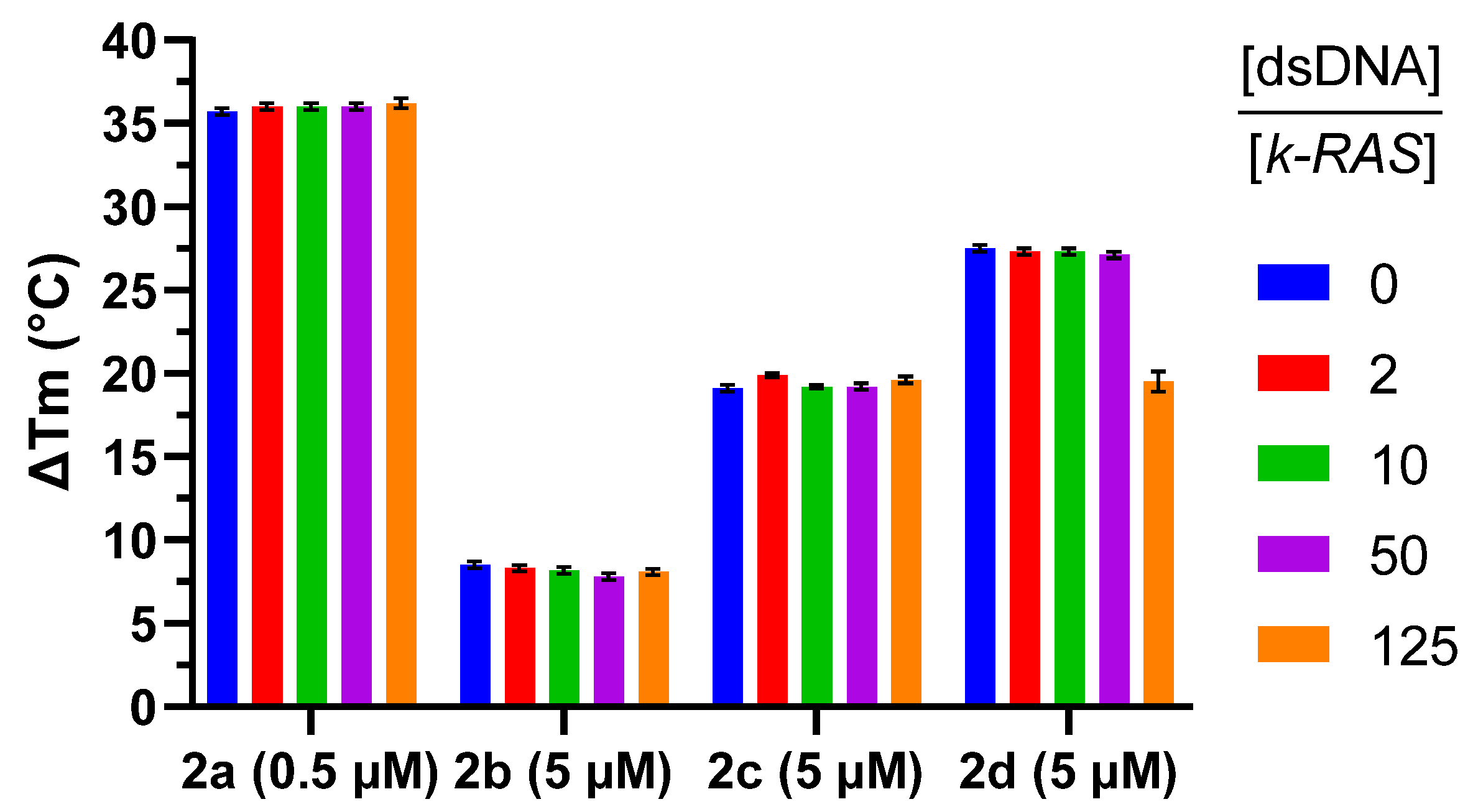
2.2. G4 Thermal Stabilization Induced by Compounds
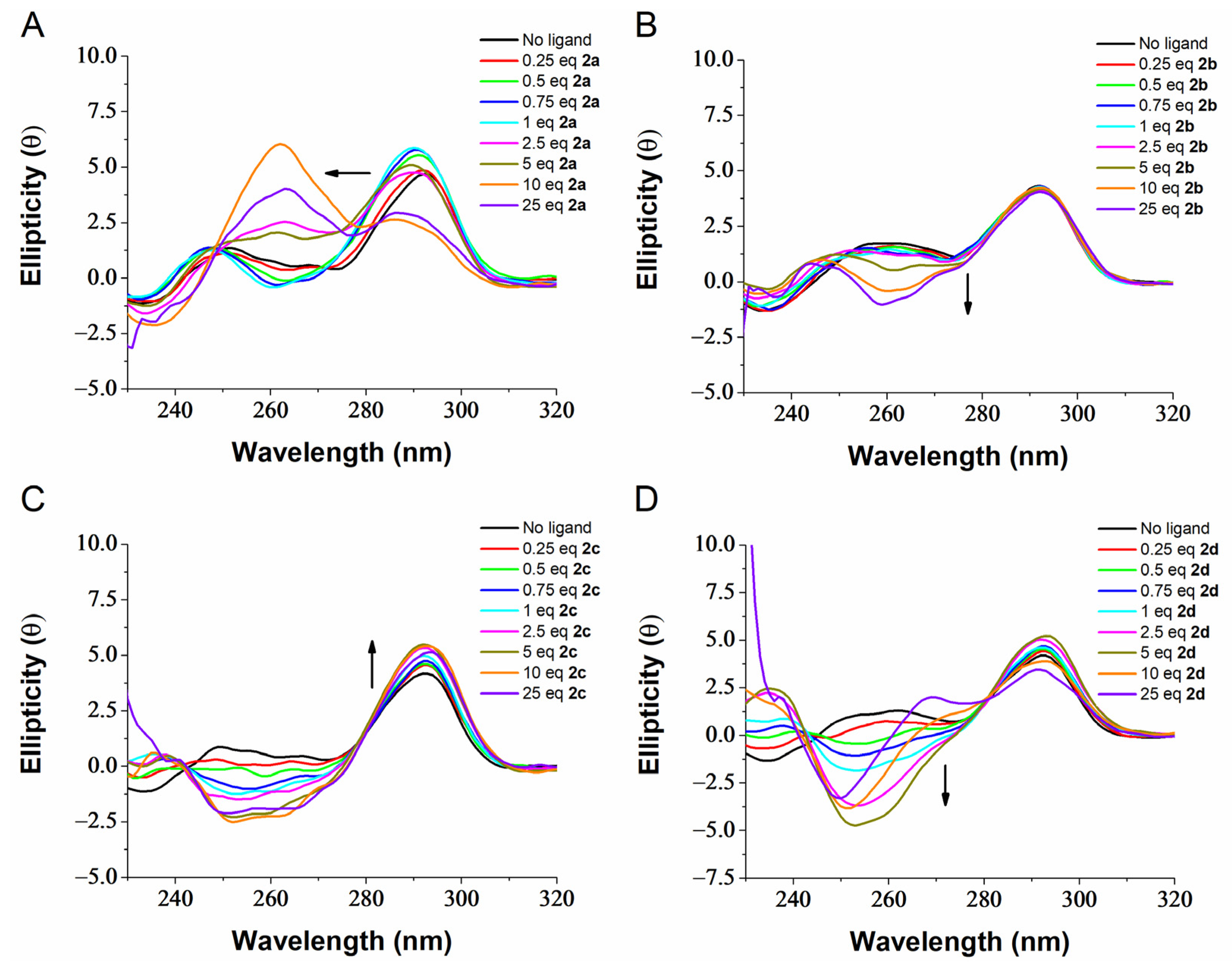
2.3. G4 Conformational Changes Induced by G4 Ligands 2a–d
2.4. Taq Polymerase Inhibition by G4-Ligands
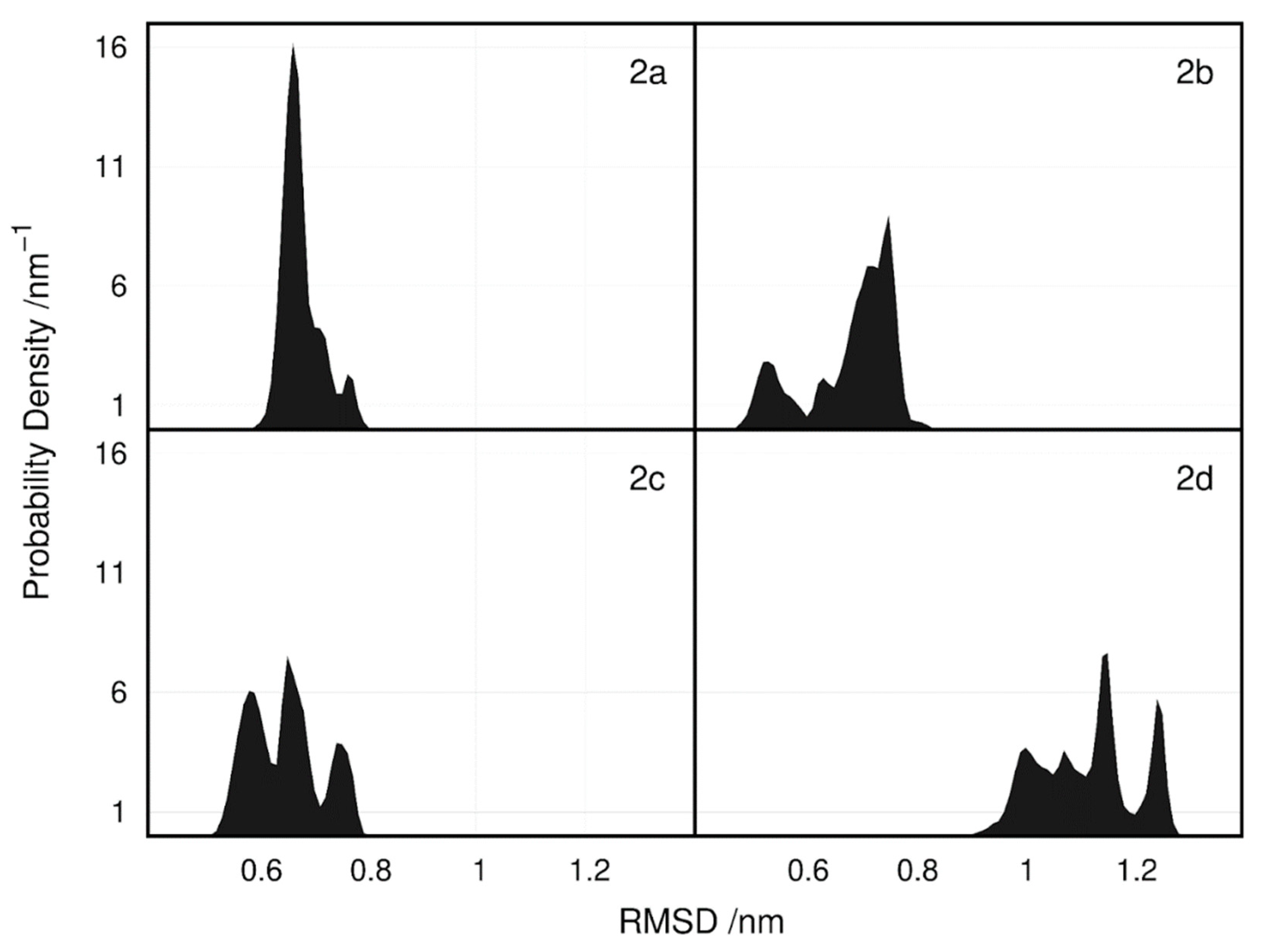
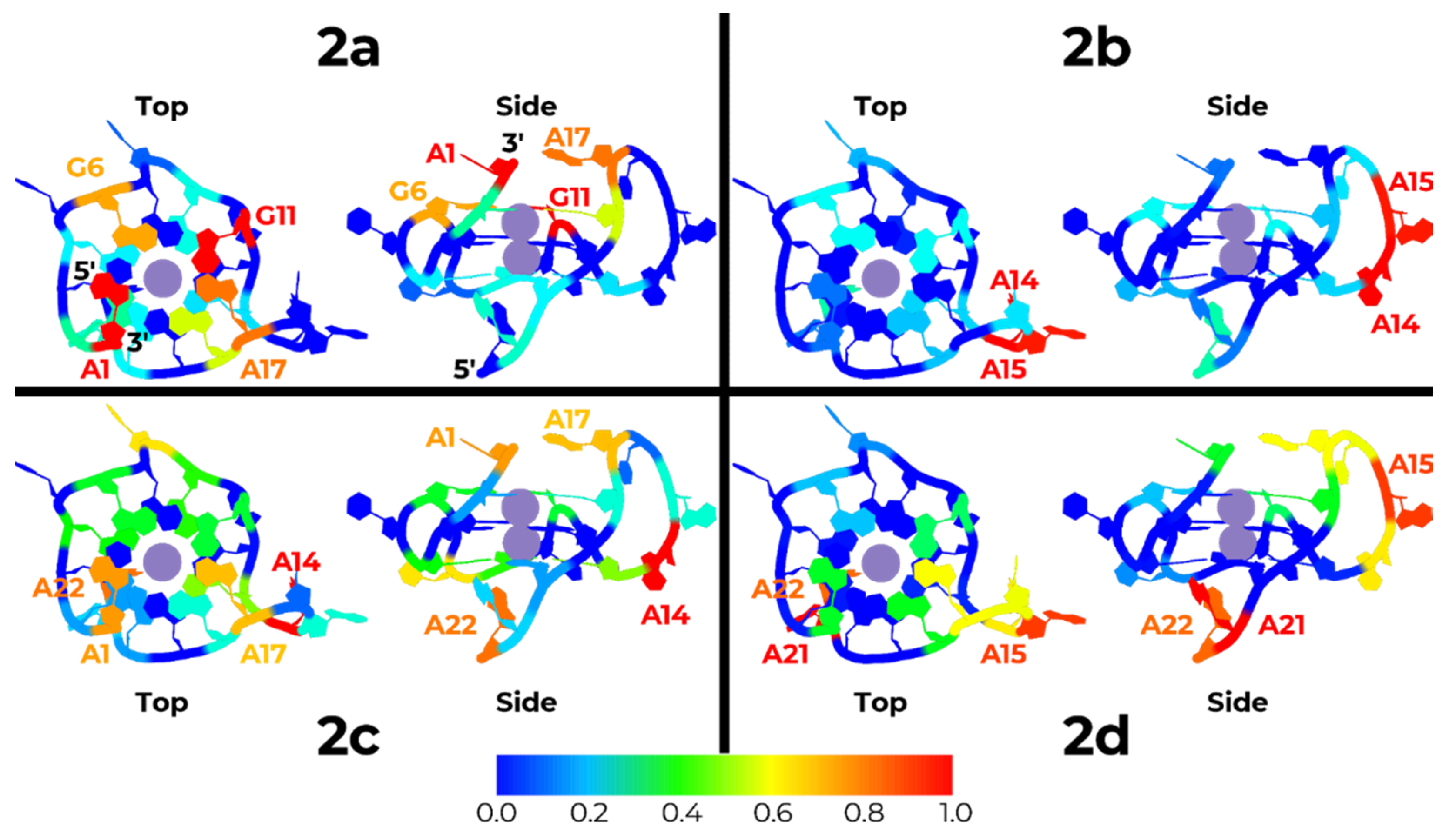
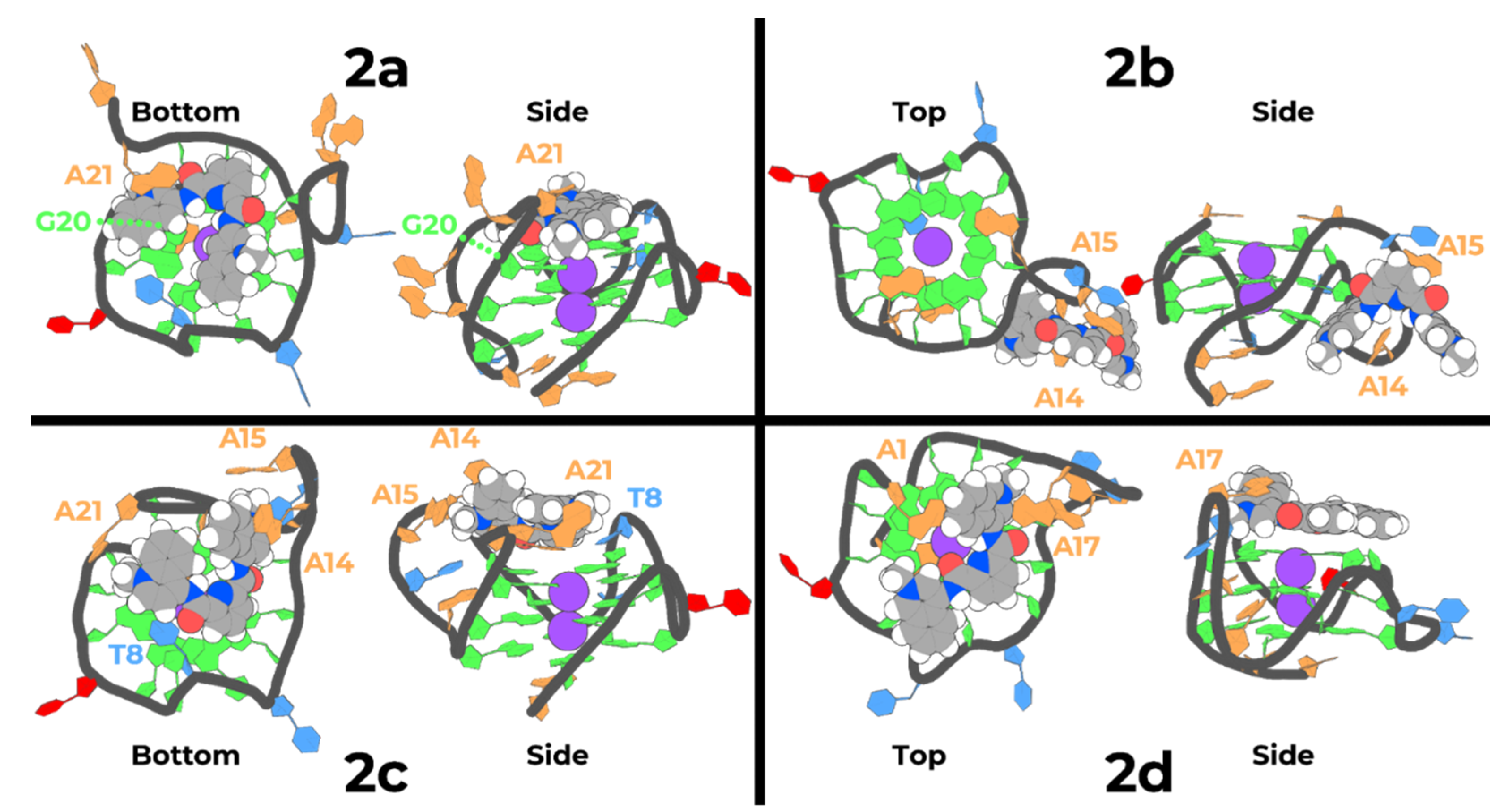
2.5. Molecular Dynamics
3. Materials and Methods
3.1. Synthesis of Compounds
3.1.1. General Procedure A for Synthesis of Bis-quinolinyl/isoquinolinyl-pyridine-2,6-dicarboxamides (1b–h)
3.1.2. General Procedure B for Synthesis of Bis-methyl-quinolinium/isoquinolinium-pyridine 2,6-Dicarboxamides (2a–d)
3.2. PCR-Stop Assay
3.3. FRET-Melting Analysis and Competition Experiments
3.4. CD Titration and Melting Analyses
3.5. Molecular Modeling
3.5.1. Compound Structure Preparation
3.5.2. Molecular Dynamics Simulations
3.5.3. MM/PBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Adjei, A.A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl.Cancer Inst. 2001, 93, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, M.; Babaei, A.; Press, D. Receptor tyrosine kinase (c-Kit ) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, W.C.; Weiss, W.A. Myc proteins as therapeutic targets. Oncogene 2010, 29, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F. KRAS as a therapeutic target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.T.; Kuryavyi, V.; Burge, S.; Neidle, S.; Patel, D.J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 2007, 129, 4386–4392. [Google Scholar] [CrossRef] [Green Version]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Paramasivam, M.; Spolaore, B.; Xodo, L.E. Structural polymorphism within a regulatory element of the human KRAS promoter: Formation of G4-DNA recognized by nuclear proteins. Nucleic Acids Res. 2008, 36, 3765–3780. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Largy, E.; Mergny, J.-L.; Gabelica, V. Role of Alkali Metal Ions in G-Quadruplex Nucleic Acid Structure and Stability. In The Alkali Metal Ions: Their Role for Life; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Metal Ions in Life Sciences; Springer International Publishing: Cham, Switzerland, 2016; Volume 16, pp. 203–258. ISBN 978-3-319-21755-0. [Google Scholar]
- Phan, A.T.; Mergny, J.-L. Human telomeric DNA: G-quadruplex, i-motif and Watson-Crick double helix. Nucleic Acids Res. 2002, 30, 4618–4625. [Google Scholar] [CrossRef]
- Pennarun, G.; Granotier, C.; Gauthier, L.R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.F.; Mergny, J.L.; Mailliet, P.; Boussin, F.D. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene 2005, 24, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Balasubramanian, S. Existence and consequences of G-quadruplex structures in DNA. Curr. Opin. Genet. Dev. 2014, 25, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hänsel-hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, E.; De Paepe, L.; Manicardi, A.; Madder, A. Beyond small molecules: Targeting G-quadruplex structures with oligonucleotides and their analogues. Nucleic Acids Res. 2021, 1–22. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Paulo, A.; Castillo, C.C.; Neidle, S. Targeting Promoter Quadruplex Nucleic Acids for Cancer Therapy. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; Volume 5, pp. 308–340. ISBN 9780124095472. [Google Scholar]
- Duarte, A.R.; Cadoni, E.; Ressurreição, A.S.; Moreira, R.; Paulo, A. Design of Modular G-quadruplex Ligands. ChemMedChem 2018, 13, 869–893. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Mergny, J.L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef]
- Summers, P.A.; Lewis, B.W.; Gonzalez-Garcia, J.; Porreca, R.M.; Lim, A.H.M.; Cadinu, P.; Martin-Pintado, N.; Mann, D.J.; Edel, J.B.; Vannier, J.B.; et al. Visualising G-quadruplex DNA dynamics in live cells by fluorescence lifetime imaging microscopy. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Biffi, G.; Di Antonio, M.; Tannahill, D.; Balasubramanian, S. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 2014, 6, 75–80. [Google Scholar] [CrossRef]
- De Cian, A.; DeLemos, E.; Mergny, J.-L.; Teulade-Fichou, M.-P.; Monchaud, D. Highly Efficient G-Quadruplex Recognition by Bisquinolinium Compounds. J. Am. Chem. Soc. 2007, 129, 1856–1857. [Google Scholar] [CrossRef]
- Dhamodharan, V.; Harikrishna, S.; Jagadeeswaran, C.; Halder, K.; Pradeepkumar, P.I. Selective G-quadruplex DNA Stabilizing Agents Based on Bisquinolinium and Bispyridinium Derivatives of 1,8-Naphthyridine. J. Org. Chem. 2012, 77, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Tan, J.-H.; Chen, S.-B.; Ou, T.-M.; Gu, L.-Q.; Huang, Z.-S. Bisaryldiketene derivatives: A new class of selective ligands for c-myc G-quadruplex DNA. Bioorganic Med. Chem. 2010, 18, 8235–8242. [Google Scholar] [CrossRef] [PubMed]
- Granotier, C.; Pennarun, G.; Riou, L.; Hoffschir, F.; Gauthier, L.R.; De Cian, A.; Gomez, D.; Mandine, E.; Riou, J.F.; Mergny, J.L.; et al. Preferential binding of a G-quadruplex ligand to human chromosome ends. Nucleic Acids Res. 2005, 33, 4182–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.R.; Granotier, C.; Hoffschir, F.; Etienne, O.; Ayouaz, A.; Desmaze, C.; Mailliet, P.; Biard, D.S.; Boussin, F.D. Rad51 and DNA-PKcs are involved in the generation of specific telomere aberrations induced by the quadruplex ligand 360A that impair mitotic cell progression and lead to cell death. Cell. Mol. Life Sci. 2012, 69, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monchaud, D.; Yang, P.; Lacroix, L.; Teulade-Fichou, M.P.; Mergny, J.L. A metal-mediated conformational switch controls G-quadruplex binding affinity. Angew. Chem. Int. Ed. 2008, 47, 4858–4861. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Granzhan, A.; Iida, K.; Tsushima, Y.; Ma, Y.; Nagasawa, K.; Teulade-Fichou, M.P.; Gabelica, V. Ligand-induced conformational changes with cation ejection upon binding to human telomeric DNA G-quadruplexes. J. Am. Chem. Soc. 2015, 137, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Saintomé, C.; Alberti, P.; Guinot, N.; Lejault, P.; Chatain, J.; Mailliet, P.; Riou, J.F.; Bugaut, A. Binding properties of mono- and dimeric pyridine dicarboxamide ligands to human telomeric higher-order G-quadruplex structures. Chem. Commun. 2018, 54, 1897–1900. [Google Scholar] [CrossRef]
- Harikrishna, S.; Kotaru, S.; Pradeepkumar, P.I. Ligand-induced conformational preorganization of loops of c-MYC G-quadruplex DNA and its implications in structure-specific drug design. Mol. Biosyst. 2017, 13, 1458–1468. [Google Scholar] [CrossRef]
- Marcel, V.; Tran, P.L.T.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.P.; Hall, J.; Mergny, J.L.; Hainaut, P.; van Dyck, E. G-quadruplex structures in TP53 intron 3: Role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 2011, 32, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Verga, D.; Hamon, F.; Poyer, F.; Bombard, S.; Teulade-Fichou, M.P. Photo-cross-linking probes for trapping G-quadruplex DNA. Angew. Chem. Int. Ed. 2014, 53, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, E.; Manicardi, A.; Fossépré, M.; Heirwegh, K.; Surin, M.; Madder, A. Teaching photosensitizers a new trick: Red light-triggered G-quadruplex alkylation by ligand co-localization. Chem. Commun. 2021, 57, 1010–1013. [Google Scholar] [CrossRef]
- Yang, P.; De Cian, A.; Teulade-Fichou, M.P.; Mergny, J.L.; Monchaud, D. Engineering bisquinolinium/thiazole orange conjugates for fluorescent sensing of G-quadruplex DNA. Angew. Chem. Int. Ed. 2009, 48, 2188–2191. [Google Scholar] [CrossRef]
- Renaud De La Faverie, A.; Hamon, F.; Di Primo, C.; Largy, E.; Dausse, E.; Delaurire, L.; Landras-Guetta, C.; Toulmé, J.J.; Teulade-Fichou, M.P.; Mergny, J.L. Nucleic acids targeted to drugs: SELEX against a quadruplex ligand. Biochimie 2011, 93, 1357–1367. [Google Scholar] [CrossRef]
- Dorazco-González, A.; Alamo, M.F.; Godoy-Alcántar, C.; Höpfl, H.; Yatsimirsky, A.K. Fluorescent anion sensing by bisquinolinium pyridine-2,6-dicarboxamide receptors in water. RSC Adv. 2014, 4, 455–466. [Google Scholar] [CrossRef]
- Hwang, I.P.; Mailliet, P.; Hossard, V.; Riou, J.F.; Bugaut, A.; Roger, L. Investigating the effect of mono- And dimeric 360A G-quadruplex ligands on telomere stability by single telomere length analysis (STELA). Molecules 2019, 24, 577. [Google Scholar] [CrossRef] [Green Version]
- Dorazco-González, A.; Höpfl, H.; Medrano, F.; Yatsimirsky, A.K. Recognition of anions and neutral guests by dicationic pyridine-2,6- dicarboxamide receptors. J. Org. Chem. 2010, 75, 2259–2273. [Google Scholar] [CrossRef] [PubMed]
- Giancola, C.; Pagano, B. Energetics of ligand binding to G-quadruplexes. Top. Curr. Chem. 2013, 330, 211–242. [Google Scholar] [CrossRef]
- Gray, R.D.; Petraccone, L.; Trent, J.O.; Chaires, J.B. Characterization of a K+-induced conformational switch in a human telomeric DNA oligonucleotide using 2-aminopurine fluorescence. Biochemistry 2010, 49, 179–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D. Structure of the hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: Insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007, 35, 4927–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrus, A.; Chen, D.; Dai, J.; Bialis, T.; Jones, R.A.; Yang, D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Mendes, E.; Cadoni, E.; Carneiro, F.; Afonso, M.B.; Brito, H.; Lavrado, J.; dos Santos, D.J.V.A.; Vítor, J.B.; Neidle, S.; Rodrigues, C.M.P.; et al. Combining 1,3-ditriazolyl-benzene and quinoline to discover a new G-quadruplex interactive small molecule active against cancer stem-like cells. ChemMedChem 2019, 14, 1325–1328. [Google Scholar] [CrossRef]
- Lemarteleur, T.; Gomez, D.; Paterski, R.; Mandine, E.; Mailliet, P.; Riou, J.F. Stabilization of the c-myc gene promoter quadruplex by specific ligands’ inhibitors of telomerase. Biochem. Biophys. Res. Commun. 2004, 323, 802–808. [Google Scholar] [CrossRef]
- Phan, T.A.; Modi, Y.S.; Patel, D.J. Propeller-Type Parallel-Stranded G-Quadruplexes in the Human c-myc Promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef] [Green Version]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5 0 -end of NHE III 1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.; Davis, N.E.; Binning, R.C.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/g09citation/ (accessed on 12 January 2021).
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.G.; Giambasu, T.; et al. AMBER 2019, University of California, San Francisco. Available online: https://ambermd.org/ (accessed on 12 January 2021).
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allnér, O.; Nilsson, L.; Villa, A. Magnesium Ion–Water Coordination and Exchange in Biomolecular Simulations. J. Chem. Theory Comput. 2012, 8, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Kerkour, A.; Marquevielle, J.; Ivashchenko, S.; Yatsunyk, L.A.; Mergny, J.-L.; Salgado, G.F. High-resolution three-dimensional NMR structure of the KRAS proto-oncogene promoter reveals key features of a G-quadruplex involved in transcriptional regulation. J. Biol. Chem. 2017, 292, 8082–8091. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Luenberger, D.G.; Ye, Y. Linear and Nonlinear Programming; Springer: Berlin/Heidelberg, Germany, 2008; Volume 228, pp. 213–262. ISBN 978-3-319-18841-6. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C.; Bersch, C. Gnuplot 5.2 An Interactive Plotting Program 2019. 2019. Available online: http://www.gnuplot.info (accessed on 1 January 2019).
- DeLano, W.L. The PyMOL Molecular Graphics System, version 2.3.1; Schrodinger LLC: New York, NY, USA, 2019; Available online: http://www.gnuplot.info (accessed on 10 January 2019).
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, R.C.; Aynechi, T.; Case, D.A.; Kuntz, I.D. Estimation of Absolute Free Energies of Hydration Using Continuum Methods: Accuracy of Partial Charge Models and Optimization of Nonpolar Contributions. J. Chem. Theory Comput. 2006, 2, 128–139. [Google Scholar] [CrossRef]
- Müller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.-F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012, 10, 6537. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FRET ΔTm (°C) a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand (5.0 μM/25 eq) | 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | |
| Sequence | k-RAS | <0.5 | 1.0 ± 0.2 | 3.0 ± 0.5 | 8.0 ± 0.2 | 8.0 ± 0.1 | 1.8 ± 0.2 | <0.5 | <0.5 |
| h-Telo | <0.5 | 1.8 ± 0.2 | 2.8 ± 0.4 | 18.0 ± 0.3 | 11.8 ± 0.2 | 4.0 ± 0.3 | <0.5 | <0.5 | |
| FRET ΔTm (°C) a | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand (eq) | 2a | 2b | 2c | 2d | |||||||||
| 0.5 | 2.5 | 5 | 5 | 12 | 25 | 5 | 12 | 25 | 5 | 12 | 25 | ||
| Sequence | k-RAS | 14.0 ± 0.3 | 32.8 ± 0.2 | 35.6 ± 0.3 | 4.0 ± 0.2 | 9.0 ± 0.1 | 11.1 ± 0.2 | 11.8 ± 0.2 | 16.6 ± 0.2 | 18.2 ± 0.2 | 15.0 ± 0.2 | 23.1 ± 0.1 | 26.1 ± 0.2 |
| h-Telo | 1.1 ± 0.2 | 13.2 ± 0.2 | 18.8 ± 0.3 | 2.3 ± 0.1 | 8.0 ± 0.3 | 10.1 ± 0.1 | 9.8 ± 0.2 | 13.1 ± 0.1 | 14.7 ± 0.1 | 15.1 ± 0.1 | 20.1 ± 0.2 | 22.7 ± 0.2 | |
| t-Loop | 2.0 ± 0.1 | 5.6 ± 0.1 | 6.5 ± 0.1 | <0.5 | <0.5 | 1.8 ± 0.2 | <0.5 | 0.7 ± 0.1 | 1.2 ± 0.1 | <0.5 | 1.5 ± 0.1 | 2.5 ± 0.1 | |
| CD ΔTm (°C) a | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand (eq) | 2a | 2b | 2c | 2d | |||||||||||||
| 1.0 | 5.0 | 10 | 25 | 1.0 | 5.0 | 10 | 25 | 1.0 | 5.0 | 10 | 25 | 1.0 | 5.0 | 10 | 25 | ||
| Sequence | k-RAS | >40 | >40 | >40 | P | 3.0 ± 0.2 | 12.2 ± 0.2 | 15.1 ± 0.2 | P | 13.8 ± 0.3 | 35.9 ± 0.1 | 37.1 ± 0.1 | >40 | 12.6 ± 0.4 | >40 | >40 | >40 |
| h-Telo | 3.5 ± 0.1 | 12.2 ± 0.3 | Topology Change | 1.7 ± 0.2 | 4.4 ± 0.2 | 6.9 ± 0.2 | 9.2 ± 0.2 | 3.3 ± 0.1 | 9.0 ± 0.1 | 9.0 ± 0.2 | 10.8 ± 0.3 | 6.8 ± 0.2 | 14.9 ± 0.2 | 19.8 ± 0.4 | 27.0 ± 1.3 | ||
| c-MYC | 35.7 ± 1.3 | >40 | >40 | >40 | 2.7 ± 0.2 | 5.3 ± 0.1 | 7.0 ± 0.2 | 9.7 ± 0.2 | 8.0 ± 0.2 | 24.1 ± 0.1 | 25.0 ± 0.1 | 29.4 ± 0.1 | 11.8 ± 0.2 | 31.2 ± 0.2 | 34.4 ± 0.2 | >40 | |
| Ligand | Average Binding Free Energy (kJ/mol) | Van der Waals Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) |
|---|---|---|---|---|---|
| 2a | −207 ± 8 | −240 ± 6 | −560 ± 4 | 612 ± 13 | −19 ± 1 |
| 2b | −215 ± 14 | −220 ± 5 | −518 ± 4 | 543 ± 17 | −19 ± 1 |
| 2c | −171 ± 8 | −248 ± 8 | −553 ± 4 | 649 ± 9 | −19 ± 1 |
| 2d | −203 ± 7 | −217 ± 7 | −521 ± 5 | 551 ± 14 | −17 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadoni, E.; Magalhães, P.R.; Emídio, R.M.; Mendes, E.; Vítor, J.; Carvalho, J.; Cruz, C.; Victor, B.L.; Paulo, A. New (Iso)quinolinyl-pyridine-2,6-dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study. Pharmaceuticals 2021, 14, 669. https://doi.org/10.3390/ph14070669
Cadoni E, Magalhães PR, Emídio RM, Mendes E, Vítor J, Carvalho J, Cruz C, Victor BL, Paulo A. New (Iso)quinolinyl-pyridine-2,6-dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study. Pharmaceuticals. 2021; 14(7):669. https://doi.org/10.3390/ph14070669
Chicago/Turabian StyleCadoni, Enrico, Pedro R. Magalhães, Rita M. Emídio, Eduarda Mendes, Jorge Vítor, Josué Carvalho, Carla Cruz, Bruno L. Victor, and Alexandra Paulo. 2021. "New (Iso)quinolinyl-pyridine-2,6-dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study" Pharmaceuticals 14, no. 7: 669. https://doi.org/10.3390/ph14070669
APA StyleCadoni, E., Magalhães, P. R., Emídio, R. M., Mendes, E., Vítor, J., Carvalho, J., Cruz, C., Victor, B. L., & Paulo, A. (2021). New (Iso)quinolinyl-pyridine-2,6-dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study. Pharmaceuticals, 14(7), 669. https://doi.org/10.3390/ph14070669