
Fulvestrant-3-Boronic Acid (ZB716) Demonstrates Oral Bioavailability and Favorable Pharmacokinetic Profile in Preclinical ADME Studies

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Absorption
2.1.1. Pharmacokinetics/Toxicokinetics after a Single Dose of ZB716 in Rats
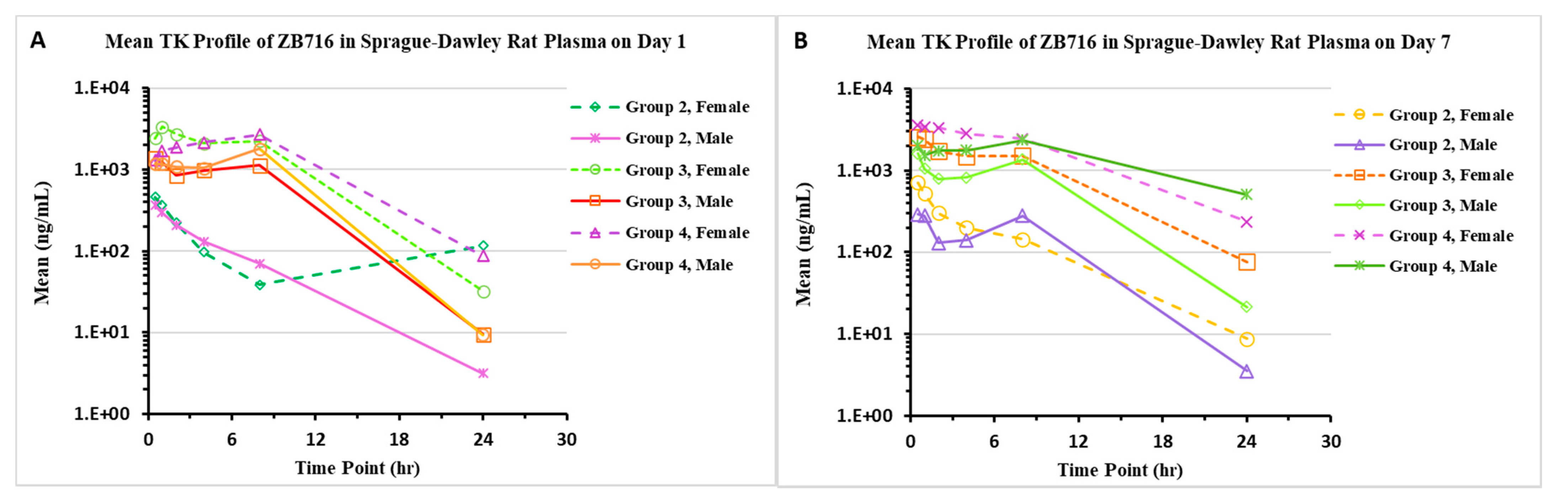
2.1.2. Pharmacokinetics/Toxicokinetics after Repeated Doses of ZB716 in Rats
2.1.3. Pharmacokinetics/Toxicokinetics after a Single Dose of ZB716 in Dogs
2.1.4. Pharmacokinetics/Toxicokinetics after Repeated Doses of ZB716 in Dogs
2.2. Distribution
2.2.1. Plasma Protein Binding
2.2.2. Intestinal Mucosal Permeation
2.3. Metabolism
2.3.1. In Vitro Metabolism Including P450 Studies
2.3.2. Enzyme Inhibition
2.3.3. Enzyme Induction
2.3.4. Metabolic Stability
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Metabolic Stability
4.4. CYP Induction
4.5. CYP Inhibition
4.6. CYP Phenotyping (CYP Enzymes Are Responsible for the Metabolism of ZB716)
4.7. Intestinal Mucosal Permeation of ZB716
4.8. Protein Binding
4.9. Pharmacokinetics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bross, P.F.; Cohen, M.H.; Williams, G.A.; Pazdur, R. FDA drug approval summaries: Fulvestrant. Oncologist 2002, 7, 477–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.astrazeneca.com/media-centre/press-releases/2017/faslodex-receives-eu-approval-as-first-line-therapy-for-advanced-breast-cancer-26072017.html (accessed on 23 July 2021).
- Available online: https://www.astrazeneca.com/media-centre/press-releases/2017/faslodex-receives-us-fda-approval-as-monotherapy-for-expanded-use-in-breast-cancer.html (accessed on 23 July 2021).
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.N.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; et al. Final overall survival: Fulvestrant 500 mg vs. 250 mg in the randomized CONFIRM trial. J. Natl. Cancer Inst. 2014, 106, djt337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.F.; Lindemann, J.; Garnett, S.; Anderson, E.; Nicholson, R.I.; Kuter, I.; Gee, J.M. A good drug made better: The fulvestrant dose-response story. Clin. Breast Cancer 2014, 14, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Fulvestrant as a reference antiestrogen and estrogen receptor (ER) degrader in preclinical studies: Treatment dosage, efficacy, and implications on development of new ER-targeting agents. Transl. Cancer Res. 2020, 9, 4464–4468. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Kang, B.; Peng, X.; Wang, G. From pure antagonists to pure degraders of the estrogen receptor: Evolving strategies for the same target. ACS Omega 2021, 6, 9334–9343. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.; Harrison, M. Fulvestrant: Pharmacokinetics and pharmacology. Br. J. Cancer 2004, 90 (Suppl. S1), S7–S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, M.; Laight, A.; Clarke, D.A.; Giles, P.; Yates, R. Pharmacokinetics and metabolism of fulvestrant after oral, intravenous and intramuscular administration in healthy volunteers. Eur. J. Cancer Suppl. 2003, 1, 564. [Google Scholar] [CrossRef]
- Chouinard, S.; Tessier, M.; Vernouillet, G.; Gauthier, S.; Labrie, F.; Barbier, O.; Bélanger, A. Inactivation of the pure antiestrogen fulvestrant and other synthetic estrogen molecules by UDPglucuronosyltransferase 1A enzymes expressed in breast tissue. Mol. Pharmacol. 2006, 69, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, S.; Akerstrom, V.L.; Yuan, C.; Ma, Y.; Zhong, Q.; Zhang, C.; Zhang, Q.; Guo, S.; Ma, P.; et al. Fulvestrant-3 Boronic Acid (ZB716): An Orally Bioavailable Selective Estrogen Receptor Downregulator (SERD). J. Med. Chem. 2016, 59, 8134–8140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Zhang, C.; Bratton, M.; Mottamal, M.; Liu, J.; Ma, P.; Zheng, S.; Zhong, Q.; Yang, L.; Wiese, T.E.; et al. ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models. Oncotarget 2018, 9, 6924–6937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, S.; Yang, L.; Liu, J.; Zheng, S.; Zhong, Q.; Zhang, Q.; Wang, G. Metabolism, pharmacokinetics, and bioavailability of ZB716, a Steroidal Selective Estrogen Receptor Downregulator (SERD). Oncotarget 2017, 8, 103874–103889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Zhong, Q.; Zhang, Q.; Zheng, S.; Wang, G. Boron-based 4-hydroxytamoxifen bioisosteres for treatment of de novo tamoxifen resistant breast cancer. ACS Med. Chem. Lett. 2012, 3, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, S.; Guo, S.; Zhang, C.; Zhong, Q.; Zhang, Q.; Ma, P.; Skripnikova, E.V.; Bratton, M.R.; Wiese, T.E.; et al. Rational design of a boron-modified triphenylethylene (GLL398) as an orally bioavailable non-steroidal selective estrogen receptor downregulator (SERD). ACS Med. Chem. Lett. 2017, 8, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
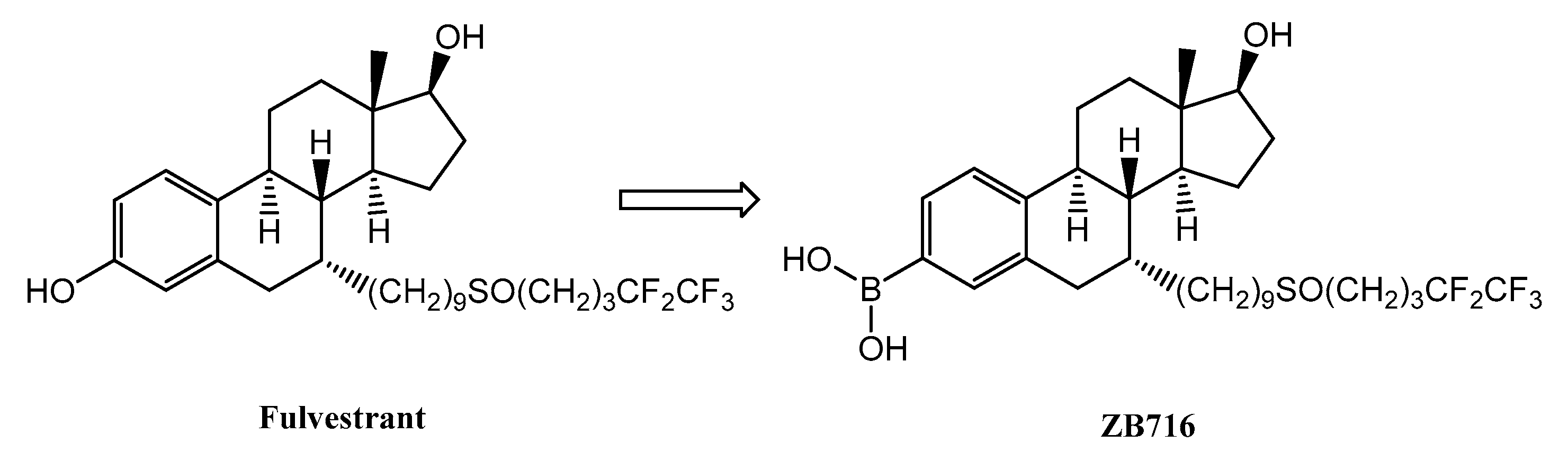


{kind=link}
{kind=link}
{kind=link}
| Group | Route | Dose Level (mg/kg/day) | Sex | Tmax (h) a | Cmax (ng/mL) | AUClast (h·ng/mL) | Bioavailability (%) |
|---|---|---|---|---|---|---|---|
| 1 | IV | 2 | M | 0.0833 | 5367 | 4906 | NA |
| F | 0.0833 | 4530 | 6443 | NA | |||
| 2 | PO | 6 | M | 0.5 | 321 | 936 | 6.36 |
| F | 0.5 | 347 | 1155 | 5.98 | |||
| 3 | PO | 30 | M | 2 | 1723 | 5410 | NA |
| F | 1 | 3523 | 15,691 | NA | |||
| 4 | PO | 100 | M | 0.5 | 3010 | 12,493 | NA |
| F | 0.5 | 3590 | 30,530 | NA | |||
| 5 | PO | 400 | M | 0.5 | 2863 | 32,608 | NA |
| F | 1 | 4310 | 55,568 | NA |
| Study Day | Group | Dose Level (mg/kg/day) | Sex | Tmax (h) a | Cmax (ng/mL) | AUClast (h·ng/mL) |
|---|---|---|---|---|---|---|
| 1 | 2 | 10 | M | 0.5 | 365 | 1324 |
| F | 0.5 | 459 | 1759 | |||
| 3 | 100 | M | 0.5 | 1420 | 11,788 | |
| F | 1 | 3350 | 26,684 | |||
| 4 | 400 | M | 8 | 1920 | 15,150 | |
| F | 8 | 2767 | 27,885 | |||
| 7 | 2 | 10 | M | 1 | 322 | 2255 |
| F | 0.5 | 710 | 2822 | |||
| 3 | 100 | M | 0.5 | 1610 | 12,884 | |
| F | 0.5 | 2753 | 20,659 | |||
| 4 | 400 | M | 8 | 2337 | 32,740 | |
| F | 0.5 | 3710 | 37,064 |
| Study Day | Group No. | Dose Level (mg/kg/day) | Sex | Tmax (h) | Cmax (ng/mL) | AUClast (h·ng/mL) |
|---|---|---|---|---|---|---|
| 1 | 2 | 25 | Male | 0.5 | 1039 | 5405 |
| Female | 1.0 | 1310 | 7695 | |||
| 3 | 100 | Male | 1.0 | 1867 | 15,708 | |
| Female | 1.0 | 3200 | 42,161 | |||
| 4 | 400 | Male | 1.0 | 3397 | 52,371 | |
| Female | 2.0 | 11,927 | 120,801 | |||
| 28 | 2 | 25 | Male | 0.5 | 1313 | 4748 |
| Female | 1.0 | 1767 | 8042 | |||
| 3 | 100 | Male | 0.5 | 2840 | 20,087 | |
| Female | 1.0 | 3847 | 24,525 | |||
| 4 | 400 | Male | 2.0 | 4063 | 50,678 | |
| Female | 2.0 | 4853 | 51,763 |
| Group | Route | Dose Level (mg/kg/day) | Sex | Tmax (h) | Cmax (ng/mL) | AUClast (h·ng/mL) | Bioavailability (%) |
|---|---|---|---|---|---|---|---|
| 1 | IV | 2 | M | 0.0833 | 2160 | 3034 | NA |
| F | 0.0833 | 2710 | 4312 | NA | |||
| 2 | PO | 6 | M | 0.5 | 157 | 563 | 6.18 |
| F | 0.5 | 213 | 872 | 6.74 | |||
| 3 | PO | 30 | M | 4 | 988 | 5522 | NA |
| F | 1 | 964 | 3456 | NA | |||
| 4 | PO | 100 | M | 2 | 1140 | 7500 | NA |
| F | 0.5 | 957 | 5841 | NA |
| Day | Group No. | Dose Level (mg/kg/day) | Sex | Tmax (h) | Animal No. | Cmax (ng/mL) | AUClast (h·ng/mL) |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 100 | M | 1 | 8,135,390 | 1350 | 5068 |
| F | 2 | 8,067,921 | 7210 | 55,649 | |||
| 3 | 200 | M | 2 | 8,121,984 | 8430 | 52,222 | |
| F | 1 | 8,135,209 | 1740 | 9785 | |||
| 7 | 2 | 100 | M | 1 | 8,135,390 | 3430 | 17,754 |
| F | 4 | 8,067,921 | 12,300 | 81,061 | |||
| 3 | 200 | M | 2 | 8,121,984 | 18,300 | 103,223 | |
| F | 2 | 8,135,209 | 8420 | 46,746 |
| Study Day | Group No. | Dose Level (mg/kg/day) | Sex | Tmax (h) | Cmax (ng/mL) | AUClast (h·ng/mL) |
|---|---|---|---|---|---|---|
| 1 | 2 | 25 | M | 0.5 | 691 | 1980 |
| F | 1.0 | 772 | 3882 | |||
| 3 | 100 | M | 1.0 | 1401 | 4572 | |
| F | 1.0 | 997 | 4492 | |||
| 4 | 200 | M | 2.0 | 1037 | 7876 | |
| F | 2.0 | 583 | 2718 | |||
| 28 | 2 | 25 | M | 1.0 | 1386 | 4327 |
| F | 1.0 | 1591 | 8001 | |||
| 3 | 100 | M | 2.0 | 3337 | 12,449 | |
| F | 2.0 | 1873 | 9647 | |||
| 4 | 200 | M | 2.0 | 2133 | 11,041 | |
| F | 2.0 | 1807 | 14,915 |
| Species | Conc. (μM) | Bound (%) | Unbound (%) | Recovery (%) | Remaining (%) |
|---|---|---|---|---|---|
| Human | 0.1 | >95.7 | <4.33 | <69.1 | 57.0 ± 7.7 |
| 1 | >99.6 | <0.392 | <64.0 | 57.9 ± 4.9 | |
| 10 | >99.96 | <0.0108 | <74.7 | 74.5 ± 3.4 | |
| Monkey | 0.1 | >96.4 | <3.63 | <68.2 | 65.3 ± 2.8 |
| 1 | >99.6 | <0.326 | <70.0 | 70.9 ± 4.8 | |
| 10 | >99.97 | <0.0308 | <87.2 | 93.9 ± 10.0 | |
| Dog | 0.1 | >95.6 | <4.04 | <63.5 | 39.1 ± 1.8 |
| 1 | >99.6 | <0.373 | <64.9 | 53.2 ± 4.4 | |
| 10 | >99.96 | <0.0412 | <68.7 | 65.2 ± 4.3 | |
| Rat | 0.1 | >97.7 | <2.35 | <94.4 | 83.9 ± 8.2 |
| 1 | >99.8 | <0.216 | <94.9 | 91.1 ± 2.1 | |
| 10 | >99.97 | <0.0281 | <82.1 | 84.7 ± 2.0 | |
| Mouse | 0.1 | >97.4 | <2.63 | <96.0 | 98.5 ± 14.5 |
| 1 | >99.7 | <0.266 | <88.8 | 92.2 ± 5.7 | |
| 10 | >99.97 | <0.0272 | <95.0 | 97.4 ± 10.7 |
| Test Article | Papp (cm/s × 10−6) | Recovery (%) | Efflux Ratio | ||
|---|---|---|---|---|---|
| A to B | B to A | A to B | B to A | ||
| ZB716 (1 μM) * | <0.778 | 0.747 ± 0.0316 | <66.6 | 66.5 ± 0.7 | >0.935 |
| ZB716 (5 μM) | <0.159 | 0.820 ± 0.127 | <67.0 | 73.7 ± 1.5 | >4.55 |
| ZB716 (15 μM) | 0.188 ± 0.021 | 0.534 ± 0.028 | 75.7 ± 2.0 | 84.2 ± 4.8 | 2.87 ± 0.49 |
| Digoxin (5 μM) | 0.687 ± 0.088 | 14.5 ± 1.68 | 92.4 ± 1.0 | 86.0 ± 6.0 | 21.2 ± 2.0 |
| Atenolol (5 μM) | 0.506 ± 0.034 | 0.635 ± 0.042 | 89.1 ± 1.2 | 97.5 ± 0.2 | 1.26 ± 0.08 |
| Minoxidil (5 μM) | 6.47 ± 0.22 | 7.44 ± 0.47 | 91.0 ± 1.4 | 94.4 ± 48 | 1.15 ± 0.09 |
| CYP Isoform | Clint (µL/min/pmol CYP) | t1/2 (min) |
|---|---|---|
| CYP1A2 | 0.866 ± 0.120 | 16.2 ± 2.2 |
| CYP2A6 | 0.836 ± 0.028 | 16.6 ± 0.6 |
| CYP2B6 | 0.343 ± 0.052 | 41.0 ± 6.8 |
| CYP2C8 | 1.25 ± 0.14 | 11.1 ± 1.2 |
| CYP2C9 | 0.746 ± 0.015 | 18.6 ± 0.4 |
| CYP2C19 | 0.646 ± 0.063 | 21.6 ± 2.0 |
| CYP2D6 | 0.773 ± 0.142 | 18.3 ± 3.3 |
| CYP2E1 | 0.716 ± 0.044 | 19.4 ± 1.2 |
| CYP3A4 | 2.35 ± 0.15 | 5.91 ± 0.39 |
| CYP3A5 | 6.86 ± 1.12 | 2.06 ± 0.33 |
| CYP Isoform (Inhibitors) | Remaining Percentage (%) at 15 min | Remaining Percentage (%) at 60 min | Inhibition % |
|---|---|---|---|
| Without Inhibitor | 18.7 ± 1.2 | 5.74 ± 0.52 | 0.00 |
| CYP1A2 (Furafylline) | 8.24 ± 0.80 | 1.43 ± 0.51 | −48.9 |
| CYP2A6 (Tranylcypromine) | 14.8 ± 2.5 | 5.24 ± 2.31 | −14.4 |
| CYP2B6 (Ticlopidine) | 15.9 ± 0.8 | 2.09 ± 0.23 | −9.46 |
| CYP2C8 (Montelukast) | 14.6 ± 2.4 | 3.41 ± 0.30 | −15.1 |
| CYP2C9 (Sulfaphenazole) | 10.9 ± 0.1 | 2.31 ± 0.18 | −31.8 |
| CYP2C19 (N-3-benzylnirvanol) | 12.8 ± 1.2 | 3.10 ± 0.44 | −22.5 |
| CYP2D6 (Quinidine) | 65.6 ± 6.0 | 3.23 ± 0.63 | 74.7 |
| CYP3A (Ketoconazole) | 74.4 ± 5.8 | 43.4 ± 2.8 | 88.2 |
| CYP Isoform | Marker Substrate | IC50 (μM) of ZB716 |
|---|---|---|
| CYP1A2 | Phenacetin | >15.0 |
| CYP2B6 | Bupropion | No Inhibition |
| CYP2C8 | Amodiaquine | 6.27 |
| CYP2C9 | Diclofenac | 4.77 |
| CYP2C19 | Mephenytoin | 2.20 |
| CYP2D6 | Dextromethorphan | 12.5 |
| CYP3A4-M | Midazolam | No Inhibition |
| CYP3A4-T | Testosterone | 11.2 |
| Donor | CYP | Compound | Conc. (µM) | Fold Induction (Mean ± SD) | Percent of Control (%) |
|---|---|---|---|---|---|
| QBU | CYP1A2 | Flumazenil | 30 | 1.06 ± 0.11 | 0.450 |
| Omeprazole | 50 | 15.0 ± 1.8 | - | ||
| ZB716 | 0.15 | 1.11 ± 0.05 | 0.777 | ||
| 1.5 | 2.17 ± 0.03 | 8.38 | |||
| 5 | 3.44 ± 0.18 | 17.4 | |||
| 15 | 2.77 ± 0.34 | 12.6 | |||
| CYP 2B6 | Flumazenil | 30 | 0.789 ± 0.070 | −3.61 | |
| Phenobarbital | 1000 | 6.86 ± 0.36 | - | ||
| ZB716 | 0.15 | 1.19 ± 0.10 | 3.16 | ||
| 1.5 | 2.16 ± 0.07 | 19.8 | |||
| 5 | 5.21 ± 0.24 | 71.7 | |||
| 15 | 6.40 ± 0.16 | 92.1 | |||
| CYP 3A4 | Flumazenil | 30 | 1.39 ± 0.04 | 4.37 | |
| Rifampicin | 10 | 9.95 ± 0.63 | - | ||
| ZB716 | 0.15 | 0.797 ± 0.100 | −2.26 | ||
| 1.5 | 1.43 ± 0.14 | 4.83 | |||
| 5 | 3.01 ± 0.33 | 22.4 | |||
| 15 | 2.92 ± 0.25 | 21.5 | |||
| ZSE | CYP 1A2 | Flumazenil | 30 | 0.807 ± 0.177 | −3.60 |
| Omeprazole | 50 | 6.37 ± 0.14 | - | ||
| ZB716 | 0.15 | 1.20 ± 0.22 | 3.78 | ||
| 1.5 | 3.42 ± 0.20 | 45.0 | |||
| 5 | 3.24 ± 0.53 | 41.6 | |||
| 15 | 2.09 ± 0.49 | 20.3 | |||
| CYP 2B6 | Flumazenil | 30 | 1.28 ± 0.20 | 5.23 | |
| Phenobarbital | 1000 | 6.29 ± 0.90 | - | ||
| ZB716 | 0.15 | 1.14 ± 0.09 | 2.61 | ||
| 1.5 | 3.07 ± 0.36 | 39.2 | |||
| 5 | 2.98 ± 0.74 | 37.4 | |||
| 15 | 1.22 ± 0.42 | 4.18 | |||
| CYP 3A4 | Flumazenil | 30 | 1.23 ± 0.09 | 3.11 | |
| Rifampicin | 10 | 8.36 ± 0.24 | - | ||
| ZB716 | 0.15 | 0.610 ± 0.043 | −5.29 | ||
| 1.5 | 0.828 ± 0.082 | −2.34 | |||
| 5 | 0.513 ± 0.081 | −6.62 | |||
| 15 | 0.318 ± 0.005 | −9.26 | |||
| VKB | CYP 1A2 | Flumazenil | 30 | 0.818 ± 0.032 | −2.20 |
| Omeprazole | 50 | 9.28 ± 0.223 | - | ||
| ZB716 | 0.15 | 1.24 ± 0.10 | 2.92 | ||
| 1.5 | 2.52 ± 0.17 | 18.4 | |||
| 5 | 7.09 ± 0.78 | 73.6 | |||
| 15 | 7.62 ± 1.63 | 80.0 | |||
| CYP2B6 | Flumazenil | 30 | 1.17 ± 0.04 | 3.77 | |
| Phenobarbital | 1000 | 5.44 ± 0.13 | - | ||
| ZB716 | 0.15 | 1.10 ± 0.05 | 2.22 | ||
| 1.5 | 1.77 ± 0.13 | 17.2 | |||
| 5 | 4.14 ± 0.06 | 70.6 | |||
| 15 | 4.24 ± 0.47 | 73.0 | |||
| CYP3A4 | Flumazenil | 30 | 1.06 ± 0.09 | 1.16 | |
| Rifampicin | 10 | 5.97 ± 0.52 | - | ||
| ZB716 | 0.15 | 0.774 ± 0.078 | −4.55 | ||
| 1.5 | 0.649 ± 0.085 | −7.08 | |||
| 5 | 1.24 ± 0.18 | 4.85 | |||
| 15 | 1.49 ± 0.21 | 9.95 |
| Species | Compound | Remaining at 120 min (%) | Estimated In Vitro t1/2 (min) | In Vitro Clint (μL/min/106 cells) | Scaled-Up Clint (mL/min/kg) |
|---|---|---|---|---|---|
| Human | Verapamil | 1.57 ± 0.18 | 20.8 ± 0.6 | 66.7 ± 2.1 | 170 ± 5 |
| ZB716 | 7.98 ± 0.7 | 34.9 ± 1.0 | 39.7 ± 1.1 | 101 ± 3 | |
| Monkey | Verapamil | 0.264 ± 0.103 | 11.35 ± 0.17 | 122 ± 2 | 440 ± 6 |
| ZB716 | 4.41 ± 0.46 | 27.6 ± 2.0 | 50.3 ± 3.6 | 181 ± 13 | |
| Dog | Verapamil | 6.02 ± 0.17 | 30.6 ± 0.4 | 45.3 ± 0.5 | 312 ± 4 |
| ZB716 | 6.99 ± 0.46 | 26.7 ± 0.4 | 51.9 ± 0.7 | 357 ± 5 | |
| Rat | Verapamil | 1.24 ± 0.38 | 18.2 ± 1.8 | 76.6 ± 8.1 | 358 ± 38 |
| ZB716 | 5.19 ± 0.32 | 28.4 ± 0.9 | 48.7 ± 1.5 | 228 ± 7 | |
| Mouse | Verapamil | 2.02 ± 0.54 | 22.0 ± 1.6 | 63.3 ± 4.8 | 747 ± 57 |
| ZB716 | 6.24 ± 0.58 | 34.4 ± 0.6 | 40.3 ± 0.7 | 476 ± 8 | |
| Medium | Verapamil | 102 | ∞ | NA | NA |
| ZB716 | 100 | ∞ | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Rajasekaran, N.; Hossain, A.; Zhang, C.; Guo, S.; Kang, B.; Jung, H.; Kim, H.; Wang, G. Fulvestrant-3-Boronic Acid (ZB716) Demonstrates Oral Bioavailability and Favorable Pharmacokinetic Profile in Preclinical ADME Studies. Pharmaceuticals 2021, 14, 719. https://doi.org/10.3390/ph14080719
Liu J, Rajasekaran N, Hossain A, Zhang C, Guo S, Kang B, Jung H, Kim H, Wang G. Fulvestrant-3-Boronic Acid (ZB716) Demonstrates Oral Bioavailability and Favorable Pharmacokinetic Profile in Preclinical ADME Studies. Pharmaceuticals. 2021; 14(8):719. https://doi.org/10.3390/ph14080719
Chicago/Turabian StyleLiu, Jiawang, Nirmal Rajasekaran, Ahamed Hossain, Changde Zhang, Shanchun Guo, Borui Kang, Hunsoon Jung, Hongjoong Kim, and Guangdi Wang. 2021. "Fulvestrant-3-Boronic Acid (ZB716) Demonstrates Oral Bioavailability and Favorable Pharmacokinetic Profile in Preclinical ADME Studies" Pharmaceuticals 14, no. 8: 719. https://doi.org/10.3390/ph14080719
APA StyleLiu, J., Rajasekaran, N., Hossain, A., Zhang, C., Guo, S., Kang, B., Jung, H., Kim, H., & Wang, G. (2021). Fulvestrant-3-Boronic Acid (ZB716) Demonstrates Oral Bioavailability and Favorable Pharmacokinetic Profile in Preclinical ADME Studies. Pharmaceuticals, 14(8), 719. https://doi.org/10.3390/ph14080719