
Exploring the Early Stages of the Amyloid Aβ(1–42) Peptide Aggregation Process: An NMR Study





Abstract
:1. Introduction
2. Results
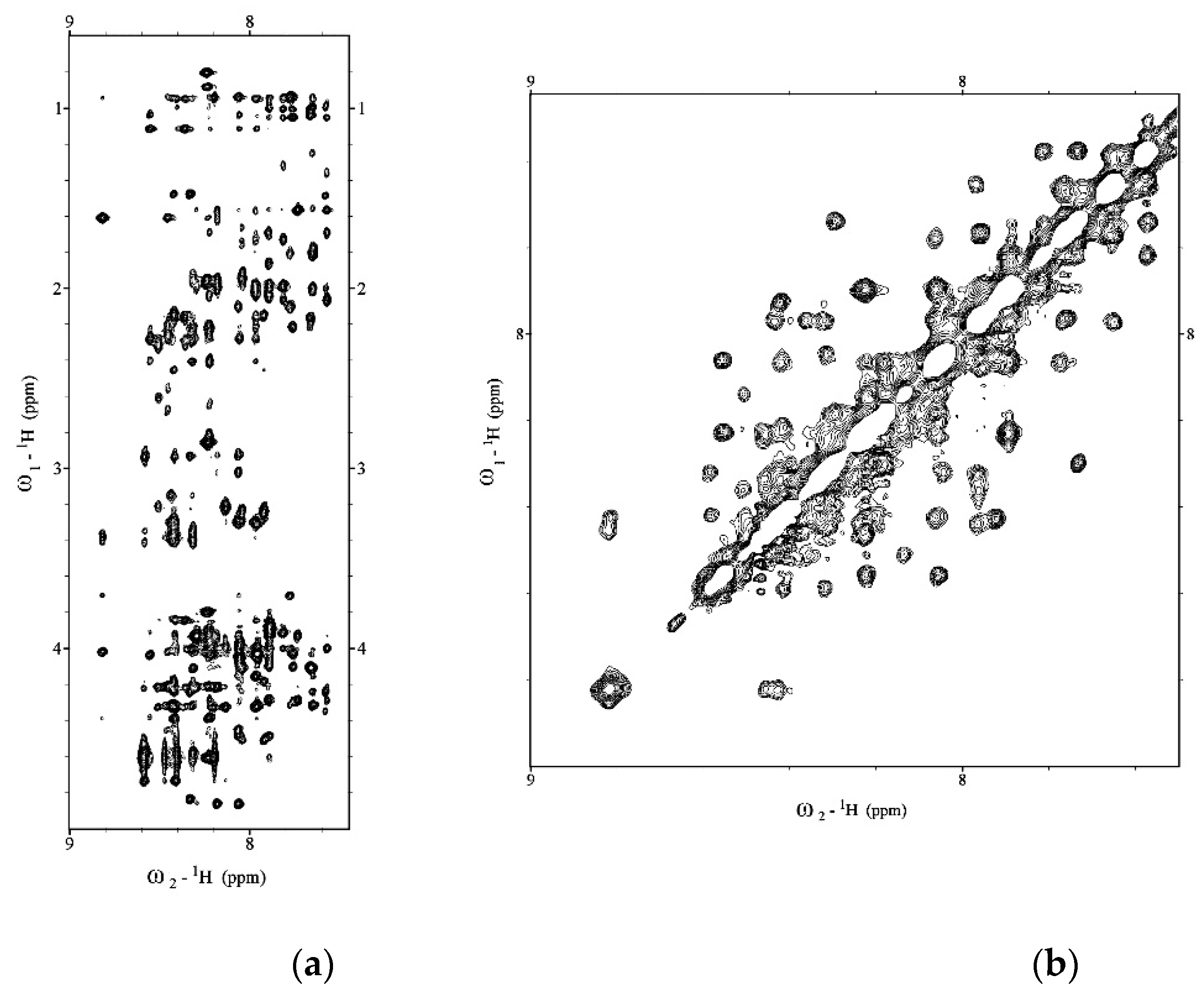
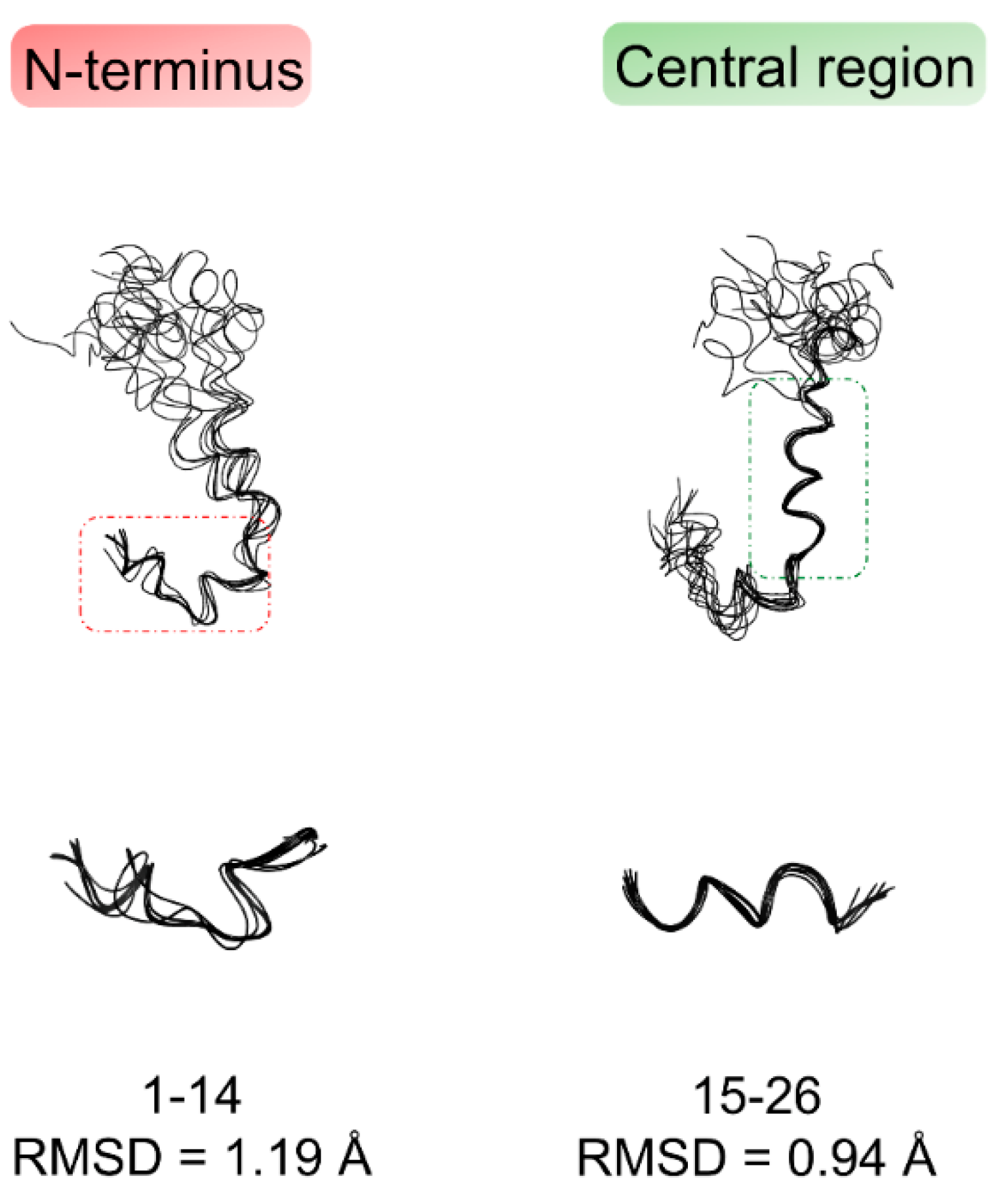
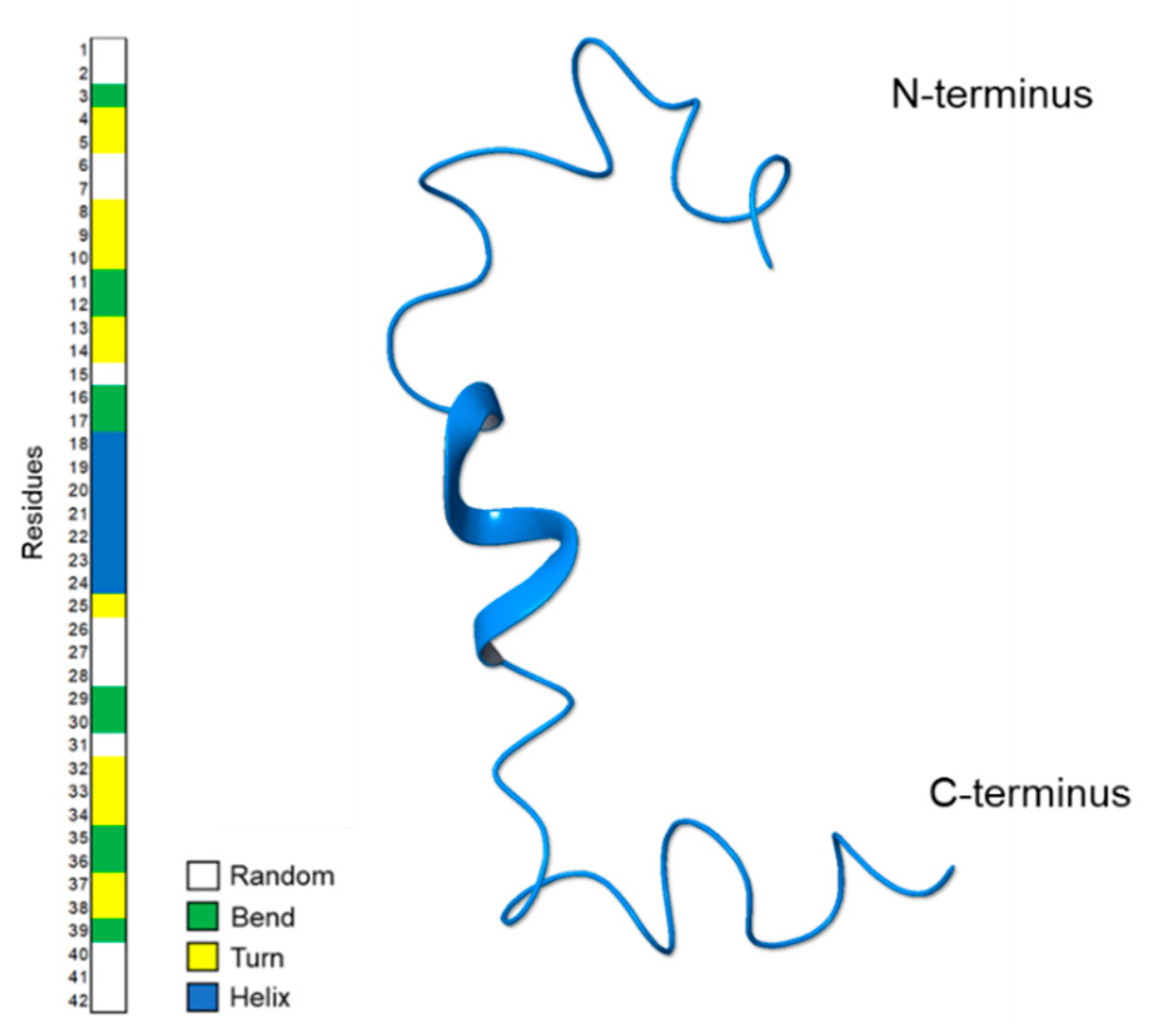
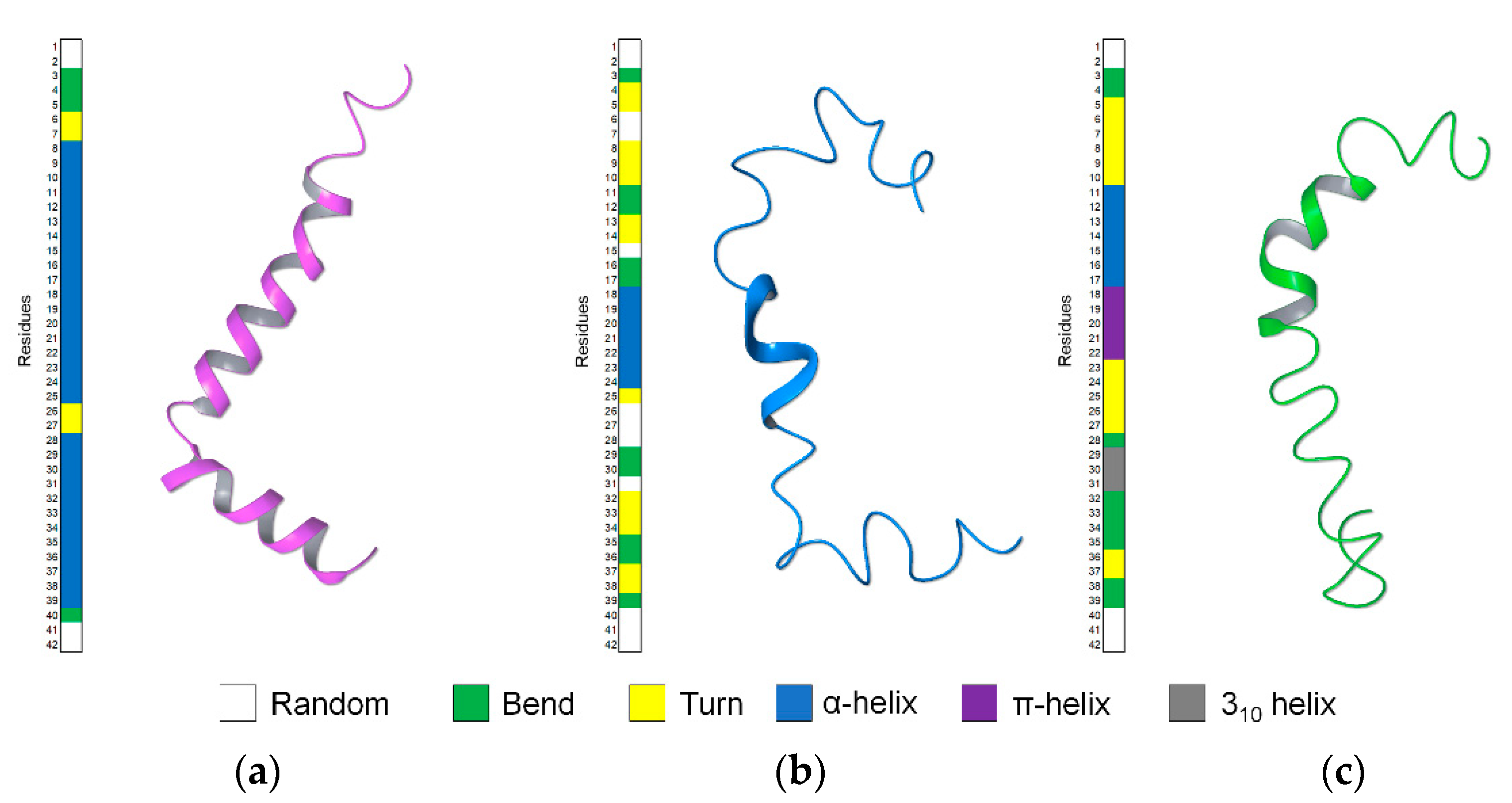
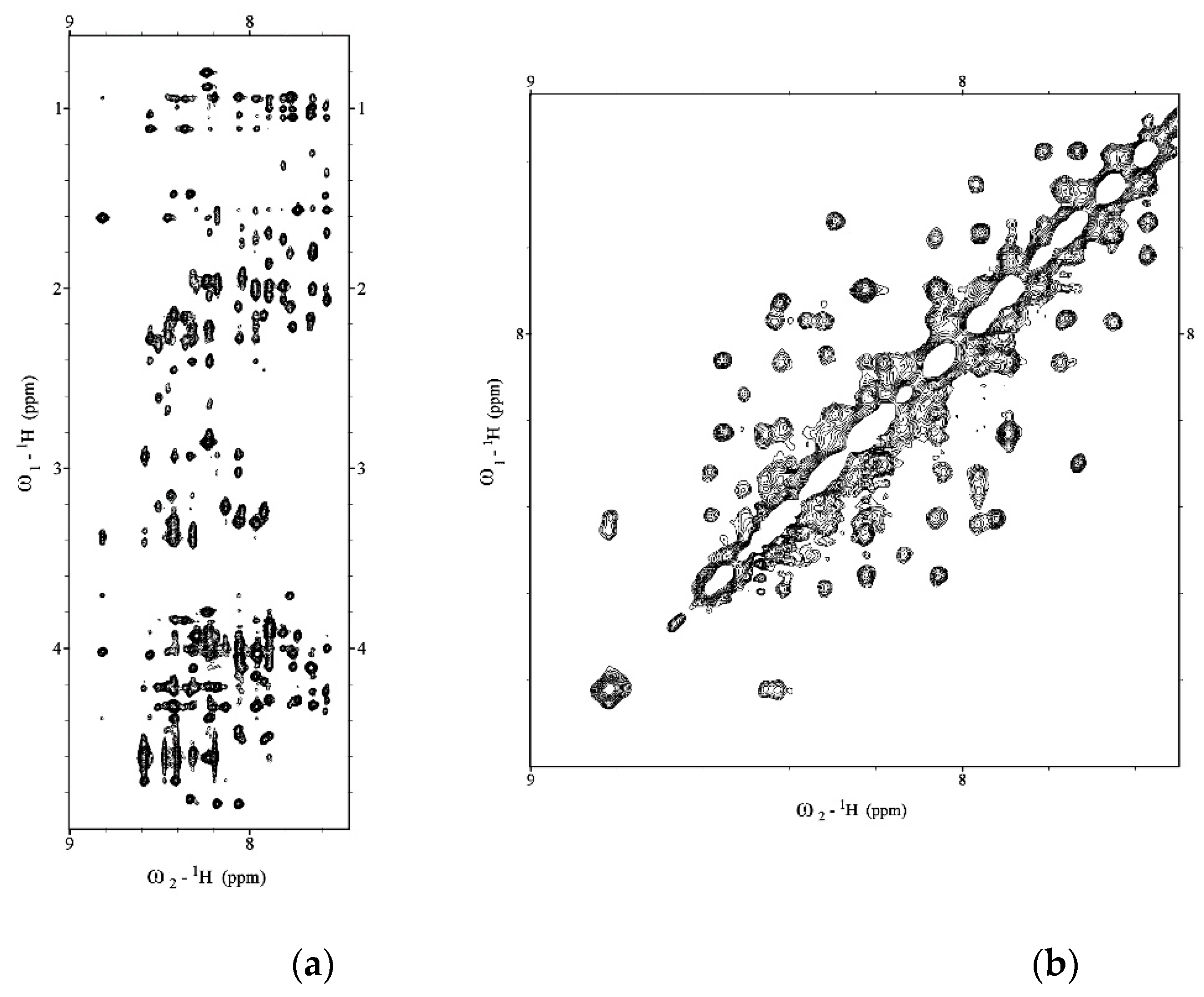
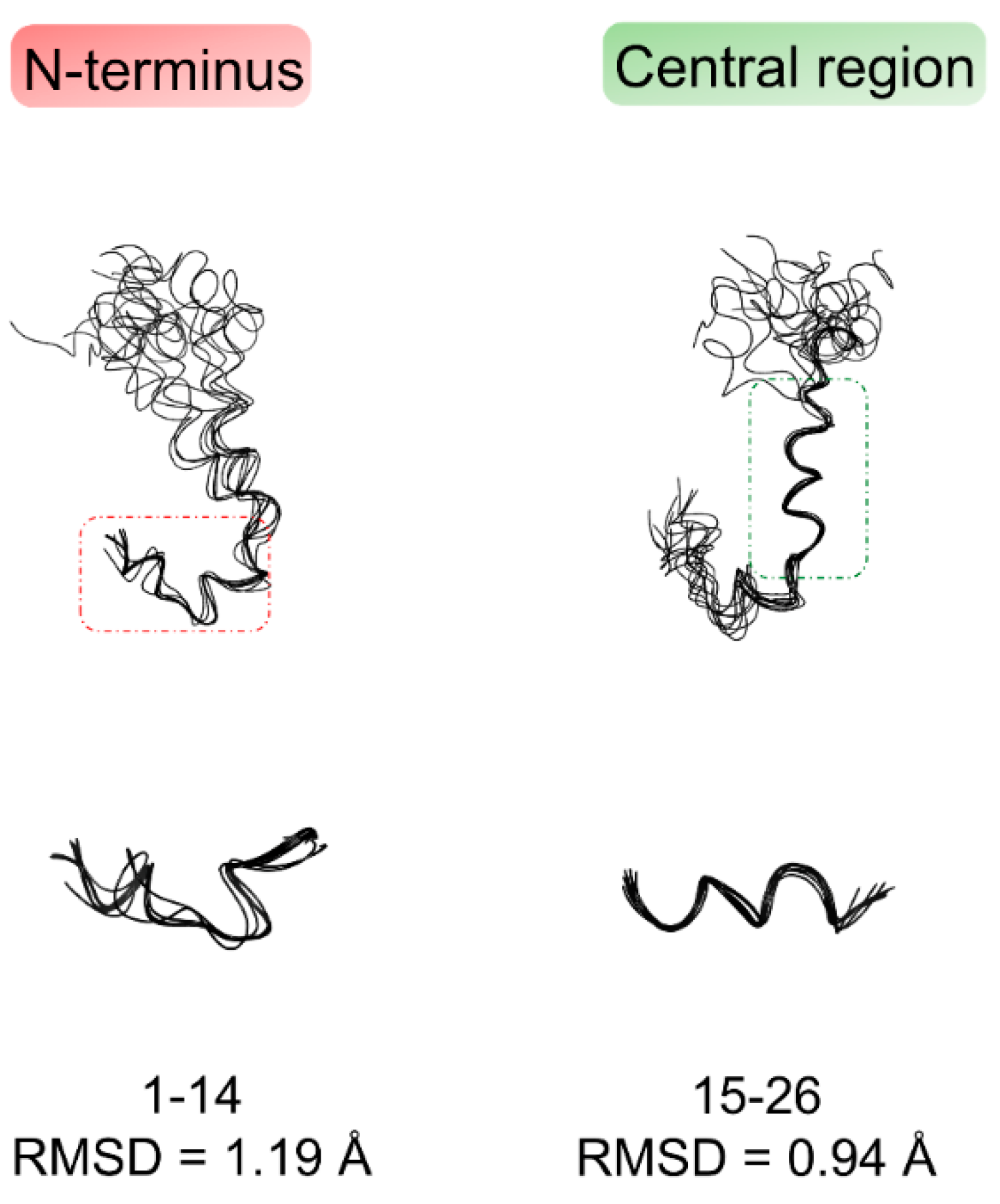
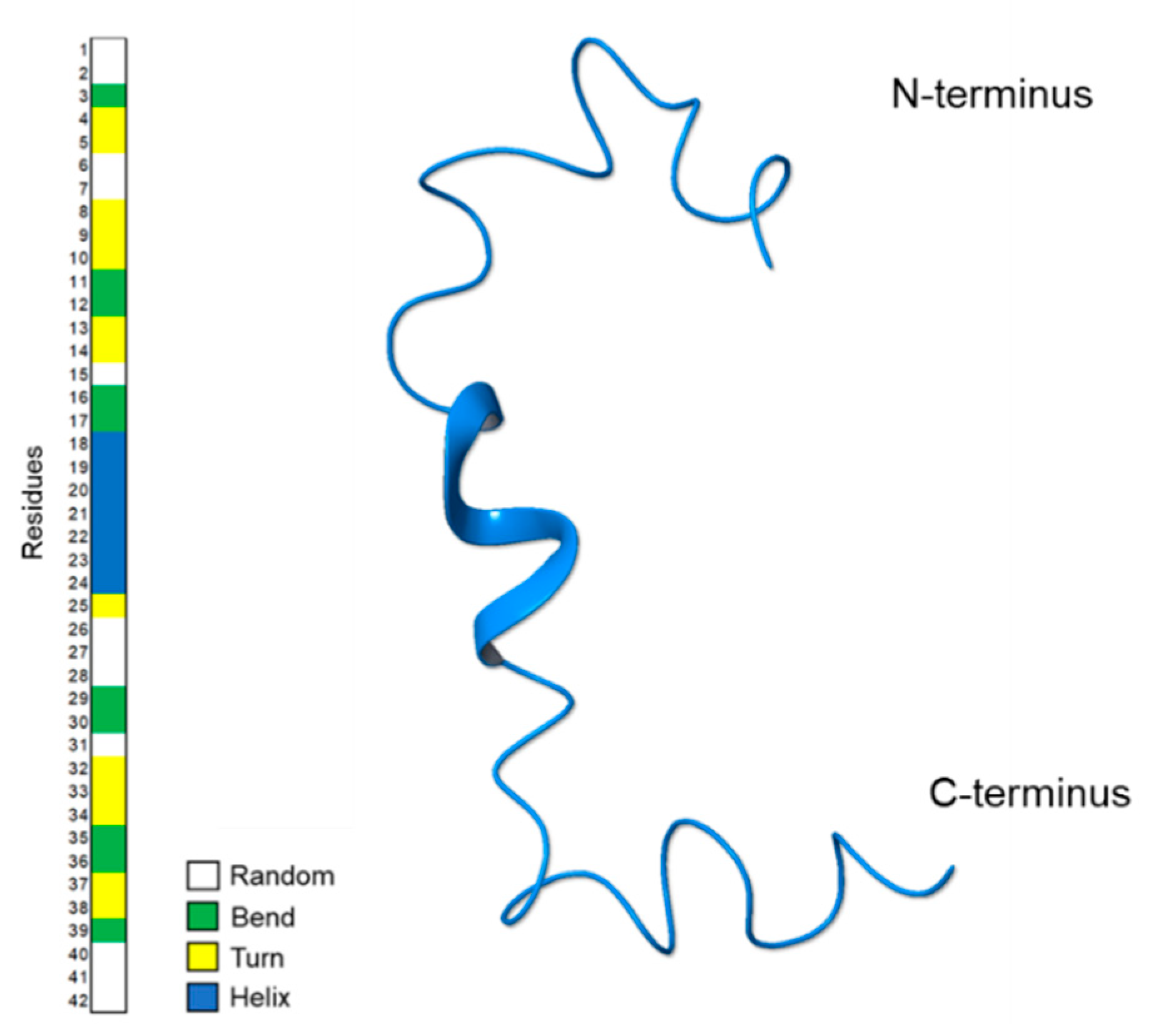
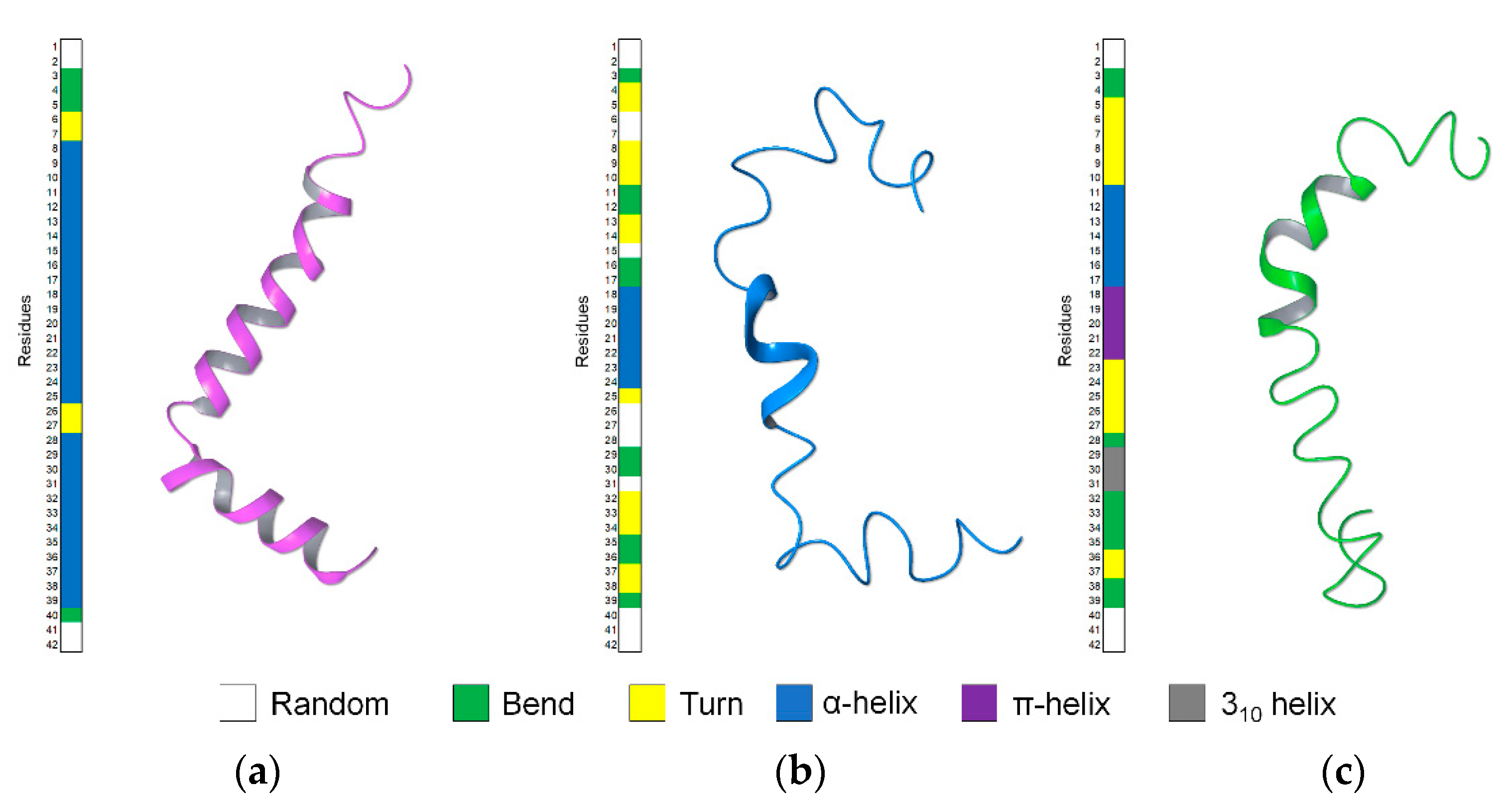
2.1. NMR Structure Determination of Aβ(1–42) Peptide
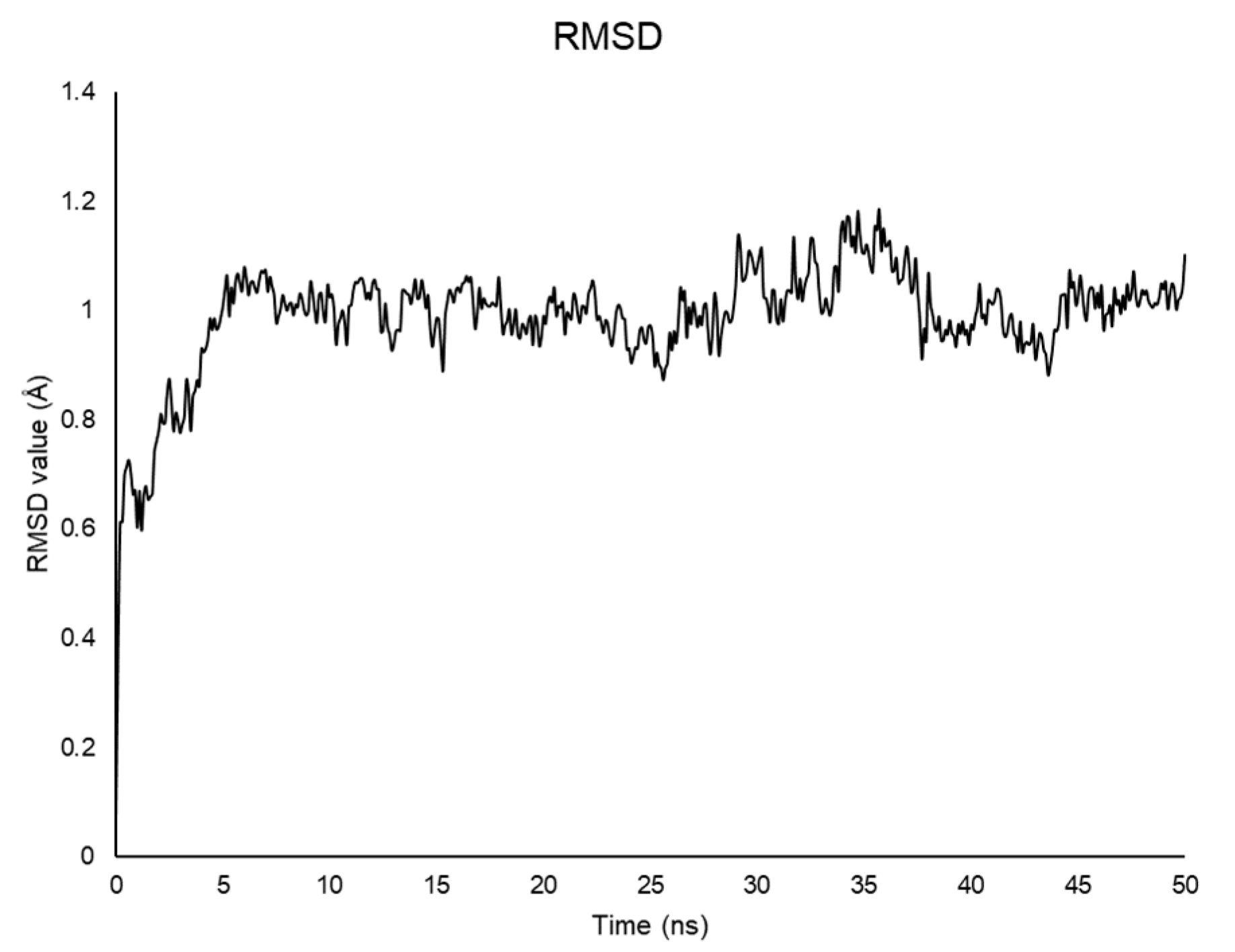
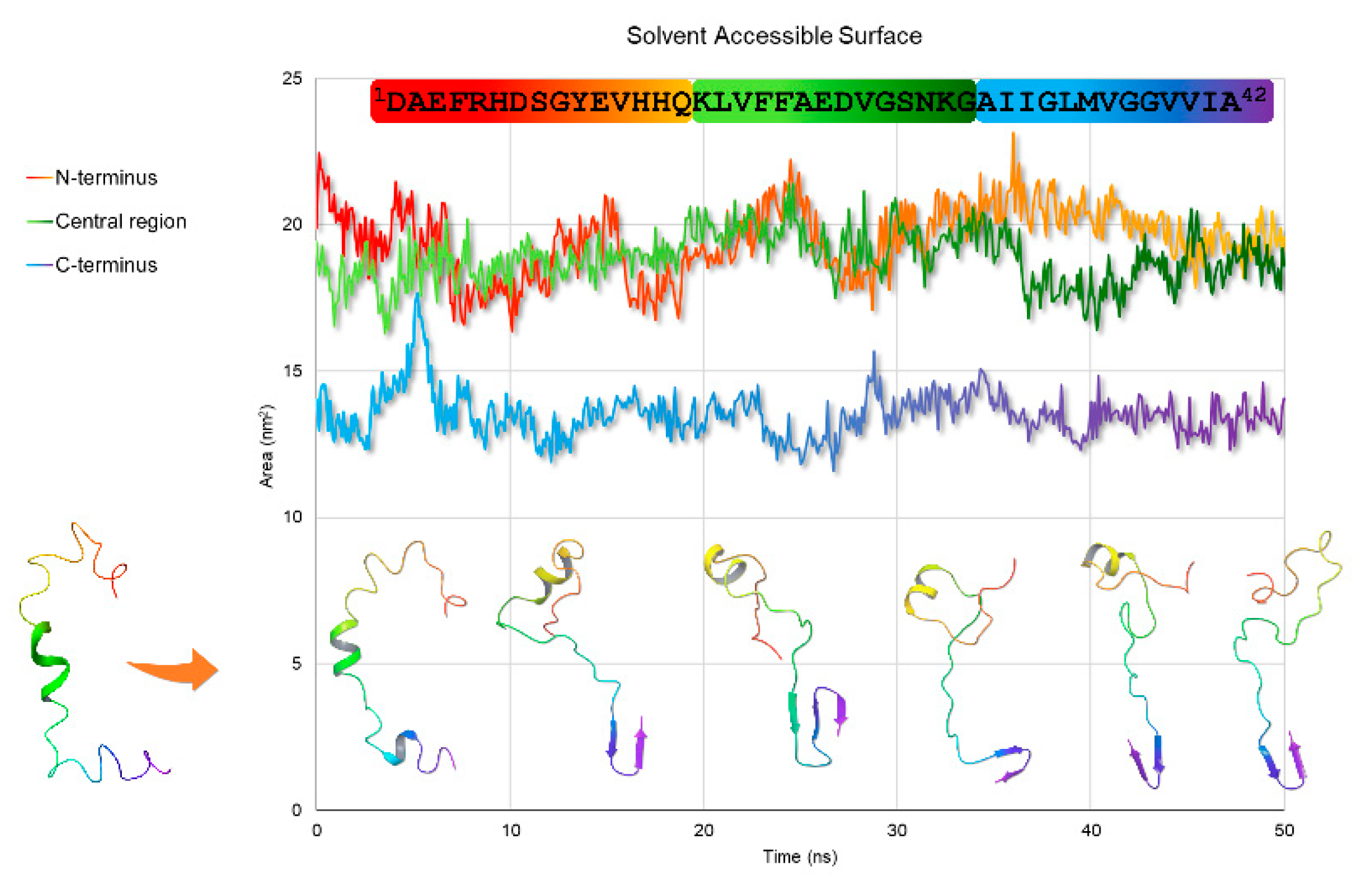
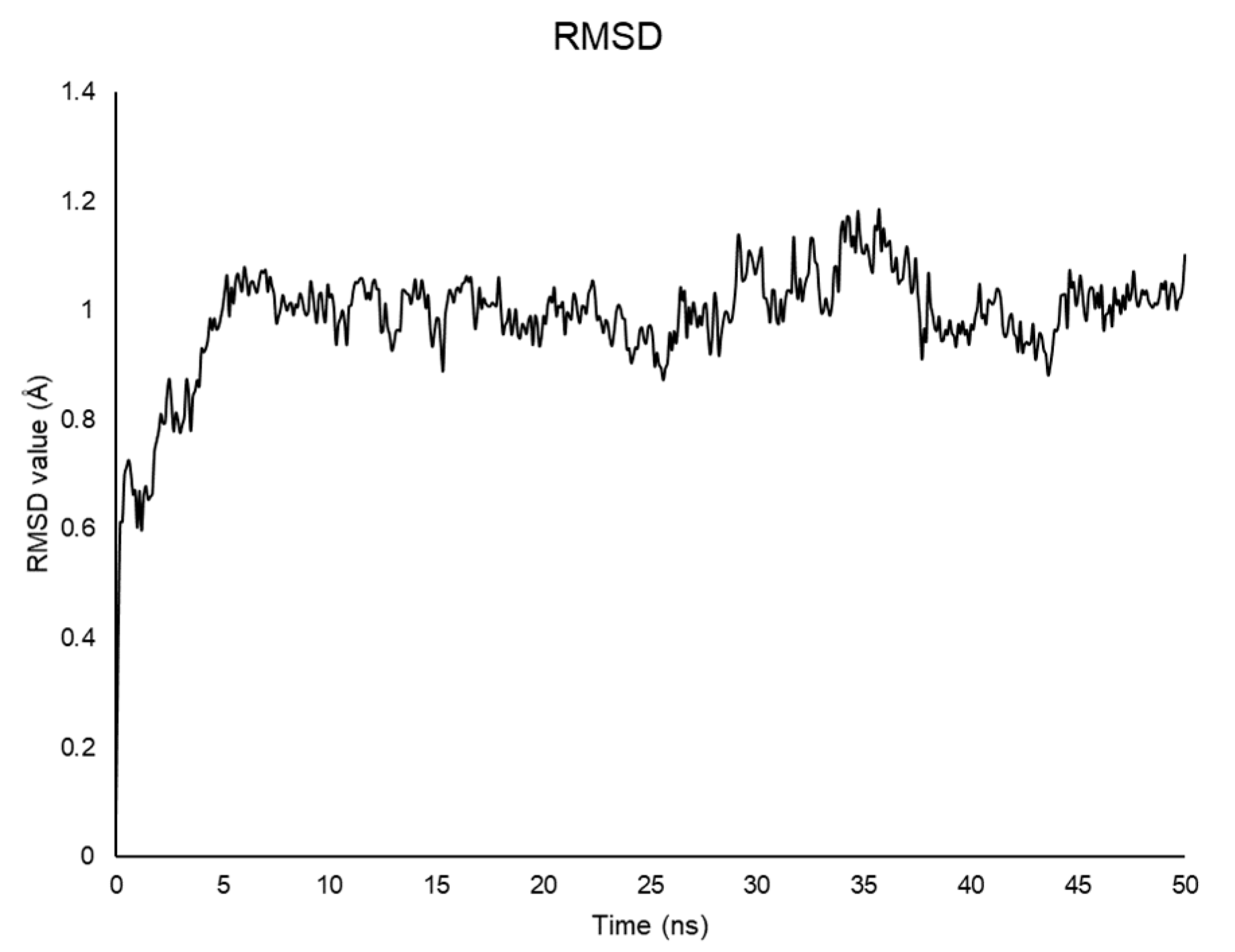
2.2. Molecular Dynamics
3. Discussion
4. Materials and Methods
4.1. Aβ(1–42) Peptide Production
4.2. NMR Sample Preparation
4.3. NMR Spectroscopy
4.3.1. Spectra Acquisition
4.3.2. Assignment of NMR Resonances
4.3.3. Structure Calculation
4.4. Molecular Dynamics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: Potential therapeutics of Alzheimer’s disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol. Ther. 2020, 9, 317–333. [Google Scholar] [CrossRef]
- Yu, J.; Xu, J. Proteolytic cleavage of neuroligins and functions of their cleavage products. J. Zhejiang Univ. Med. Sci. 2020, 49, 514–523. [Google Scholar]
- Tsang, J.Y.; Lee, M.A.; Chan, T.-H.; Li, J.; Ni, Y.-B.; Shao, Y.; Chan, S.-K.; Cheungc, S.-Y.; Lau, K.-F.; Gary, M. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMedicine 2018, 38, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Taylor, A.J.; Verchere, C.B. Islet prohormone processing in health and disease. Diabetes Obes. Metab. 2018, 20, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.-G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.-H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Gong, J.-S.; Yanagisawa, K.; Michikawa, M. A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002, 22, 4833–4841. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, M.; Stillitano, I.; Amodio, G.; Santoro, A.; Buonocore, M.; Moltedo, O.; Remondelli, P.; D’Ursi, A.M. Structural basis of antiviral activity of peptides from MPER of FIV gp36. PLoS ONE 2018, 13, e0204042. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.; Bicknell, W.; Watson, A.A.; Fairlie, D.P.; Craik, D.J. Solution Structure of Amyloid β-Peptide (1–40) in a Water–Micelle Environment. Is the Membrane-Spanning Domain Where We Think It Is? Biochemistry 1998, 37, 11064–11077. [Google Scholar] [CrossRef]
- Shao, H.; Jao, S.-C.; Ma, K.; Zagorski, M.G. Solution structures of micelle-bound amyloid β-(1-40) and β-(1-42) peptides of Alzheimer’s disease. J. Mol. Biol. 1999, 285, 755–773. [Google Scholar] [CrossRef]
- Jarvet, J.; Damberg, P.; Bodell, K.; Göran Eriksson, L.; Graeslund, A. Reversible random coil to β-sheet transition and the early stage of aggregation of the Aβ (12–28) fragment from the Alzheimer peptide. J. Am. Chem. Soc. 2000, 122, 4261–4268. [Google Scholar] [CrossRef]
- Talafous, J.; Marcinowski, K.J.; Klopman, G.; Zagorski, M.G. Solution Structure of Residues 1-28 of the Amyloid. beta.-Peptide. Biochemistry 1994, 33, 7788–7796. [Google Scholar] [CrossRef]
- Kohno, T.; Kobayashi, K.; Maeda, T.; Sato, K.; Takashima, A. Three-dimensional structures of the amyloid β peptide (25–35) in membrane-mimicking environment. Biochemistry 1996, 35, 16094–16104. [Google Scholar] [CrossRef]
- Fletcher, T.G.; Keire, D.A. The interaction of β-amyloid protein fragment (12-28) with lipid environments. Protein Sci. 1997, 6, 666–675. [Google Scholar] [CrossRef]
- Grimaldi, M.; Scrima, M.; Esposito, C.; Vitiello, G.; Ramunno, A.; Limongelli, V.; D’Errico, G.; Novellino, E.; D’Ursi, A.M. Membrane charge dependent states of the β-amyloid fragment Aβ (16–35) with differently charged micelle aggregates. Biochim. Biophys. Acta Biomembr. 2010, 1798, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment: Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Janek, K.; Rothemund, S.; Gast, K.; Beyermann, M.; Zipper, J.; Fabian, H.; Bienert, M.; Krause, E. Study of the conformational transition of Aβ (1–42) using d-amino acid replacement analogues. Biochemistry 2001, 40, 5457–5463. [Google Scholar] [CrossRef]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.; Bonvin, A.M.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The α-to-β conformational transition of Alzheimer’s Aβ-(1–42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of β conformation seeding. ChemBioChem 2006, 7, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Pachahara, S.K.; Chaudhary, N.; Subbalakshmi, C.; Nagaraj, R. Hexafluoroisopropanol induces self-assembly of β-amyloid peptides into highly ordered nanostructures. J. Pept. Sci. 2012, 18, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Pachahara, S.K.; Adicherla, H.; Nagaraj, R. Self-Assembly of Aβ40, Aβ42 and Aβ43 peptides in aqueous mixtures of fluorinated alcohols. PLoS ONE 2015, 10, e0136567. [Google Scholar] [CrossRef]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; Teplow, D.B.; Linse, S. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid β-peptide. FEBS J. 2009, 276, 1266–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stilbs, P. Molecular self-diffusion coefficients in Fourier transform nuclear magnetic resonance spectrometric analysis of complex mixtures. Anal. Chem. 1981, 53, 2135–2137. [Google Scholar] [CrossRef]
- Goddard, T.; Kneller, D. SPARKY 3; University of California: San Francisco, CA, USA, 2004; Volume 15. [Google Scholar]
- Wüthrich, K. NMR with proteins and nucleic acids. Europhys. News 1986, 17, 11–13. [Google Scholar] [CrossRef]
- Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [PubMed]
- Dongre, R.; Folkers, G.E.; Gualerzi, C.O.; Boelens, R.; Wienk, H. A model for the interaction of the G3-subdomain of Geobacillus stearothermophilus IF2 with the 30S ribosomal subunit. Protein Sci. 2016, 25, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Granata, D.; Baftizadeh, F.; Habchi, J.; Galvagnion, C.; De Simone, A.; Camilloni, C.; Laio, A.; Vendruscolo, M. The inverted free energy landscape of an intrinsically disordered peptide by simulations and experiments. Sci. Rep. 2015, 5, 15449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanan, K.M.; Zhang, H.; Zhang, H.; Xi, W.; Wei, Y. On the conformational dynamics of β-amyloid forming peptides: A computational perspective. Front. Bioeng. Biotechnol. 2020, 8, 532. [Google Scholar] [CrossRef]
- Tran, L.; Ha-Duong, T. Exploring the Alzheimer amyloid-β peptide conformational ensemble: A review of molecular dynamics approaches. Peptides 2015, 69, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Danani, A. Molecular simulations of amyloid beta assemblies. Adv. Phys. X 2020, 5, 1770627. [Google Scholar]
- Yang, C.; Li, J.; Li, Y.; Zhu, X. The effect of solvents on the conformations of Amyloid β-peptide (1–42) studied by molecular dynamics simulation. J. Mol. Struct. THEOCHEM 2009, 895, 1–8. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; van Drunen, R.; der Spoel, D.; Bekker, H.; Dijkstra, E.; Van Der Spoel, D.; Sijbers, A. Gromacs: A parallel computer for molecular dynamics simulations. In Proceedings of the 4th International Conference on Computational Physics, Prague, Czech Republic, 24–28 August 1992; World Scientific Publishing Co.: Singapore, 1993. [Google Scholar]
- Safarizadeh, H.; Garkani-Nejad, Z. Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer’s amyloid-beta aggregation: Insight into mechanism of interactions and parameters for design of new inhibitors. J. Mol. Graph. Model. 2019, 87, 129–143. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef]
- Schenk, D. Amyloid-β immunotherapy for Alzheimer’s disease: The end of the beginning. Nat. Rev. Neurosci. 2002, 3, 824–828. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β 1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer disease: A review. JAMA Neurol. 2016, 73, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Österlund, N.; Luo, J.; Wärmländer, S.K.; Gräslund, A. Membrane-mimetic systems for biophysical studies of the amyloid-β peptide. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 492–501. [Google Scholar] [CrossRef]
- Jao, S.-C.; Ma, K.; Talafous, J.; Orlando, R.; Zagorski, M.G. Trifluoroacetic acid pretreatment reproducibly disaggregates the amyloid β-peptide. Amyloid 1997, 4, 240–252. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.; Bachmann, P.; Ernst, R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Piantini, U.; Sorensen, O.; Ernst, R.R. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar] [CrossRef]
- Parella, T.; Adell, P.; Sánchez-Ferrando, F.; Virgili, A. Effective multiple-solvent suppression scheme using the excitation sculpting principle. Magn. Res. Chem. 1998, 36, 245–249. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.P.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Release 2021-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; The R Development Core Team: Vienna, Austria, 2013. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Experimental Restraints after CYANA | |
| Total NOEs | 585 |
| Intra-residual | 348 |
| Sequential | 143 |
| Long-range | 94 |
| RMSD | |
| bb/heavy Å | 2.92/3.11 |
| Ramachandran Analysis | |
| Favorable regions | 50.60% |
| Additional allowed regions | 37.90% |
| Generously allowed regions | 9.10% |
| Disallowed regions | 2.4% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoro, A.; Grimaldi, M.; Buonocore, M.; Stillitano, I.; D’Ursi, A.M. Exploring the Early Stages of the Amyloid Aβ(1–42) Peptide Aggregation Process: An NMR Study. Pharmaceuticals 2021, 14, 732. https://doi.org/10.3390/ph14080732
Santoro A, Grimaldi M, Buonocore M, Stillitano I, D’Ursi AM. Exploring the Early Stages of the Amyloid Aβ(1–42) Peptide Aggregation Process: An NMR Study. Pharmaceuticals. 2021; 14(8):732. https://doi.org/10.3390/ph14080732
Chicago/Turabian StyleSantoro, Angelo, Manuela Grimaldi, Michela Buonocore, Ilaria Stillitano, and Anna Maria D’Ursi. 2021. "Exploring the Early Stages of the Amyloid Aβ(1–42) Peptide Aggregation Process: An NMR Study" Pharmaceuticals 14, no. 8: 732. https://doi.org/10.3390/ph14080732
APA StyleSantoro, A., Grimaldi, M., Buonocore, M., Stillitano, I., & D’Ursi, A. M. (2021). Exploring the Early Stages of the Amyloid Aβ(1–42) Peptide Aggregation Process: An NMR Study. Pharmaceuticals, 14(8), 732. https://doi.org/10.3390/ph14080732