
Pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines as Potential Cytotoxic Agents against Human Neuroblastoma

 , , ,
, , ,  ,
,  and
and 
Abstract
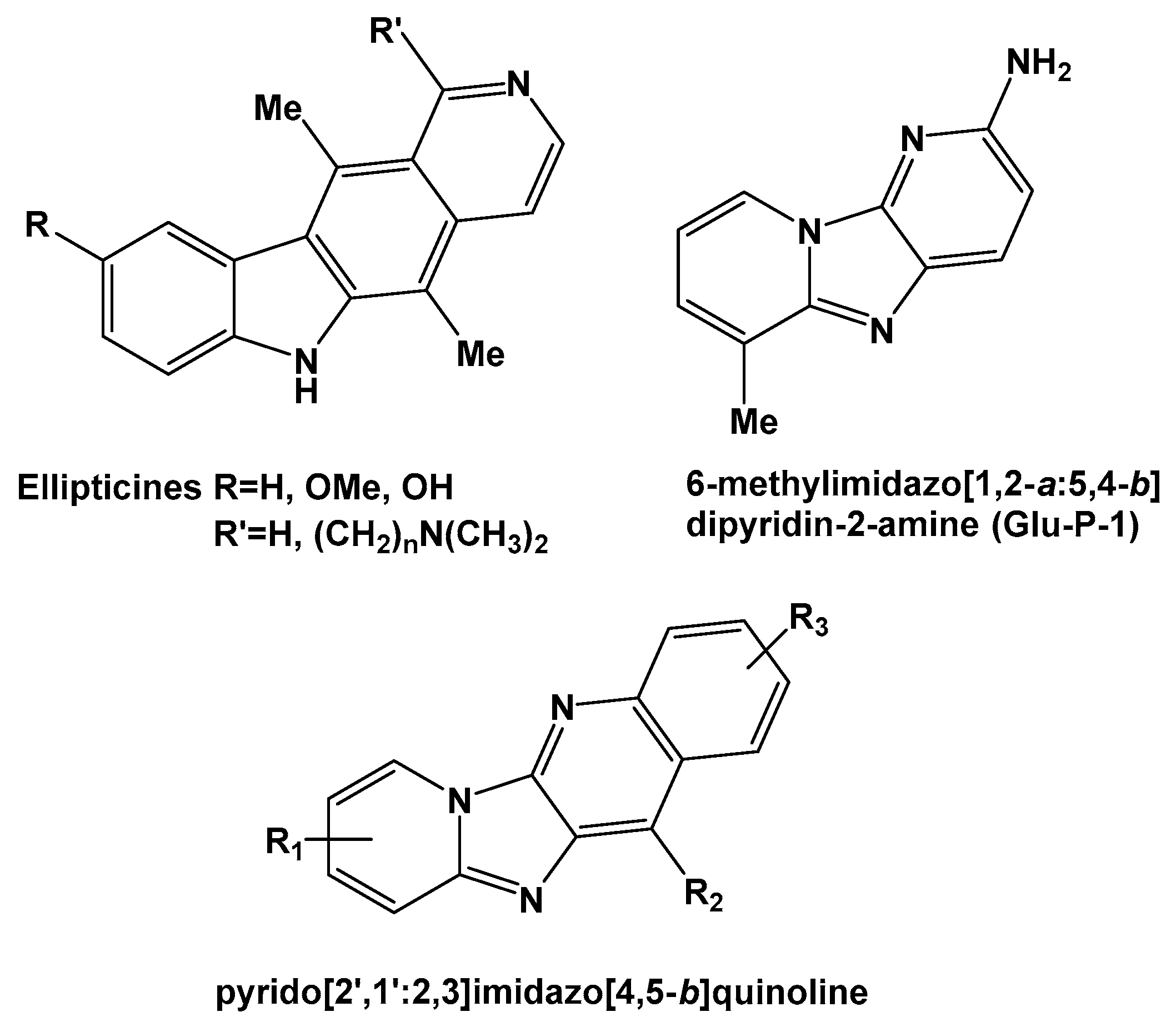
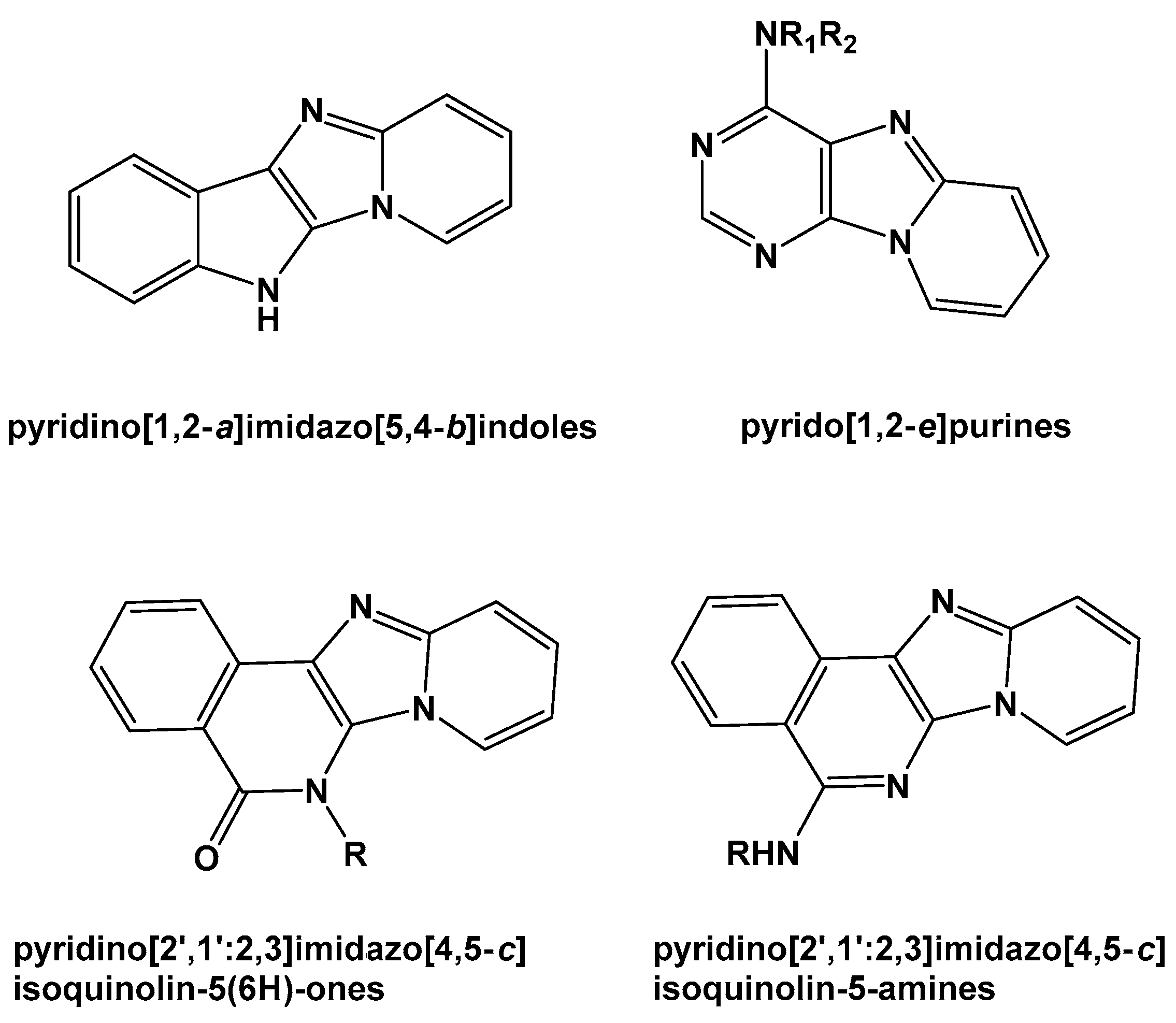
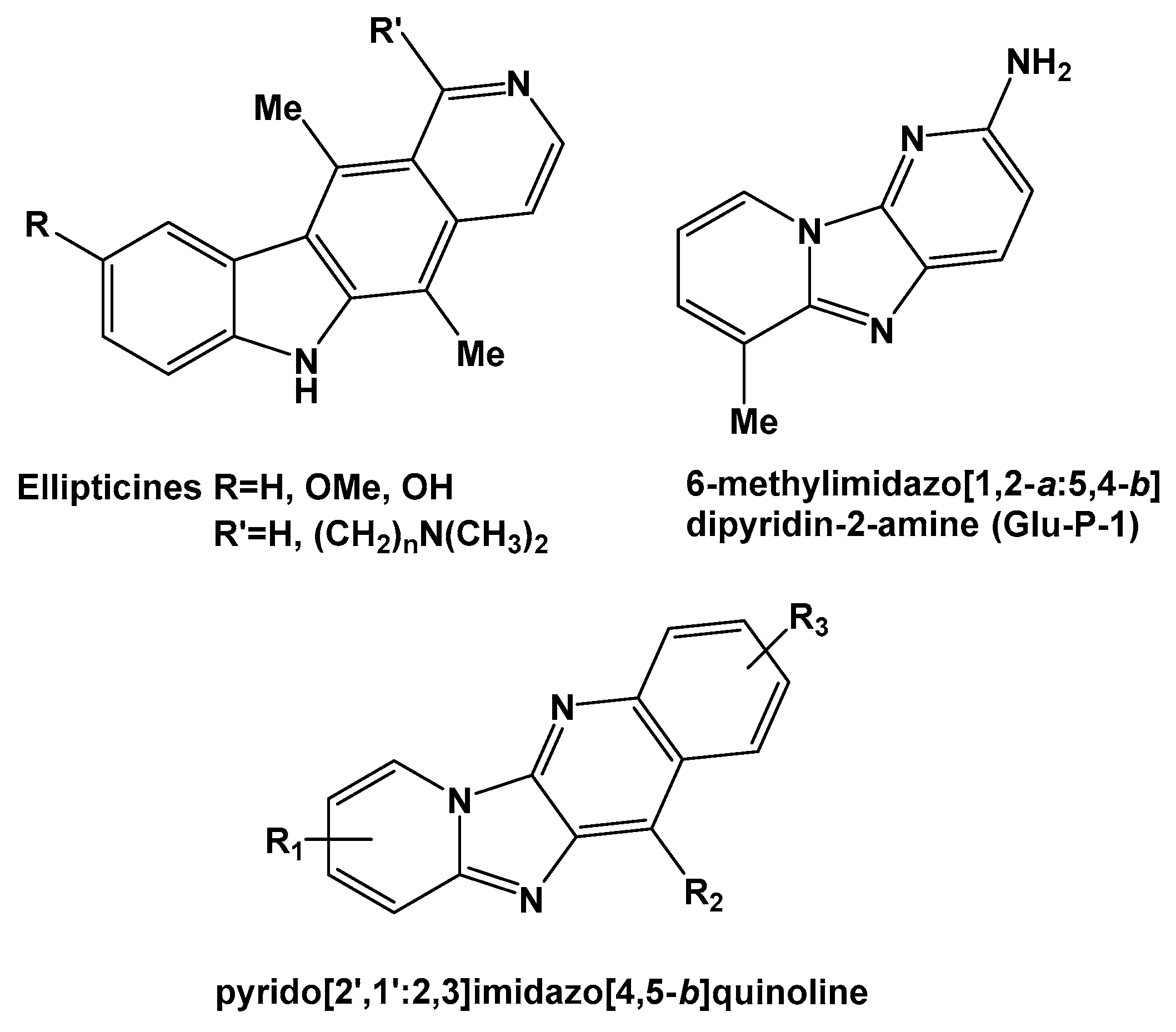
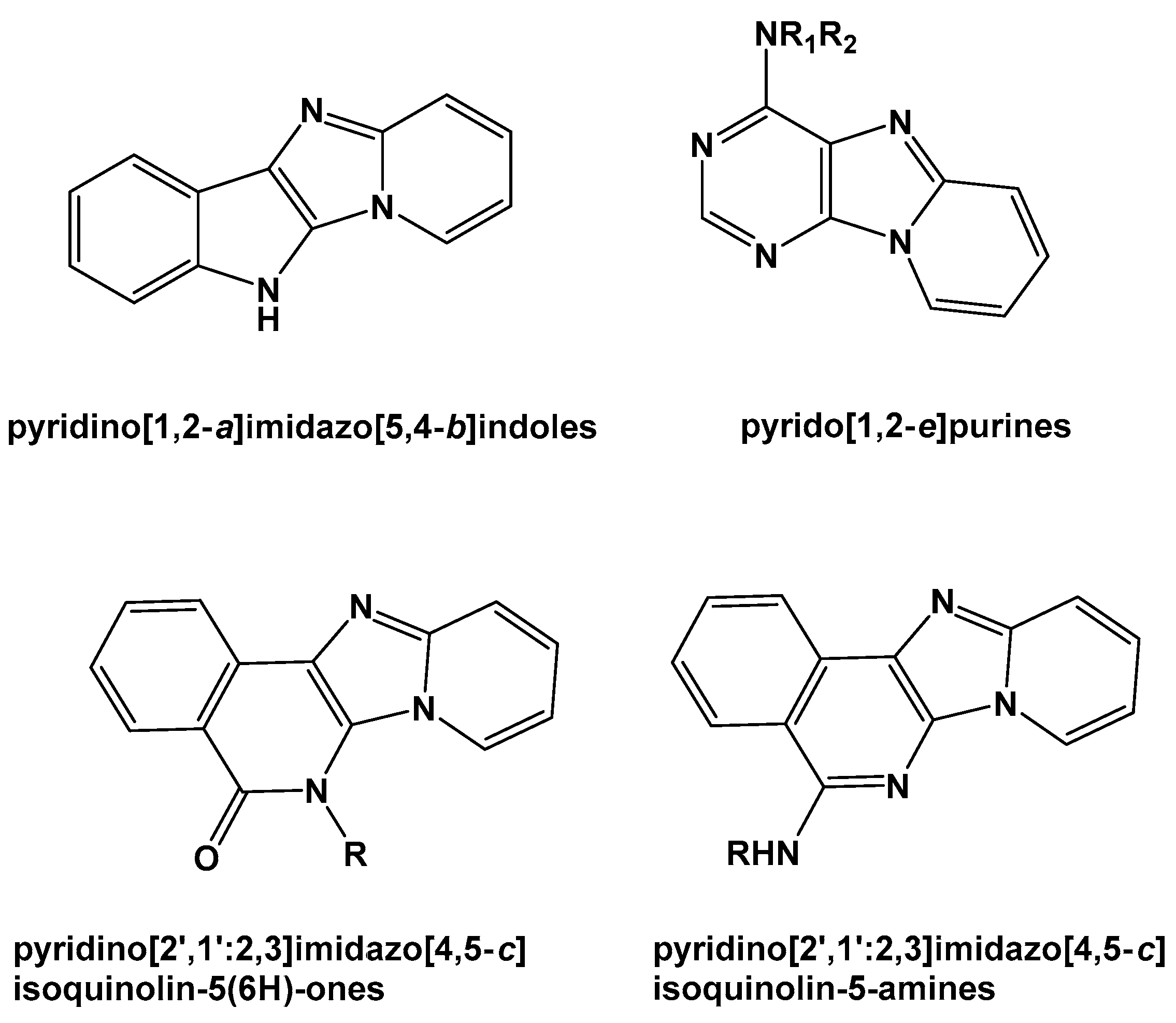
:1. Introduction
2. Results and Discussion
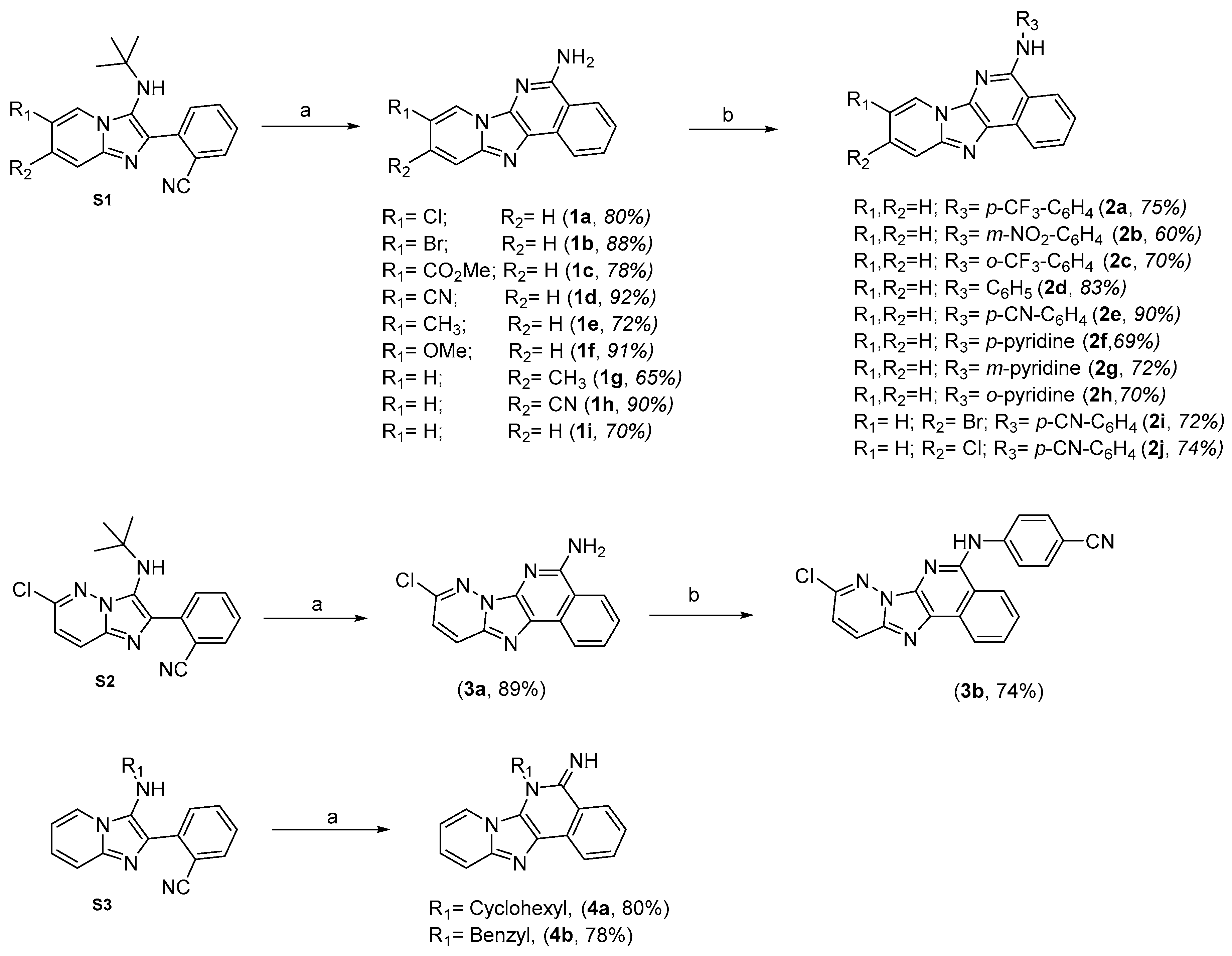
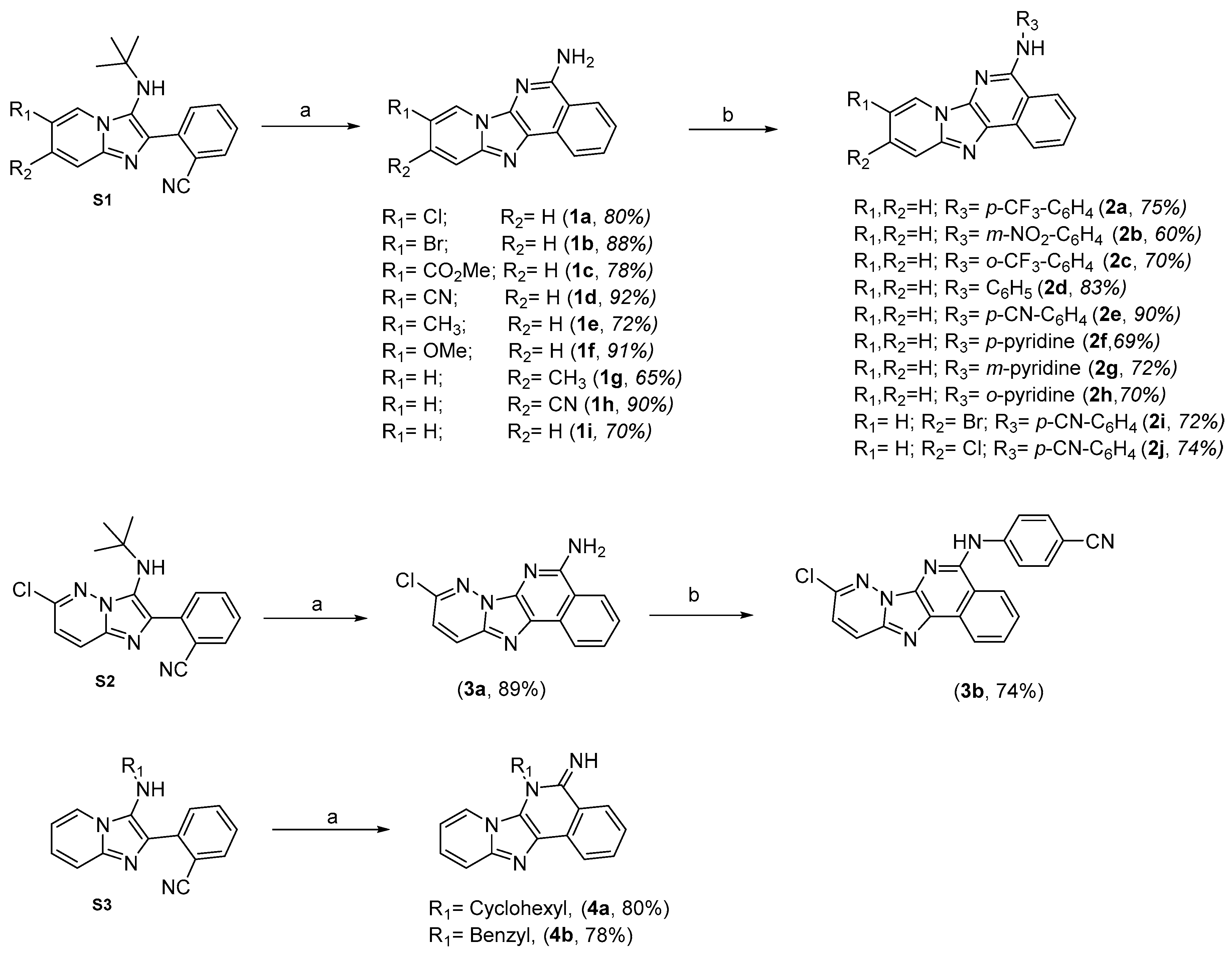
2.1. Chemistry
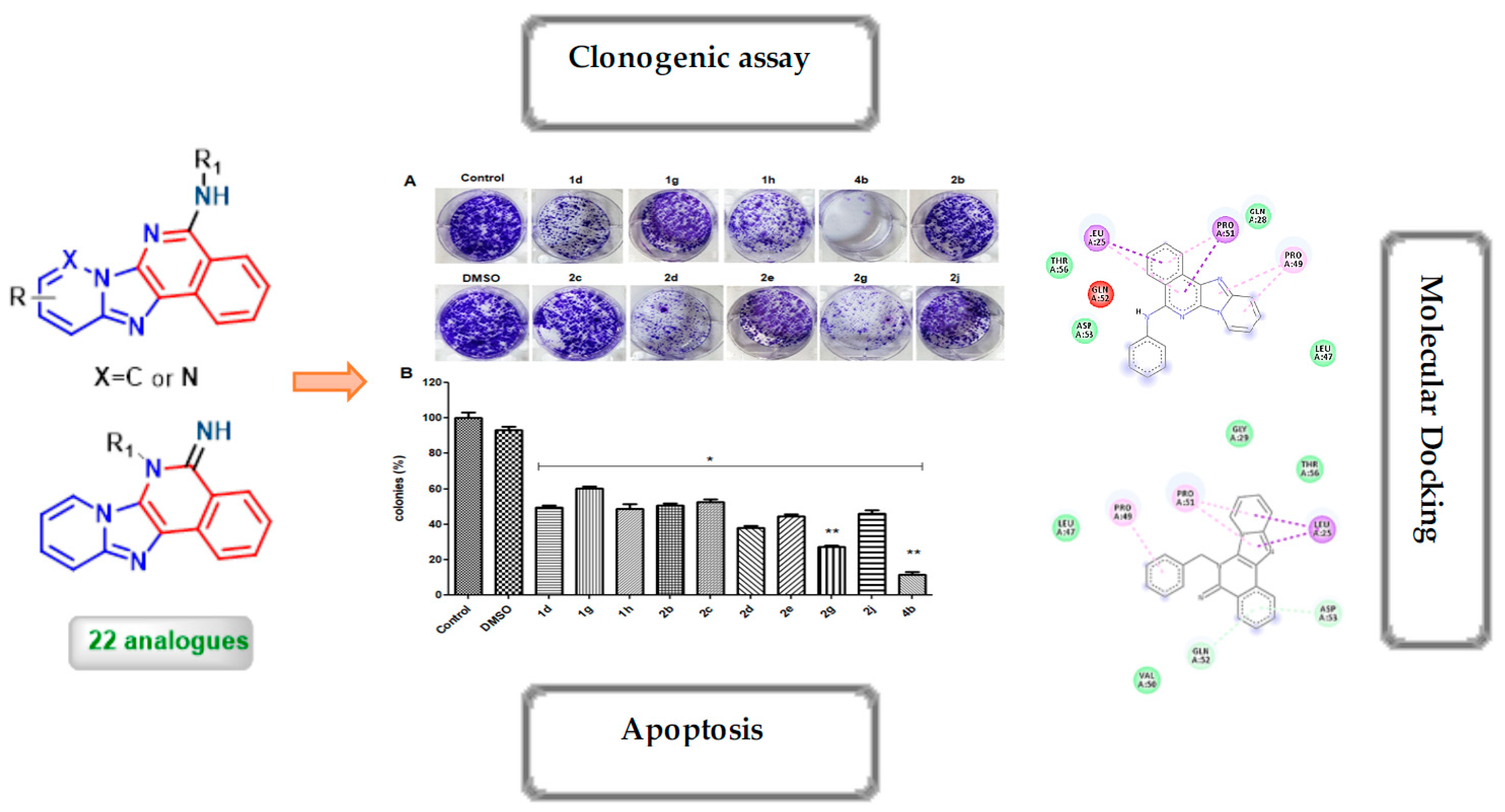
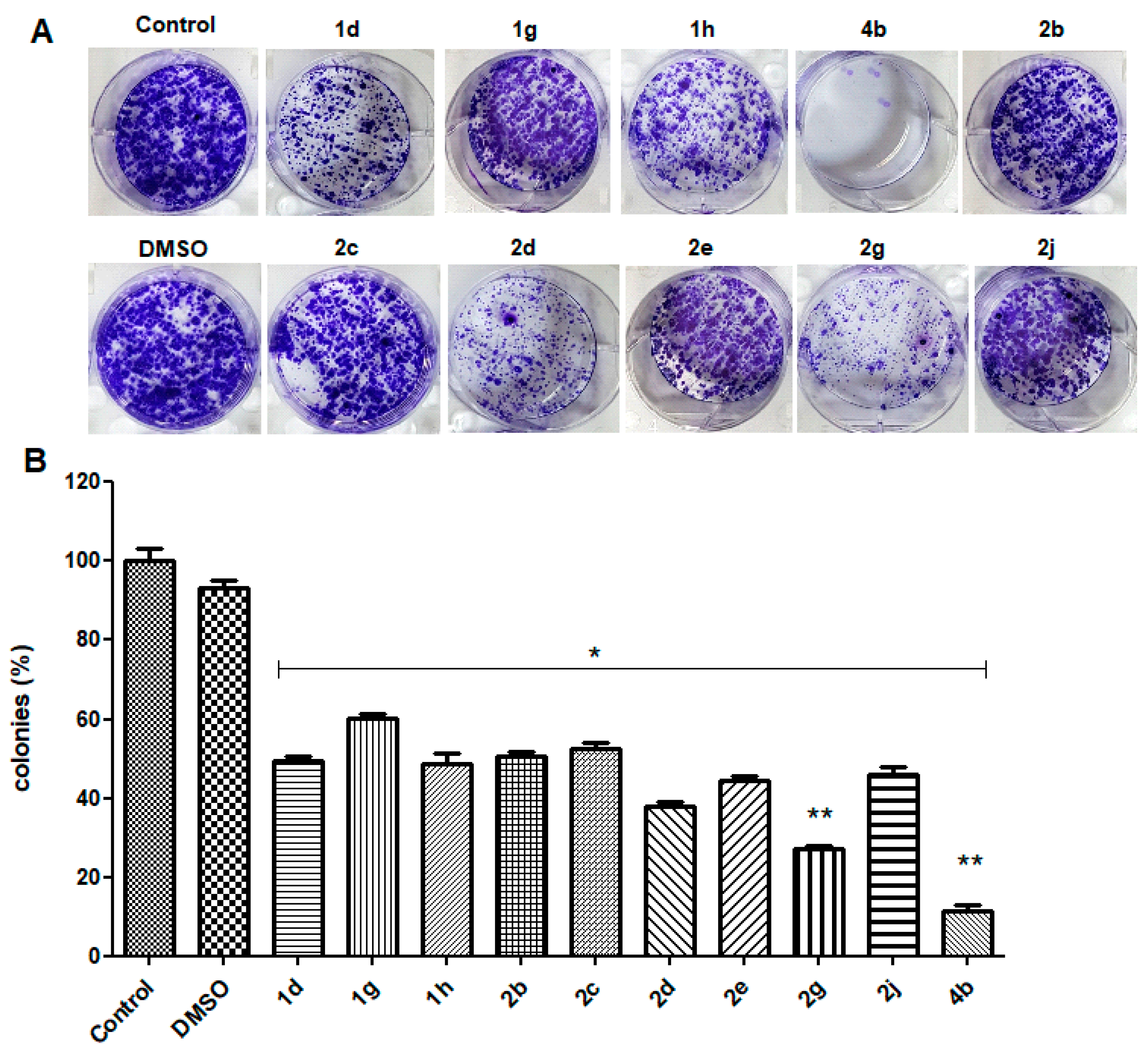
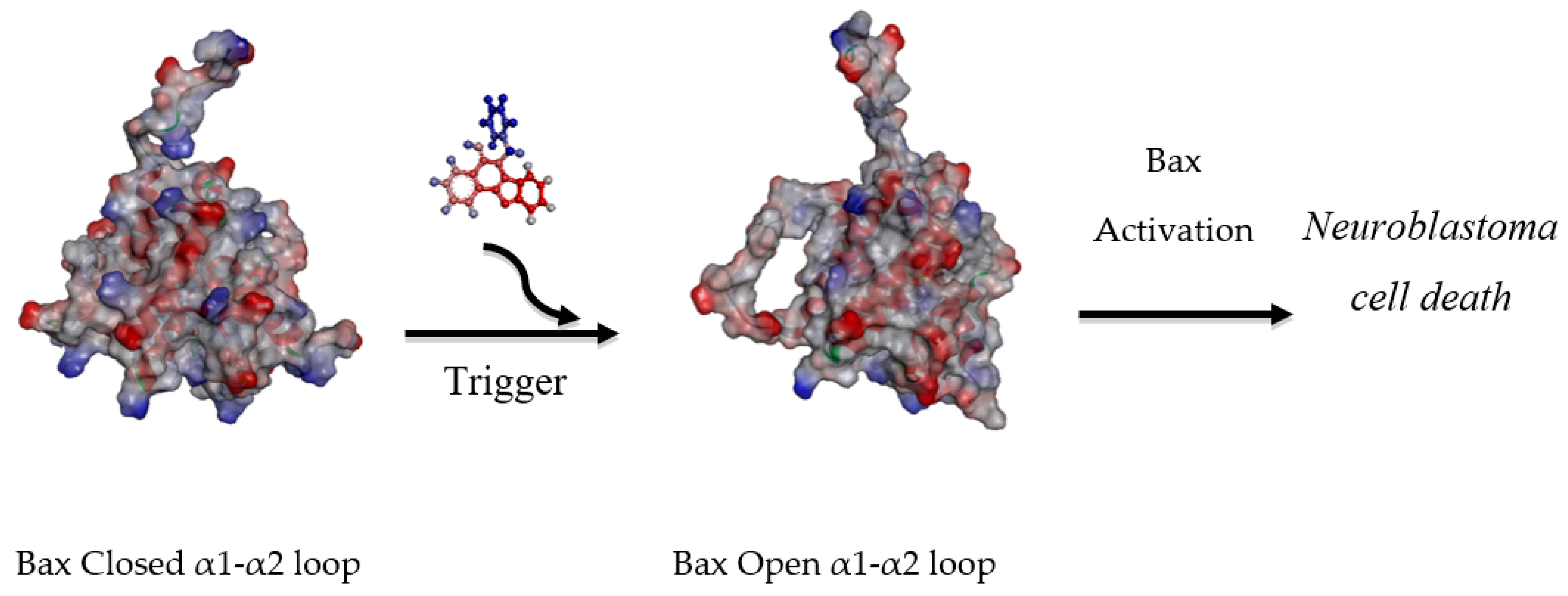
2.2. Biological Studies
2.2.1. Cell Viability Studies
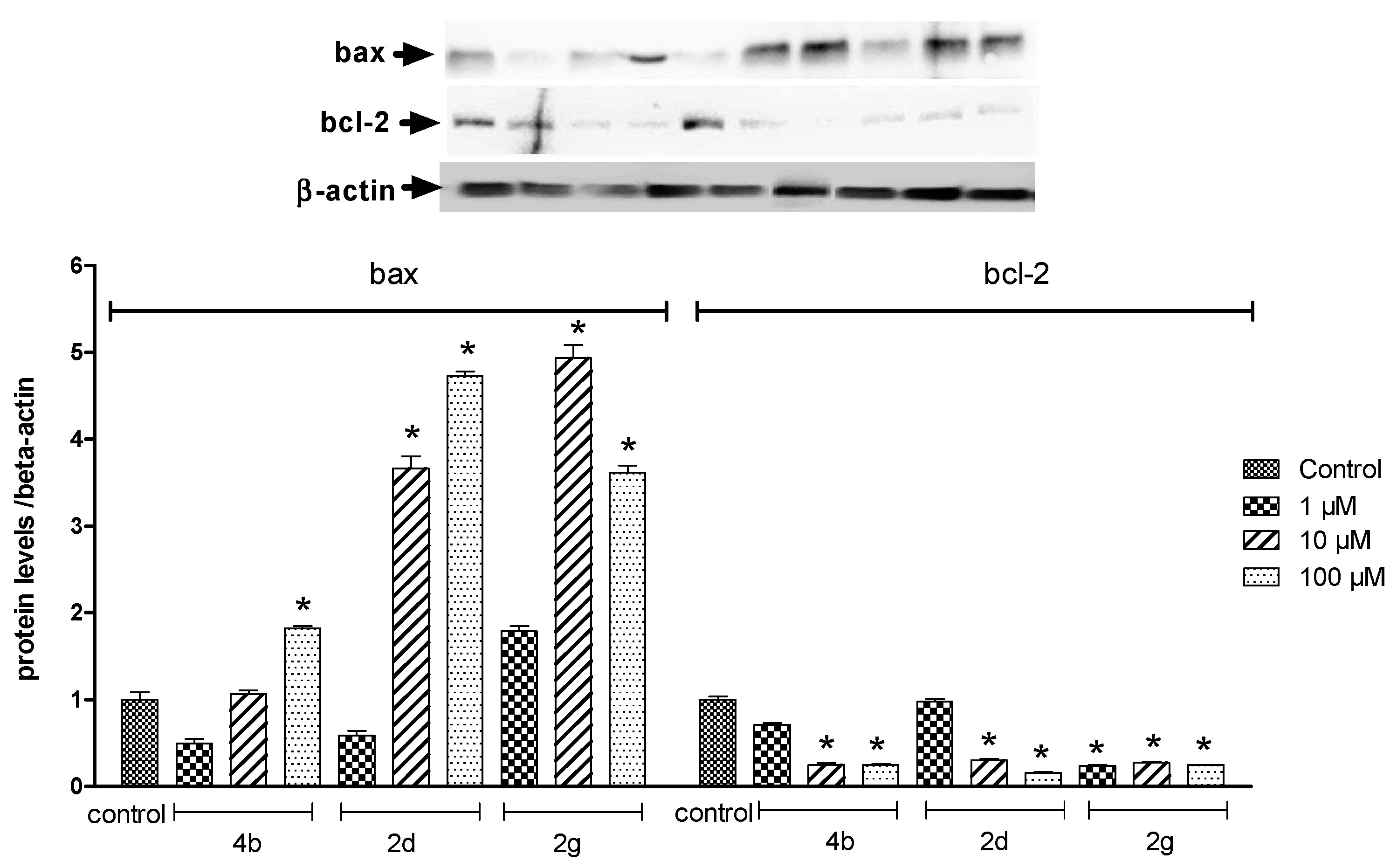
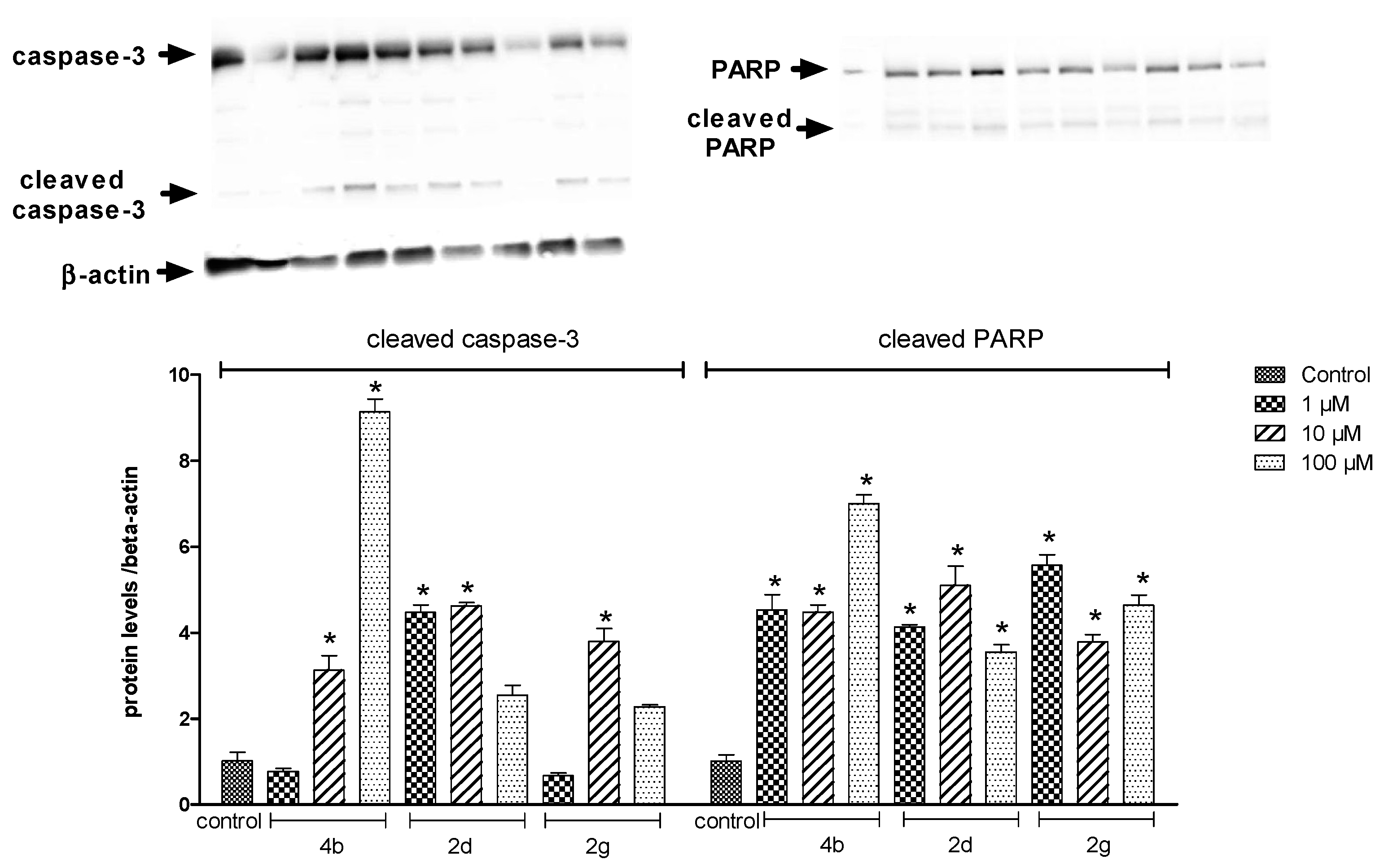
2.2.2. Protein Analysis
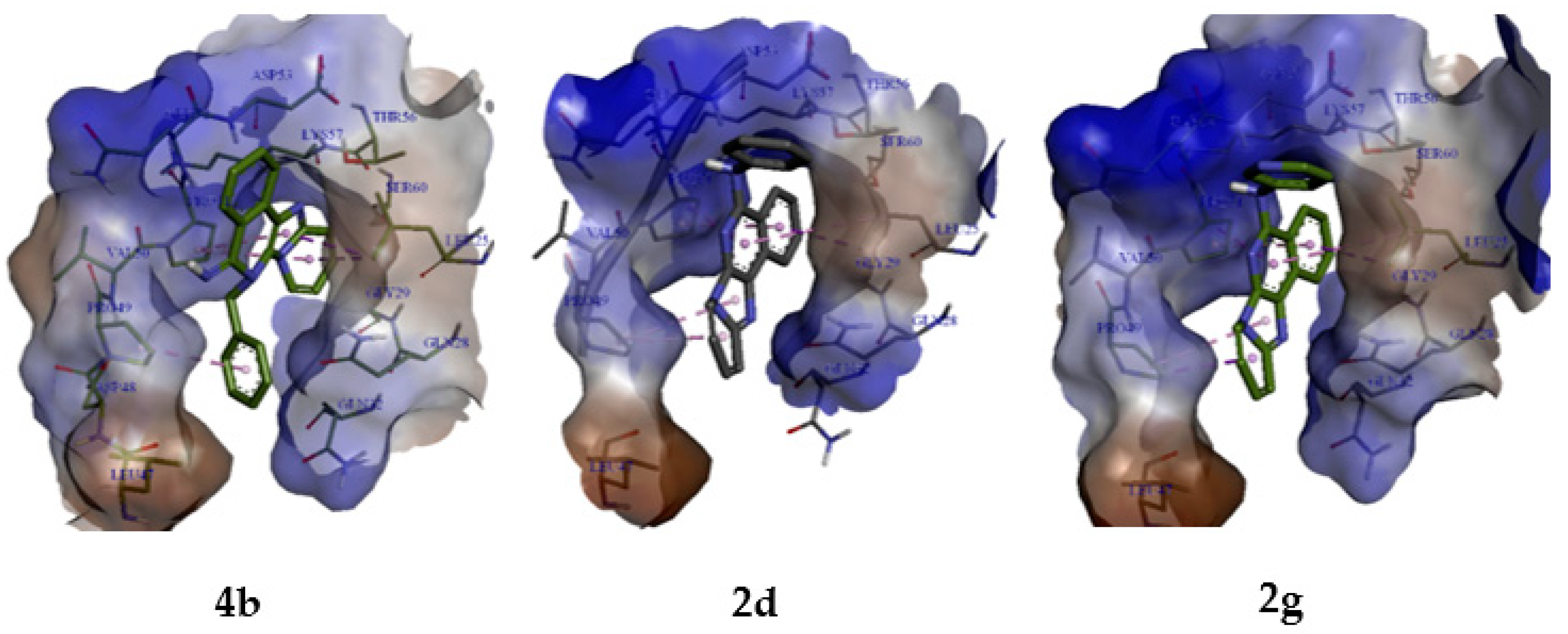
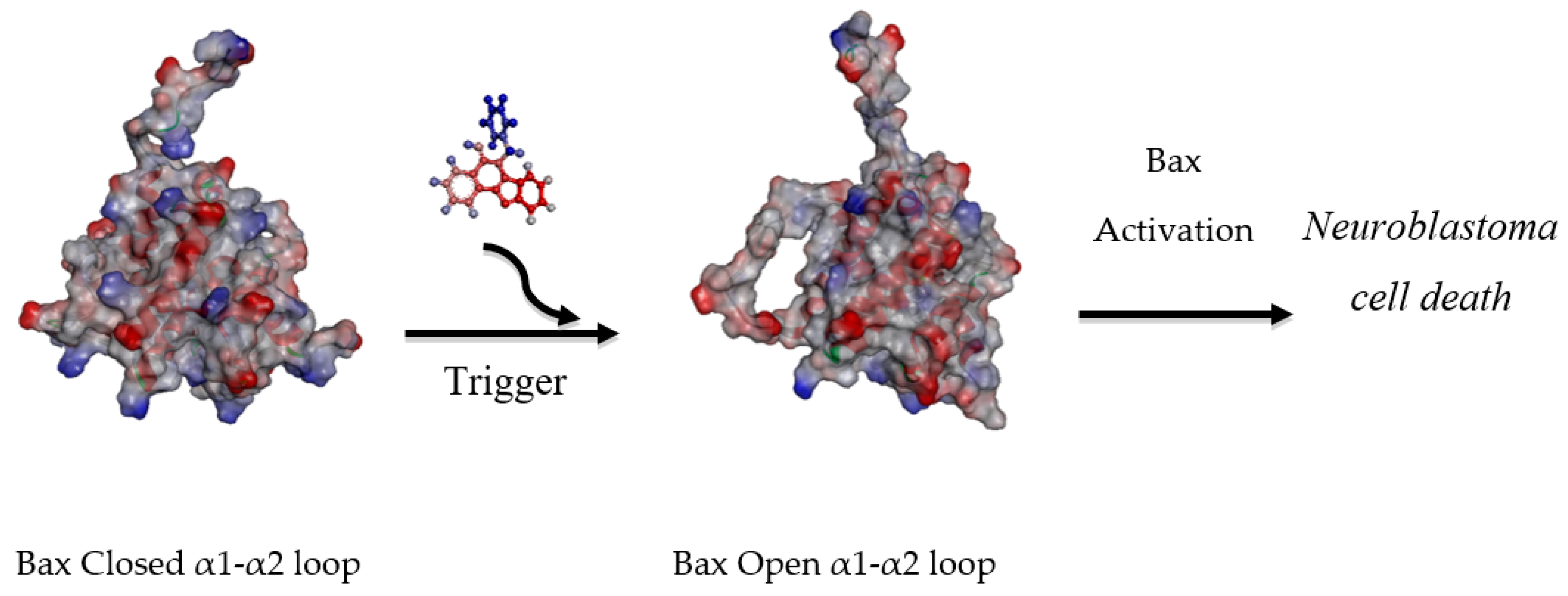
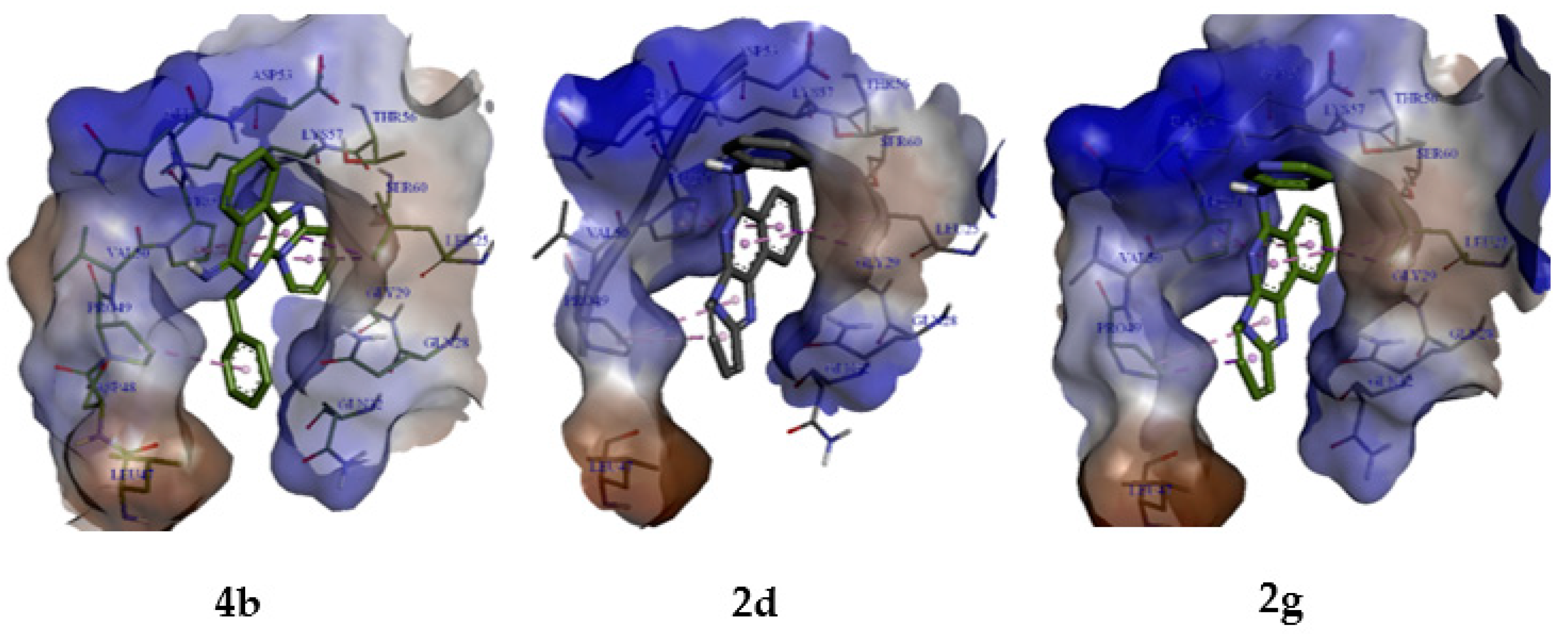
2.3. Molecular Modelling Studies
2.3.1. Protein and Ligand Preparation
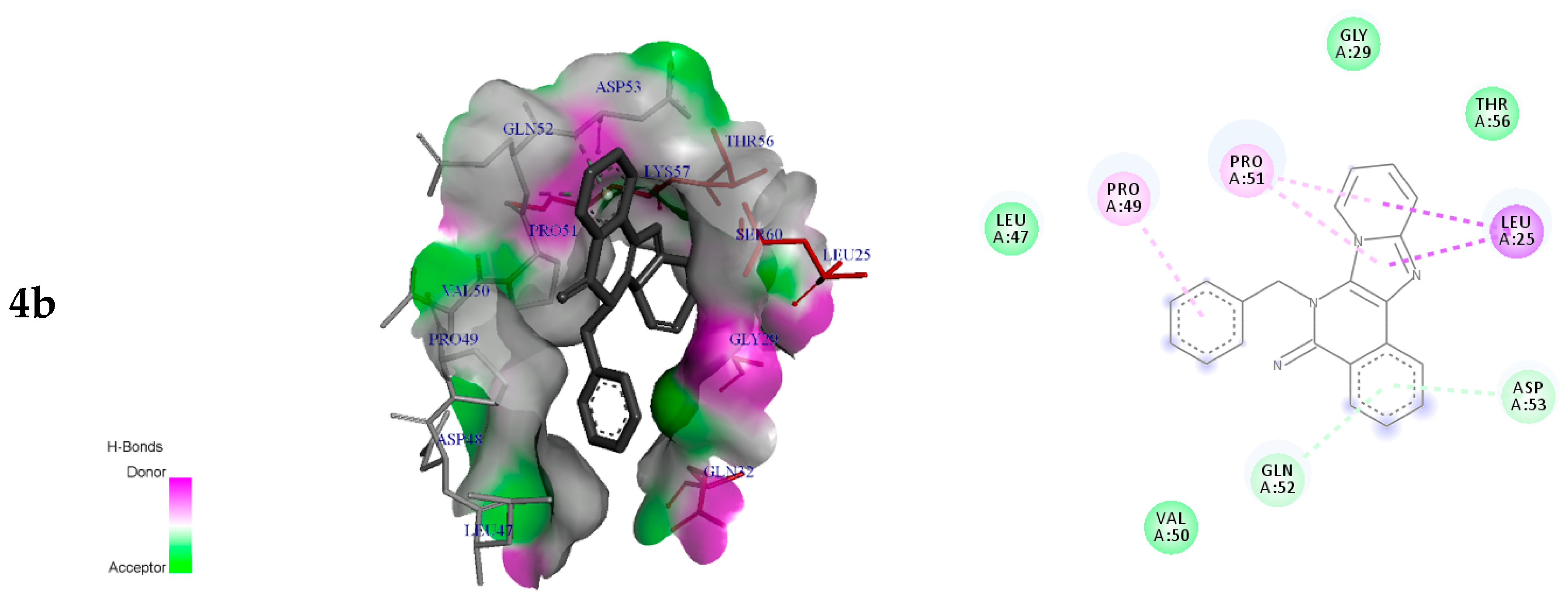
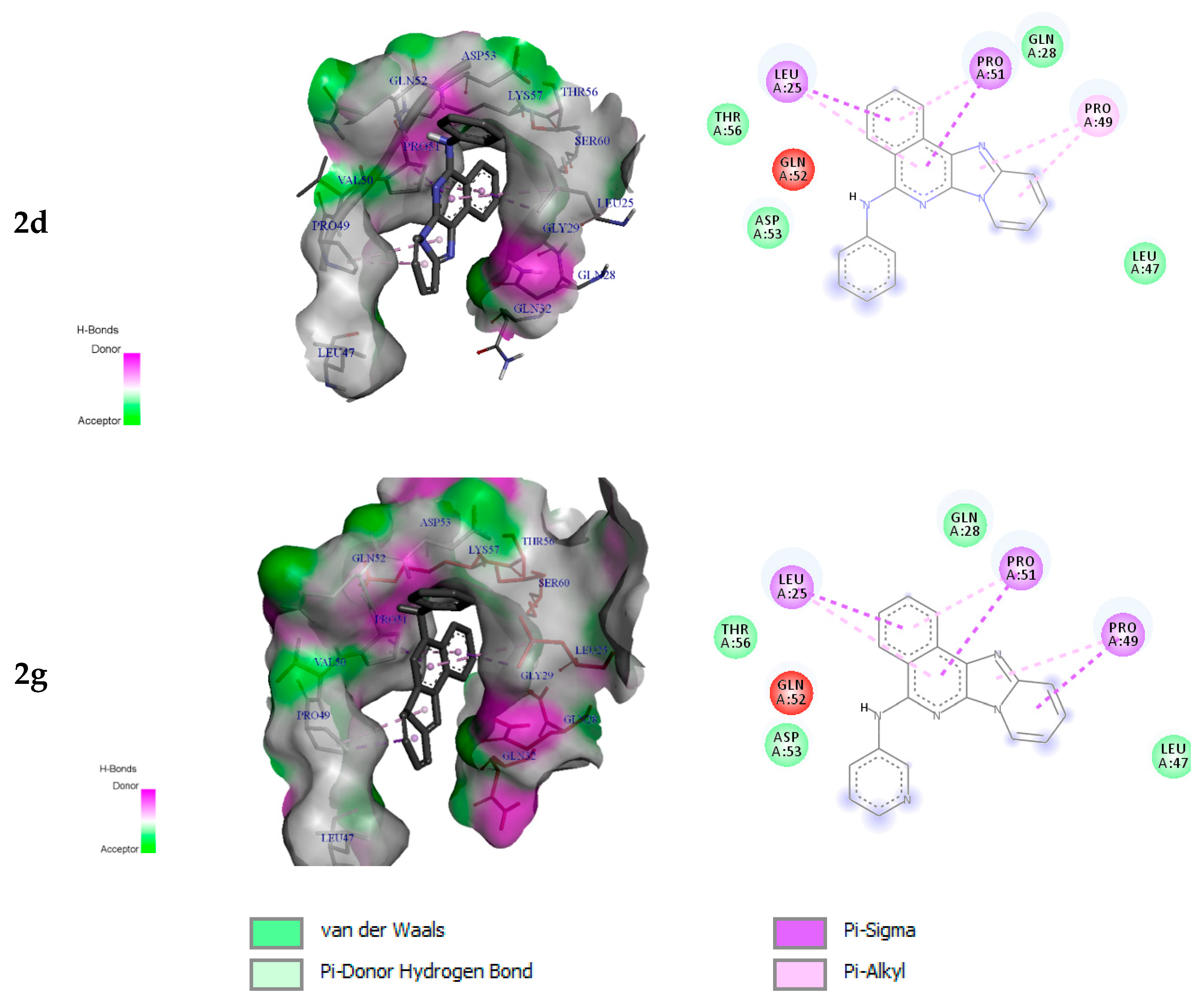
2.3.2. Molecular Docking Analysis
3. Materials and Methods
3.1. Chemistry Methods
3.1.1. General Information
3.1.2. General Groebke–Blackburn–Bienaymé MCR Procedure for the Synthesis of (S1, S2 and S3)
3.1.3. General N-Deprotection-cyclization Procedure
3.1.4. Buchwald–Hartwig Cross-Coupling Procedure
3.2. Biological Methods
3.2.1. Cell Culture and Treatments
3.2.2. Cell Proliferation Assay
3.2.3. Clonogenic Survival Assay
3.2.4. Protein Analysis
3.2.5. Western Blotting
3.2.6. Statistical Analysis
3.3. Computational Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mueller, S.; Matthay, K.K. Neuroblastoma: Biology and staging. Curr. Oncol. Rep. 2009, 11, 431–438. [Google Scholar] [CrossRef]
- Weinstein, J.L.; Katzenstein, H.M.; Cohn, S.L. Advances in the Diagnosis and Treatment of Neuroblastoma. Oncologist 2003, 8, 278–292. [Google Scholar] [CrossRef]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, J.I.; Dyberg, C.; Wickström, M. Neuroblastoma—A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nile, D.L.; Rae, C.; Hyndman, I.J.; Gaze, M.N.; Mairs, R.J. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitizers for the treatment of neuroblastoma. BMC Cancer 2016, 16, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse, S.; Mathieu, V.; Pillard, C.; Massip, S.; Marchivie, M.; Jarry, C.; Bernard, P.; Kiss, R.; Guillaumet, G. New imidazo[1,2-b]pyrazoles as anticancer agents: Synthesis, biological evaluation and structure activity relationship analysis. Eur. J. Med. Chem. 2014, 84, 718–730. [Google Scholar] [CrossRef]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused Imidazoles As Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef]
- Guchhait, S.K.; Chaudhary, V. Desilylative activation of TMSCN in chemoselective Strecker-Ugi type reaction: Functional fused imidazoles as building blocks as an entry route to annulated purines. Org. Biomol. Chem. 2014, 12, 6694–6705. [Google Scholar] [CrossRef] [PubMed]
- Marzouk, V.H.R.; Hennum, M.; Gundersen, L.L. Efficient synthesis of cytotoxic pyrido[1,2-e]purines from purines employing direct C-allylation and RCM-oxidation as key steps. Tetrahedron Lett. 2013, 54, 3437–3439. [Google Scholar] [CrossRef]
- Favier, A.; Blackledge, M.; Simorre, J.P.; Crouzy, S.; Dabouis, V.; Gueiffier, A.; Marion, D.; Debouzy, J.C. Solution structure of 2-(pyrido[1,2-e]purin-4-yl)amino-ethanol intercalated in the DNA duplex d(CGATCG) 2. Biochemistry 2001, 40, 8717–8726. [Google Scholar] [CrossRef]
- Debouzy, J.C.; Gueiffier, A.; Fauvelle, F.; Viols, H.; Dejean, E.; Neirinck, V.; Peinnequin, A.; Bachelet, C.; Perly, B.; Chapat, J.P. Synthetic pyridopurines derived from food pyrolysis products: Intercalation, interactions with membranes, cyclodextrin complexation, and biological mitogenic properties. J. Pharm. Sci. 1996, 85, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Brullo, C.; Spisani, S.; Selvatici, R.; Bruno, O. N-Aryl-2-phenyl-2,3-dihydro-imidazo[1,2-b]pyrazole-1-carboxamides 7-substituted strongly inhibiting both fMLP-OMe- and IL-8-induced human neutrophil chemotaxis. Eur. J. Med. Chem. 2012, 47, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Bruno, O.; Brullo, C.; Bondavalli, F.; Ranise, A.; Schenone, S.; Falzarano, M.S.; Varani, K.; Spisani, S. 2-Phenyl-2,3-dihydro-1H-imidazo[1,2-b]pyrazole derivatives: New potent inhibitors of fMLP-induced neutrophil chemotaxis. Bioorganic Med. Chem. Lett. 2007, 17, 3696–3701. [Google Scholar] [CrossRef]
- Tyagi, V.; Khan, S.; Bajpai, V.; Gauniyal, H.M.; Kumar, B.; Chauhan, P.M.S. Skeletal diverse synthesis of N-fused polycyclic heterocycles via the sequence of Ugi-type MCR and CuI-catalyzed coupling/tandem Pictet-Spengler reaction. J. Org. Chem. 2012, 77, 1414–1421. [Google Scholar] [CrossRef]
- Deady, L.W.; Rodemann, T. Reduced Benzimidazo[2,1-a]isoquinolines. Synthesis and Cytotoxicity Studies. Aust. J. Chem. 2001, 54, 529–534. [Google Scholar] [CrossRef]
- Vanotti, E.; Fiorentini, F.; Villa, M. Synthesis of novel derivatives of 1 H-imidazo[1,2- b]pyrazole as potential CNS-agents. J. Heterocycl. Chem. 1994, 31, 737–743. [Google Scholar] [CrossRef]
- Reddyrajula, R.; Dalimba, U.K. Structural modification of zolpidem led to potent antimicrobial activity in imidazo[1,2-a]pyridine/pyrimidine-1,2,3-triazoles. New J. Chem. 2019, 43, 16281–16299. [Google Scholar] [CrossRef]
- Guetzoyan, L.; Nikbin, N.; Baxendale, I.R.; Ley, S.V. Flow chemistry synthesis of zolpidem, alpidem and other GABAA agonists and their biological evaluation through the use of in-line frontal affinity chromatography. Chem. Sci. 2013, 4, 764–769. [Google Scholar] [CrossRef]
- Arnould, M.; Hiebel, M.A.; Massip, S.; Léger, J.M.; Jarry, C.; Berteina-Raboin, S.; Guillaumet, G. Efficient Metal-Free Synthesis of Various Pyrido[2′,1′:2,3]imidazo′[4,5-b]quinolines. Chem. Eur. J. 2013, 19, 12249–12253. [Google Scholar] [CrossRef] [PubMed]
- Bonjean, K.; De Pauw-Gillet, M.C.; Defresne, M.P.; Colson, P.; Houssier, C.; Dassonneville, L.; Bailly, C.; Greimers, R.; Wright, C.; Quetin-Leclercq, J.; et al. The DNA Intercalating Alkaloid cryptolepine Interferes with Topoisomerase II and Inhibits Primarily DNA Synthesis in B16 Melanoma Cells. Biochemistry 1998, 37, 5136–5146. [Google Scholar] [CrossRef] [PubMed]
- Arzel, E.; Rocca, P.; Grellier, P.; Labaeïd, M.; Frappier, F.; Gueritte, F.; Gaspard, C.; Marsais, F.; Godard, A.; Quéguiner, G. New Synthesis of Benzo-δ-carbolines, Cryptolepines and their Salts: In vitro Cytotoxic, Antiplasmodial and Antitrypanosomal Activities of δ -Carbolines, Benzo-δ -carbolines and Cryptolepines. J. Med. Chem. 2001, 44, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Chewonarin, T.; Kinouchi, T.; Kataoka, K.; Arimochi, H.; Kuwahara, T.; Vinitketkumnuen, T.; Ohnishi, T. Effects of roselle (Hibiscus sabdariffa Linn.), a Thai medicinal plant, on the mutagenicity of various known mutagens in salmonella typhimurium and of formation of Aberrant Cryp Foci Induced by a Colon Carcinogens Azoxymethane and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in F344 rats. Food Chem. Toxicol. 1999, 37, 591–601. [Google Scholar] [CrossRef]
- Groebke, K.; Weber, L.; Mehlin, F. Synthesis of Imidazo[1,2-a]annulated Pyridines, Pyrazines and Pyrimidines by a novel Three-Component Condensation. Synlett 1998, 1998, 661–663. [Google Scholar] [CrossRef]
- Blackburn, F.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Parallel synthesis of 3-aminoimidazo[1,2-a]pyridines and pyrazines by a new Three-Component Condensation. Tetrahedron Lett. 1998, 39, 3635–3638. [Google Scholar] [CrossRef]
- Bienaymé, H.; Bouzid, K. A New Heterocyclic Multicomponent Reaction for the Combinatorial Synthesis of Fused 3-Aminoimidazoles. Angew. Chem. Int. Ed. 1998, 37, 2234–2237. [Google Scholar] [CrossRef]
- Tber, Z.; Hiebel, M.A.; Allouchi, H.; El Hakmaoui, A.; Akssira, M.; Guillaumet, G.; Berteina-Raboin, S. Metal free direct formation of various substituted pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines and their further functionalization. RSC Adv. 2015, 5, 35201–35210. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Loubidi, M.; Lorion, M.; El Hakmaoui, A.; Bernard, P.; Akssira, M.; Guillaumet, G. One-Step Synthesis of 1H-Imidazo[1,5-a]imidazole Scaffolds and Access to their Polyheterocycles. Synthesis 2019, 51, 3973–3980. [Google Scholar] [CrossRef]
- Sugimoto, T.; Jin, H.; Fijita, M.; Fukunaga, T.; Nagaoka, N.; Yamaai, T.; Ichikawa, H. Induction of activated caspase-3-immunoreactivity and apoptosis in the trigeminal ganglion neurons by neonatal peripheral nerve injury. Brain Res. 2004, 1017, 238–243. [Google Scholar] [CrossRef]
- Ten Berge, R.L.; Meijer, C.J.L.M.; Dukers, D.F.; Alain Kummer, J.; Bladergroen, B.A.; Vos, W.; Erik Hack, C.; Ossenkoppele, G.J.; Oudejans, J.J. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood 2002, 99, 4540–4546. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Castle, V.P. Alternative NHEJ pathway components are therapeutic targets in high-risk neuroblastoma. Mol. Cancer Res. 2005, 13, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly(ADP-ribose) Polymerase: An Early Marker of Chemotherapy-induced Apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Cheung, N.K.V.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Plumb, J.A. Cell sensitivity assays: Clonogenic assay. Methods Mol. Med. 2004, 88, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Akar, U.; Mehta, K.; Lopei-Berestein, G. PKCδ and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy 2007, 3, 480–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.J.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted Silencing of Elongation Factor 2 Kinase Suppresses Growth and Sensitizes Tumors to Doxorubicin in an Orthotopic Model of Breast Cancer. PLoS ONE 2012, 7, e41171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009; Available online: http://gaussian.com/ (accessed on 1 November 2016).
- Systèmes, B.D. Discovery Studio Modeling Environment, San Diego Dassault Systèmes. (n.d.). Available online: http://accelrys.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 15 April 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | % Cell Viability ± S.D. | |||
|---|---|---|---|---|---|
| 0.1 µM | 1 µM | 10 µM | 100 µM | ||
| 1a | 30.92 | 79.26 ± 7.30 | 74.37 ± 3.07 | 56.05 ± 1.49 | 42.28 ± 3.54 |
| 1b | 20.22 | 78.13 ± 7.12 | 82.00 ± 7.82 | 80.76 ± 17.03 | 2.82 ± 0.68 |
| 1c | 63.57 | 73.13 ± 6.09 | 64.93 ± 8.65 | 59.36 ± 3.36 | 47.06 ± 2.76 |
| 1d | >100 | 67.87 ± 1.59 | 74.17 ± 1.80 | 68.67 ± 1.23 | 58.14 ± 5.16 |
| 1e | 29.72 | 74.28 ± 7.46 | 76.26 ± 9.43 | 69.99 ± 5.20 | 32.25 ± 1.75 |
| 1f | 62.75 | 95.60 ± 7.30 | 83.16 ± 2.45 | 61.48 ± 3.22 | 48.50 ± 1.59 |
| 1g | 4.31 | 67.22 ± 2.46 | 77.39 ± 13.70 | 48.98 ± 2.88 | 2.8 ± 2.13 |
| 1h | >100 | 98.76 ± 18.97 | 88.84 ± 9.17 | 63.66 ± 3.69 | 77.20 ± 10.89 |
| 2a | >100 | 103.32 ± 7.22 | 124.52 ± 24.18 | 122.45 ± 3.75 | 53.93 ± 10.50 |
| 2b | 75.51 | 89.66 ± 5.59 | 77.28 ± 6.50 | 58.54 ± 2.45 | 41.20 ± 6.35 |
| 2c | 65.03 | 77.25 ± 0.46 | 73.04 ± 1.71 | 68.01 ± 6.44 | 42.65 ± 2.82 |
| 2d | 9.68 | 79.99 ± 2.81 | 71.43 ± 1.99 | 69.37 ± 6.58 | 2.68 ± 0.78 |
| 2e | 21.09 | 81.18 ± 2.57 | 76.57 ± 7.64 | 61.70 ± 3.10 | 32.39 ± 11.92 |
| 2f | >100 | 89.52 ± 10.33 | 83.36 ± 10.20 | 70.19 ± 4.08 | 52.03 ± 0.82 |
| 2g | 20.54 | 96.81 ± 11.13 | 81.72 ± 2.09 | 59.98 ± 3.53 | 30.02 ± 3.13 |
| 2h | 91.71 | 85.05 ± 8.52 | 77.79 ± 5.59 | 72.48 ± 4.91 | 45.71 ± 1.66 |
| 2i | >100 | 99.39 ± 5.10 | 82.60 ± 3.65 | 89.10 ± 8.44 | 85.14 ± 6.14 |
| 2j | >100 | 96.16 ± 17.53 | 82.43 ± 14.56 | 81.94 ± 8.51 | 109.05 ± 9.13 |
| 3a | 43.51 | 98.72 ± 6.89 | 90.64 ± 6.24 | 68.55 ± 4.49 | 38.98 ± 2.26 |
| 3b | >100 | 79.29 ± 4.73 | 83.81 ± 2.62 | 85.39 ± 1.74 | 106.26 ± 10.35 |
| 4a | 15.53 | 69.08 ± 5.18 | 72.08 ± 3.46 | 76.77 ± 8.51 | 17.89 ± 2.70 |
| 4b | 1.68 | 73.29 ± 2.02 | 63.74 ± 5.20 | 27.16 ± 2.43 | 9.24 ± 2.60 |
| Compound | Binding Free Energies (Kcal/mol) | Residues in Contact | Interaction Type | Distance in Å |
|---|---|---|---|---|
| 4b | −6.9 | Leu25 Pro49 Pro51 Gln52 Asp53 | π-sigma π-alkyl π-alkyl π-donor H-bond π-donor H-bond | 3.89 4.65 4.34 3.95 4.16 |
| 2d | −6.7 | Leu25 Pro49 Pro51 | π-sigma π-alkyl π-sigma | 3.74 4.03 3.91 |
| 2g | −6.6 | Leu25 Pro49 Pro51 | π-sigma π-sigma π-sigma | 3.74 3.56 3.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tber, Z.; Loubidi, M.; Jouha, J.; Hdoufane, I.; Erdogan, M.A.; Saso, L.; Armagan, G.; Berteina-Raboin, S. Pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines as Potential Cytotoxic Agents against Human Neuroblastoma. Pharmaceuticals 2021, 14, 750. https://doi.org/10.3390/ph14080750
Tber Z, Loubidi M, Jouha J, Hdoufane I, Erdogan MA, Saso L, Armagan G, Berteina-Raboin S. Pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines as Potential Cytotoxic Agents against Human Neuroblastoma. Pharmaceuticals. 2021; 14(8):750. https://doi.org/10.3390/ph14080750
Chicago/Turabian StyleTber, Zahira, Mohammed Loubidi, Jabrane Jouha, Ismail Hdoufane, Mümin Alper Erdogan, Luciano Saso, Güliz Armagan, and Sabine Berteina-Raboin. 2021. "Pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines as Potential Cytotoxic Agents against Human Neuroblastoma" Pharmaceuticals 14, no. 8: 750. https://doi.org/10.3390/ph14080750
APA StyleTber, Z., Loubidi, M., Jouha, J., Hdoufane, I., Erdogan, M. A., Saso, L., Armagan, G., & Berteina-Raboin, S. (2021). Pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5-amines as Potential Cytotoxic Agents against Human Neuroblastoma. Pharmaceuticals, 14(8), 750. https://doi.org/10.3390/ph14080750