
Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
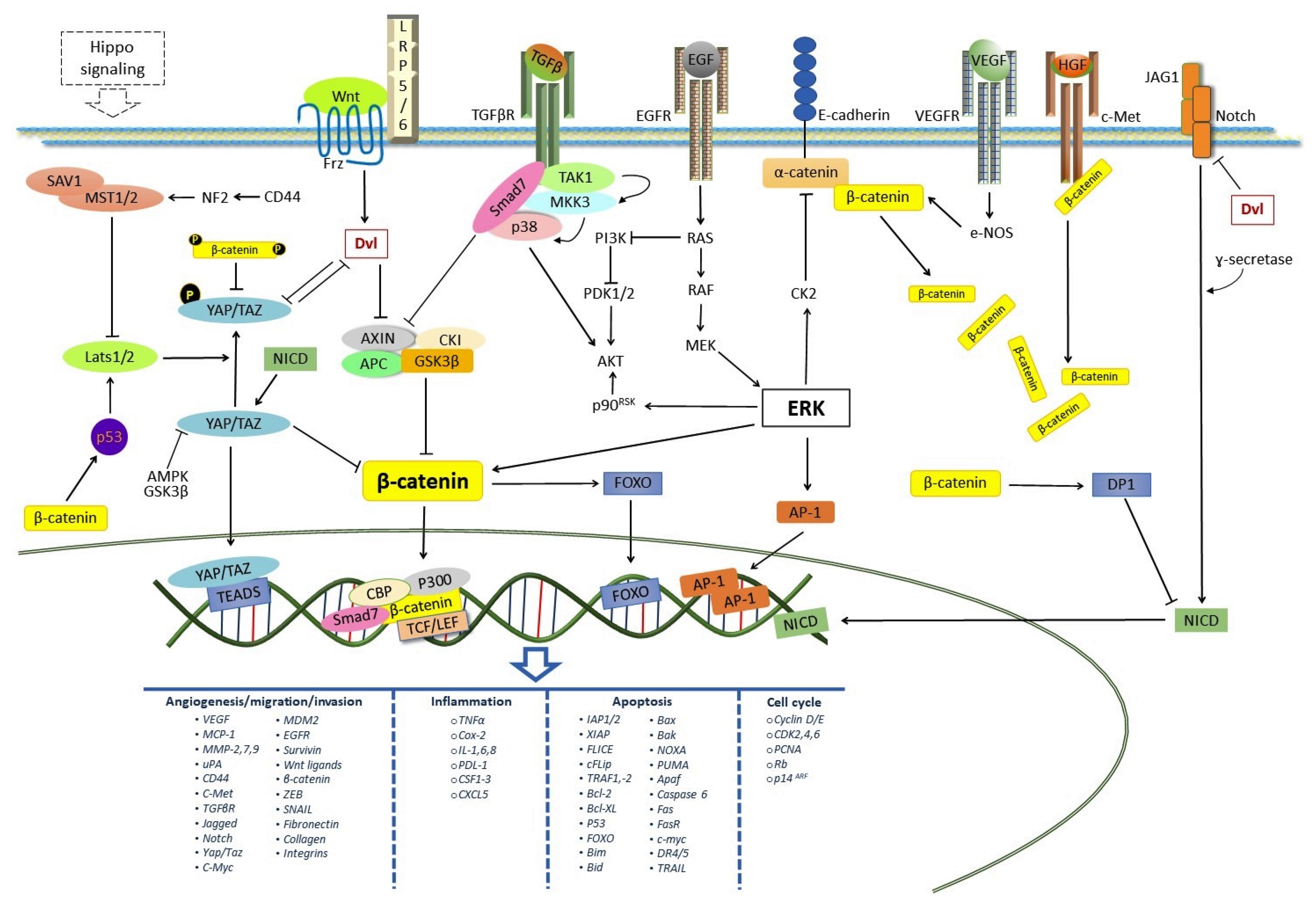
:1. Introduction
2. The Wnt Pathways
2.1. Wnt/Planar Cell Polarity (PCP) Pathway
2.2. Wnt/Ca2+ Pathway
2.3. β-Catenin Canonical Pathway
2.4. Crosstalk of β-Catenin Canonical Pathway
2.4.1. Transcriptional Factors YAP and TAZ
2.4.2. Notch Signaling and Receptor Tyrosine Kinase Pathways
3. Apoptosis
3.1. Extrinsic Apoptotic Pathway
3.2. Intrinsic Apoptotic Pathway
3.3. Apoptosis Regulation via p53
4. Proapoptotic Effect of the Wnt/β-Catenin Pathway in Cancer Cells
4.1. Glioblastoma
4.2. Colorectal Cancer
4.3. Osteosarcoma
4.4. Melanoma
4.5. Medulloblastoma
4.6. Hepatocellular Cancer
4.7. Hematologic Neoplasms
4.8. Other Neoplasms
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5′-AMP-activated protein kinase | AMPK |
| Adenomatous polyposis coli | APC |
| Adenosine monophosphate | AMP |
| Adenosine triphosphate | ATP |
| Apoptotic protease-activating factor 1 | apaf-1 |
| Apoptosis inhibitor | IAP |
| BCL2 Associated X | BAX |
| B-cell lymphoma 2/B-cell lymphoma 9 | Bcl-2 / Bcl-9 |
| B-cell lymphoma-extra-large | Bcl-xL |
| Bcl-2-like protein 11 | BIM |
| BH3-interacting domain death agonist | BID |
| Brain tumor initiating cell | BTIC |
| Central nervous system | CNS |
| Chaperone-mediated autophagy | CMA |
| c-Jun N-terminal kinase | JNK |
| Colony-stimulating factor-1 | CSF1R |
| C vacuolar protein | C-VPS |
| Cytochrome c | cyt c |
| Dapper homolog 2 | DACT2 |
| Dishevelled | Dvl |
| Death domain | DD |
| Death effector domain | DED |
| Death-inducing signaling complex | DISC |
| Deoxyribonucleic acid | DNA |
| Docosahexaenoic acid | DHA |
| Element-binding protein | CBP |
| Epidermal growth factor receptor | EGFR |
| Extracellular-signal-regulated kinase | ERK |
| Fas-associated death domain-containing | FADD |
| Forkhead box class O | FOXO |
| Glycogen synthase kinase 3β | GSK-3β |
| Glioblastoma multiforme | GBM |
| Glioma Stem Cells | GSC |
| GSK-3β binding protein | GBP |
| Guanosine triphosphatases | GTPases |
| High-temperature requirement A2 | Omi/HtrA2 |
| Histone deacetylase | HDAC |
| Liver kinase B1 | LKB1 |
| Low-density lipoprotein receptor-related proteins 5/6 | LRP5/6 |
| Mammalian sterile-20 kinase 1 and 2 | MST1/2 |
| Matrix metallopeptidases | MMP |
| Methylguanine-O6-methyltransferase | MGMT |
| Mitochondrial outer membrane permeabilization | MOMP |
| Mitogen-activated protein kinase | MAPK |
| Mammalian target of rapamycin | mTOR |
| Myeloid cell leukemia 1 | Mcl-1 |
| Nuclear factor kappa-light-chain-enhancer of activated B cells | NF-κB |
| Overall survival | OS |
| Planar cell polarity | PCP |
| Phorbol-12-myristate-13-acetate-induced protein 1 | NOXA (PMAIP1) |
| Protein endoplasmic reticulum kinase | PERK |
| p53-induced death-domain-containing protein | PIDD |
| p53 upregulated modulator of apoptosis | PUMA |
| Pyruvate kinase M2 | PKM2 |
| RAS-related C3 botulinum toxin substrate 1 | RAC1 |
| Reactive oxygen species | ROS |
| Rho-associated kinase | ROCK |
| Salvadoran homoloid 1 | SAV1 |
| Second Mitochondria-derived Activator of Caspases | SMAC |
| Sequestosome 1 | SQSTM1 |
| Tafazzin | TAZ |
| Target of rapamycin complex 1 | TORC1 |
| Truncated Bid | tBID |
| T cell factor/lymphocyte enhancer | Tcf/Lef |
| Temozolomide | TMZ |
| Transcription factor EB | TFEB |
| Tumor Necrosis Factor receptors | TNFR |
| Tumor necrosis factor-related apoptosis-inducing ligand | TRAIL |
| Tumor necrosis factor-related apoptosis-inducing receptor | TRAIR |
| yes –associated protein | YAP |
References
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharmacol. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Murillo-Garzón, V.; Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Multi-layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/β-catenin signaling activation (Review). Int. J. Mol. Med. 2018, 42, 713–725. [Google Scholar] [CrossRef]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.M.; Rank, K.B.; Zeng, Y.; Capen, A.; Yadav, V.; Manro, J.R.; Engler, T.A.; Chedid, M. Activating the Wnt/β-Catenin Pathway for the Treatment of Melanoma--Application of LY2090314, a Novel Selective Inhibitor of Glycogen Synthase Kinase-3. PLoS ONE 2015, 10, e0125028. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; DeRan, M.T.; Ignatius, M.S.; Grandinetti, K.B.; Clagg, R.; McCarthy, K.M.; Lobbardi, R.M.; Brockmann, J.; Keller, C.; Wu, X.; et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/β-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5349–5354. [Google Scholar] [CrossRef] [Green Version]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- McDonald, S.L.; Silver, A. The opposing roles of Wnt-5a in cancer. Br. J. Cancer 2009, 101, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Pinedo, E.C.; Durham, A.C.; Stewart, K.M.; Goss, A.M.; Lu, M.M.; Demayo, F.J.; Morrisey, E.E. Wnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J. Clin. Investig. 2011, 121, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007, 21, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Halasz, M.; Iljin, K.; Fey, D.; Haley, B.; Whilde, J.; Haapa-Paananen, S.; Fey, V.; et al. Wnt signalling is a bi-directional vulnerability of cancer cells. Oncotarget 2016, 7, 60310–60331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, C.; Mendoza, C.; Trejo, C.; Custodio, V.; Rubio-Osornio, M.; Hernández, L.; González, E.; Paz, C. Activation of the Extrinsic and Intrinsic Apoptotic Pathways in Cerebellum of Kindled Rats. Cerebellum 2019, 18, 750–760. [Google Scholar] [CrossRef]
- Rubio-Osornio, C.; Eguiluz-Meléndez, A.; Trejo-Solís, C.; Custodio, V.; Rubio-Osornio, M.; Rosiles-Abonce, A.; Martínez-Lazcano, J.C.; González, E.; Paz, C. Decreased Expression of Sox-1 in Cerebellum of Rat with Generalized Seizures Induced by Kindling Model. CNS Neurol. Disord. Drug Targets 2016, 15, 723–729. [Google Scholar] [CrossRef]
- Rubio, C.; Rosiles-Abonce, A.; Trejo-Solis, C.; Rubio-Osornio, M.; Mendoza, C.; Custodio, V.; Martinez-Lazcano, J.C.; Gonzalez, E.; Paz, C. Increase Signaling of Wnt/β-Catenin Pathway and Presence of Apoptosis in Cerebellum of Kindled Rats. CNS Neurol. Disord. Drug Targets 2017, 16, 772–780. [Google Scholar] [CrossRef]
- Rosiles, A.; Rubio, C.; Trejo, C.; Gutierrez, J.; Hernández, L.; Paz, C. Commentary: Participation of Sox-1 Expression and Signaling of β-Catenin in the Pathophysiology of Generalized Seizures in Cerebellum of Rat. CNS Neurol. Disord. Drug Targets 2016, 15, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Zhang, J.; Wang, J.; Wang, H.; Zheng, F.; Peng, J.; Xu, Y.; Yan, M.; Liu, B.; Cui, B.; et al. SOX1 down-regulates β-catenin and reverses malignant phenotype in nasopharyngeal carcinoma. Mol. Cancer 2014, 13, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damalas, A.; Ben-Ze’ev, A.; Simcha, I.; Shtutman, M.; Leal, J.F.; Zhurinsky, J.; Geiger, B.; Oren, M. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999, 18, 3054–3063. [Google Scholar] [CrossRef]
- Charpin, C.; Garcia, S.; Bouvier, C.; Devictor, B.; Andrac, L.; Choux, R.; Lavaut, M. E-cadherin quantitative immunocytochemical assays in breast carcinomas. J. Pathol. 1997, 181, 294–300. [Google Scholar] [CrossRef]
- Michaelidis, T.M.; Lie, D.C. Wnt signaling and neural stem cells: Caught in the Wnt web. Cell Tissue Res. 2008, 331, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. WNT/PCP signaling pathway and human cancer (review). Oncol. Rep. 2005, 14, 1583–1588. [Google Scholar] [CrossRef]
- VanderVorst, K.; Hatakeyama, J.; Berg, A.; Lee, H.; Carraway, K.L., 3rd. Cellular and molecular mechanisms underlying planar cell polarity pathway contributions to cancer malignancy. Semin. Cell Dev. Biol. 2018, 81, 78–87. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.E.J.; Nourse, C.C.; Cooper, H.M. The tangled web of non-canonical Wnt signalling in neural migration. Neurosignals 2012, 20, 202–220. [Google Scholar] [CrossRef]
- Peifer, M.; McCrea, P.D.; Green, K.J.; Wieschaus, E.; Gumbiner, B.M. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J. Cell Biol. 1992, 118, 681–691. [Google Scholar] [CrossRef]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Major, M.B.; Camp, N.D.; Berndt, J.D.; Yi, X.; Goldenberg, S.J.; Hubbert, C.; Biechele, T.L.; Gingras, A.-C.; Zheng, N.; Maccoss, M.J.; et al. Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 2007, 316, 1043–1046. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Li, V.S.W.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Kramps, T.; Peter, O.; Brunner, E.; Nellen, D.; Froesch, B.; Chatterjee, S.; Murone, M.; Züllig, S.; Basler, K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 2002, 109, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S.; Pietsch, T.; Karsten, U.; van de Wetering, M.; Clevers, H.; Schlag, P.M.; Birchmeier, W.; et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002, 22, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Saucedo, L.J.; Edgar, B.A. Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 2007, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Ma, L.-G.; Bian, S.-B.; Cui, J.-X.; Xi, H.-Q.; Zhang, K.-C.; Qin, H.-Z.; Zhu, X.-M.; Chen, L. LKB1 inhibits the proliferation of gastric cancer cells by suppressing the nuclear translocation of Yap and β-catenin. Int. J. Mol. Med. 2016, 37, 1039–1048. [Google Scholar] [CrossRef]
- Kim, N.H.; Lee, Y.; Yook, J.I. Dishevelling Wnt and Hippo. BMB Rep. 2018, 51, 425–426. [Google Scholar] [CrossRef] [Green Version]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Konsavage, W.M.J.; Yochum, G.S. Intersection of Hippo/YAP and Wnt/β-catenin signaling pathways. Acta Biochim. Biophys. Sin. 2013, 45, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef] [Green Version]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Zeng, G.; Apte, U.; Micsenyi, A.; Bell, A.; Monga, S.P.S. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp. Cell Res. 2006, 312, 3620–3630. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Liu, Z.; Zhao, L.; Clemens, T.L.; Cao, X. Smad7 stabilizes beta-catenin binding to E-cadherin complex and promotes cell-cell adhesion. J. Biol. Chem. 2008, 283, 23956–23963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.-P. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell 2010, 39, 468–476. [Google Scholar] [CrossRef]
- Hayward, P.; Brennan, K.; Sanders, P.; Balayo, T.; DasGupta, R.; Perrimon, N.; Martinez Arias, A. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 2005, 132, 1819–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelrod, J.D.; Matsuno, K.; Artavanis-Tsakonas, S.; Perrimon, N. Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science 1996, 271, 1826–1832. [Google Scholar] [CrossRef]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.-H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P.S. Epidermal growth factor receptor: A novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef] [Green Version]
- Boon, E.M.J.; van der Neut, R.; van de Wetering, M.; Clevers, H.; Pals, S.T. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer Res. 2002, 62, 5126–5128. [Google Scholar]
- Katoh, M.; Katoh, M. Transcriptional regulation of WNT2B based on the balance of Hedgehog, Notch, BMP and WNT signals. Int. J. Oncol. 2009, 34, 1411–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjar, A.M.; DiGiacomo, R.F.; Carpenter, J.K.; Bingel, S.A.; Moazed, T.C. Chronic sialodacryoadenitis virus (SDAV) infection in athymic rats. Lab. Anim. Sci. 1991, 41, 22–25. [Google Scholar]
- Müllauer, L.; Mosberger, I.; Grusch, M.; Rudas, M.; Chott, A. Fas ligand is expressed in normal breast epithelial cells and is frequently up-regulated in breast cancer. J. Pathol. 2000, 190, 20–30. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Corre, J.; Avet-Loiseau, H. Risk-Based Therapeutic Strategies. Cancer J. 2019, 25, 54–58. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Cavaleri, J.M.; Sutera, P.A.; Rajasekar, T.; Pollack, I.F. Dinaciclib, a Cyclin-Dependent Kinase Inhibitor Promotes Proteasomal Degradation of Mcl-1 and Enhances ABT-737-Mediated Cell Death in Malignant Human Glioma Cell Lines. J. Pharmacol. Exp. Ther. 2016, 356, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- MacEwan, D.J. TNF ligands and receptors-a matter of life and death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-S.; Chen, G.-S.; Lin, P.-Y.; Pan, I.-H.; Wang, S.-T.; Lin, S.H.; Yu, H.-S.; Lin, C.-C. Tazarotene induces apoptosis in human basal cell carcinoma via activation of caspase-8/t-Bid and the reactive oxygen species-dependent mitochondrial pathway. DNA Cell Biol. 2014, 33, 652–666. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 92, 57–70. [Google Scholar] [CrossRef]
- Ashe, P.C.; Berry, M.D. Apoptotic signaling cascades. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 199–214. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Mendrysa, S.M.; Perry, M.E. The p53 tumor suppressor protein does not regulate expression of its own inhibitor, MDM2, except under conditions of stress. Mol. Cell. Biol. 2000, 20, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef]
- Burns, T.F.; Bernhard, E.J.; El-Deiry, W.S. Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo. Oncogene 2001, 20, 4601–4612. [Google Scholar] [CrossRef] [Green Version]
- MacLachlan, T.K.; El-Deiry, W.S. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc. Natl. Acad. Sci. USA 2002, 99, 9492–9497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieschke, E.; Wang, Z.; Kelly, G.L.; Strasser, A. Discussion of some “knowns” and some “unknowns” about the tumour suppressor p53. J. Mol. Cell Biol. 2019, 11, 212–223. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Fortin, A.; Cregan, S.P.; MacLaurin, J.G.; Kushwaha, N.; Hickman, E.S.; Thompson, C.S.; Hakim, A.; Albert, P.R.; Cecconi, F.; Helin, K.; et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J. Cell Biol. 2001, 155, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budhram-Mahadeo, V.; Morris, P.J.; Smith, M.D.; Midgley, C.A.; Boxer, L.M.; Latchman, D.S. P53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J. Biol. Chem. 1999, 274, 15237–15244. [Google Scholar] [CrossRef] [Green Version]
- Tagscherer, K.E.; Fassl, A.; Sinkovic, T.; Combs, S.E.; Roth, W. P53-dependent regulation of Mcl-1 contributes to synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL inhibitor ABT-737. Apoptosis 2012, 17, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. P53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Leu, J.I.-J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Blosser, J.C.; McCreedy, S.; Parker, R.B.; Kinsolving, C.R.; Watkins, B.E.; Wu, E.S. Flavodilol: A new antihypertensive agent that preferentially depletes peripheral biogenic amines. J. Cardiovasc. Pharmacol. 1989, 14, 142–156. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Askew, D.S.; Ashmun, R.A.; Simmons, B.C.; Cleveland, J.L. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991, 6, 1915–1922. [Google Scholar] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.H.; Keller, U.B.; Rodriguez-Galindo, C.; Nilsson, J.A.; Cleveland, J.L. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol. Cell. Biol. 2003, 23, 7256–7270. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.S.; Jin, Z.; Dews, M.; Yu, D.; Thomas-Tikhonenko, A.; Dicker, D.T.; El-Deiry, W.S. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol. Cell. Biol. 2004, 24, 8541–8555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, K.O.; Ricci, M.S.; Miyashita, T.; Dicker, D.T.; Jin, Z.; Reed, J.C.; El-Deiry, W.S. Bax is a transcriptional target and mediator of c-myc-induced apoptosis. Cancer Res. 2000, 60, 6318–6325. [Google Scholar] [PubMed]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, M.A.; Riblett, M.; Tang, W.-H.; Gratchouck, V.; Zhuang, D.; Fernandez, Y.; Verhaegen, M.; Varambally, S.; Chinnaiyan, A.M.; Jakubowiak, A.J.; et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA 2007, 104, 19488–19493. [Google Scholar] [CrossRef] [Green Version]
- Kangas, A.; Nicholson, D.W.; Hölttä, E. Involvement of CPP32/Caspase-3 in c-Myc-induced apoptosis. Oncogene 1998, 16, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Hueber, A.O.; Zörnig, M.; Lyon, D.; Suda, T.; Nagata, S.; Evan, G.I. Requirement for the CD95 receptor-ligand pathway in c-Myc-induced apoptosis. Science 1997, 278, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Klefstrom, J.; Västrik, I.; Saksela, E.; Valle, J.; Eilers, M.; Alitalo, K. c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 1994, 13, 5442–5450. [Google Scholar] [CrossRef]
- Nieminen, A.I.; Partanen, J.I.; Klefstrom, J. c-Myc blazing a trail of death: Coupling of the mitochondrial and death receptor apoptosis pathways by c-Myc. Cell Cycle 2007, 6, 2464–2472. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A. Glioblastoma and Other Malignant Gliomas. JAMA 2013. [Google Scholar] [CrossRef]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a potential therapy for malignant glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Gao, L.; Chen, B.; Li, J.; Yang, F.; Cen, X.; Liao, Z.; Long, X. Wnt/β-catenin signaling pathway inhibits the proliferation and apoptosis of U87 glioma cells via different mechanisms. PLoS ONE 2017, 12, e0181346. [Google Scholar] [CrossRef]
- Wang, K.; Wang, X.; Zou, J.; Zhang, A.; Wan, Y.; Pu, P.; Song, Z.; Qian, C.; Chen, Y.; Yang, S.; et al. miR-92b controls glioma proliferation and invasion through regulating Wnt/beta-catenin signaling via Nemo-like kinase. Neuro. Oncol. 2013, 15, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Yang, Y.; Feng, D.; Zheng, S.; Meng, R.; Fa, P.; Zhao, C.; Liu, H.; Song, R.; Tao, T.; et al. MAGI3 negatively regulates Wnt/β-catenin signaling and suppresses malignant phenotypes of glioma cells. Oncotarget 2015, 6, 35851–35865. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Li, Q.; Shi, Z.; Li, C.; Wang, L.; Liu, X.; Jiang, C.; Qian, X.; You, Y.; Liu, N.; et al. GSK-3β regulates tumor growth and angiogenesis in human glioma cells. Oncotarget 2015, 6, 31901–31915. [Google Scholar] [CrossRef] [Green Version]
- Sita, G.; Hrelia, P.; Graziosi, A.; Morroni, F. Sulforaphane from Cruciferous Vegetables: Recent Advances to Improve Glioblastoma Treatment. Nutrients 2018, 10, 1755. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Li, H.; Yi, D.; Bai, Y.; Zhong, S.; Liu, Q.; Chen, Y.; Zhao, G. β-catenin contributes to cordycepin-induced MGMT inhibition and reduction of temozolomide resistance in glioma cells by increasing intracellular reactive oxygen species. Cancer Lett. 2018, 435, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, K.; Kawakami, K.; Nakada, M.; Mai, W.; Shakoori, A.; Fujisawa, H.; Hayashi, Y.; Hamada, J.; Minamoto, T. Potential therapeutic effect of glycogen synthase kinase 3beta inhibition against human glioblastoma. Clin. Cancer Res. 2009, 15, 887–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyko, I.V.; Nakada, M.; Sabit, H.; Teng, L.; Furuyama, N.; Hayashi, Y.; Kawakami, K.; Minamoto, T.; Fedulau, A.S.; Hamada, J. Glycogen synthase kinase 3β inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 2013, 34, 2206–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Chu, C.-W.; Ko, H.-J.; Chou, C.-H.; Cheng, T.-S.; Cheng, H.-W.; Liang, Y.-H.; Lai, Y.-L.; Lin, C.-Y.; Wang, C.; Loh, J.-K.; et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro. Oncol. 2008, 10, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Aras, Y.; Erguven, M.; Aktas, E.; Yazihan, N.; Bilir, A. Antagonist activity of the antipsychotic drug lithium chloride and the antileukemic drug imatinib mesylate during glioblastoma treatment in vitro. Neurol. Res. 2016, 38, 766–774. [Google Scholar] [CrossRef]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef]
- Tan, Y.; Li, Q.-M.; Huang, N.; Cheng, S.; Zhao, G.-J.; Chen, H.; Chen, S.; Tang, Z.-H.; Zhang, W.-Q.; Huang, Q.; et al. Upregulation of DACT2 suppresses proliferation and enhances apoptosis of glioma cell via inactivation of YAP signaling pathway. Cell Death Dis. 2017, 8, e2981. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Du, S.; Lei, T.; Wang, H.; He, X.; Tong, R.; Wang, Y. Multifaceted regulation and functions of YAP/TAZ in tumors (Review). Oncol. Rep. 2018, 40, 16–28. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclikova, P.; et al. Dishevelled has a YAP nuclear export function in a tumor suppressor context-dependent manner. Nat. Commun. 2018, 9, 2301. [Google Scholar] [CrossRef] [Green Version]
- Orr, B.A.; Bai, H.; Odia, Y.; Jain, D.; Anders, R.A.; Eberhart, C.G. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J. Neuropathol. Exp. Neurol. 2011, 70, 568–577. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarova, D.L.; Chiaro, C.; Bordonaro, M. Butyrate induced changes in Wnt-signaling specific gene expression in colorectal cancer cells. BMC Res. Notes 2014, 7, 226. [Google Scholar] [CrossRef] [Green Version]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Hyperinduction of Wnt activity: A new paradigm for the treatment of colorectal cancer? Oncol. Res. 2008, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp. Cell Res. 2007, 313, 1652–1666. [Google Scholar] [CrossRef] [Green Version]
- Drago, E.; Bordonaro, M.; Lee, S.; Atamna, W.; Lazarova, D.L. Propolis augments apoptosis induced by butyrate via targeting cell survival pathways. PLoS ONE 2013, 8, e73151. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.-L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarova, D.L.; Wong, T.; Chiaro, C.; Drago, E.; Bordonaro, M. P300 Influences Butyrate-Mediated WNT Hyperactivation In Colorectal Cancer Cells. J. Cancer 2013, 4, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.; Lee, A.; Wong, T.; Marian, B.; Chiaro, C.; Rainey, C.; Bordonaro, M. Modulation of Wnt Activity and Cell Physiology by Butyrate in LT97 Microadenoma Cells. J. Cancer 2014, 5, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Lazarova, D.L.; Chiaro, C.; Wong, T.; Drago, E.; Rainey, A.; O’Malley, S.; Bordonaro, M. CBP Activity Mediates Effects of the Histone Deacetylase Inhibitor Butyrate on WNT Activity and Apoptosis in Colon Cancer Cells. J. Cancer 2013, 4, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Nguyen, C.; Lee, K.-S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarova, D.L.; Bordonaro, M. P300 Knockout Promotes Butyrate Resistance. J. Cancer 2017, 8, 3405–3409. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.W.; Kim, H.C.; Kim, S.H.; Park, Y.A.; Cho, Y.B.; Yun, S.H.; Lee, W.Y.; Chun, H.-K. Prognostic impact of p300 expression in patients with colorectal cancer. J. Surg. Oncol. 2013, 108, 374–377. [Google Scholar] [CrossRef]
- Lazarova, D.; Bordonaro, M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J. Cancer 2017, 8, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Liao, Y.; Hsu, F.-N.; Zhang, R.; Searle, J.S.; Pei, X.; Li, X.; Ryoo, H.D.; Ji, J.-Y.; Du, W. Hyperactivated Wnt signaling induces synthetic lethal interaction with Rb inactivation by elevating TORC1 activities. PLoS Genet. 2014, 10, e1004357. [Google Scholar] [CrossRef] [Green Version]
- Bordonaro, M. Hypothesis: Retinoblastoma protein inactivation mediates effects of histone deacetylase inhibitor-induced Wnt hyperactivation in colorectal cancer cells. J. Cancer 2020, 11, 668–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.G.; Xian, J.; Chin, S.-F.; Bannister, A.J.; Daigo, Y.; Aparicio, S.; Kouzarides, T.; Caldas, C. P300 is required for orderly G1/S transition in human cancer cells. Oncogene 2007, 26, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionov, Y.; Matsui, S.-I.; Cowell, J.K. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proc. Natl. Acad. Sci. USA 2004, 101, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Bordonaro, M.; Shirasawa, S.; Lazarova, D.L. In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth. Cancers 2016, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [Green Version]
- Bordonaro, M.; Drago, E.; Atamna, W.; Lazarova, D.L. Comprehensive suppression of all apoptosis-induced proliferation pathways as a proposed approach to colorectal cancer prevention and therapy. PLoS ONE 2014, 9, e115068. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Sasaki, M.; Sugio, K.; Sasazuki, T. K-ras activation in colorectal tumors from patients with familial polyposis coli. Cancer 1990, 65, 2576–2579. [Google Scholar] [CrossRef]
- Jalving, M.; Heijink, D.M.; Koornstra, J.J.; Boersma-van Ek, W.; Zwart, N.; Wesseling, J.; Sluiter, W.J.; de Vries, E.G.E.; Kleibeuker, J.H.; de Jong, S. Regulation of TRAIL receptor expression by β-catenin in colorectal tumours. Carcinogenesis 2014, 35, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Hiscox, S.; Jiang, W.G. Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem. Biophys. Res. Commun. 1999, 261, 406–411. [Google Scholar] [CrossRef]
- Li, Y.; Bharti, A.; Chen, D.; Gong, J.; Kufe, D. Interaction of glycogen synthase kinase 3beta with the DF3/MUC1 carcinoma-associated antigen and beta-catenin. Mol. Cell. Biol. 1998, 18, 7216–7224. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Ochiai, A.; Shibata, T.; Oyama, T.; Ushijima, S.; Akimoto, S.; Hirohashi, S. c-erbB-2 gene product directly associates with beta-catenin and plakoglobin. Biochem. Biophys. Res. Commun. 1995, 208, 1067–1072. [Google Scholar] [CrossRef]
- Li, L.; Ji, S.-Y.; Yang, J.-L.; Li, X.-X.; Zhang, J.; Zhang, Y.; Hu, Z.-Y.; Liu, Y.-X. Wnt/β-catenin signaling regulates follicular development by modulating the expression of Foxo3a signaling components. Mol. Cell. Endocrinol. 2014, 382, 915–925. [Google Scholar] [CrossRef]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Chen, Q.; Ganapathy, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PLoS ONE 2010, 5, e15288. [Google Scholar] [CrossRef]
- Marrone, T.J.; Merz, K.M. Molecular Recognition of Potassium Ion by the Naturally Occurring Antibiotic Ionophore Nonactin. J. Am. Chem. Soc. 1992, 114, 7542–7549. [Google Scholar] [CrossRef]
- Shikata, Y.; Kiga, M.; Futamura, Y.; Aono, H.; Inoue, H.; Kawada, M.; Osada, H.; Imoto, M. Mitochondrial uncoupler exerts a synthetic lethal effect against β-catenin mutant tumor cells. Cancer Sci. 2017, 108, 772–784. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the surveillance, epidemiology, and end results program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Mohseny, A.B.; Karperien, M.; Hogendoorn, P.C.W.; Zhou, G.; Cleton-Jansen, A.-M. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J. Pathol. 2010, 220, 24–33. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.; Niu, J.; Nie, Z.; Liu, Q.; Lv, C. Zinc promotes cell apoptosis via activating the Wnt-3a/β-catenin signaling pathway in osteosarcoma. J. Orthop. Surg. Res. 2020, 15, 57. [Google Scholar] [CrossRef] [Green Version]
- Khayyatzadeh, S.S.; Maghsoudi, Z.; Foroughi, M.; Askari, G.; Ghiasvand, R. Dietary intake of Zinc, serum levels of Zinc and risk of gastric cancer: A review of studies. Adv. Biomed. Res. 2015, 4, 118. [Google Scholar] [CrossRef]
- Jüllig, M.; Zhang, W.V.; Ferreira, A.; Stott, N.S. MG132 induced apoptosis is associated with p53-independent induction of pro-apoptotic Noxa and transcriptional activity of beta-catenin. Apoptosis 2006, 11, 627–641. [Google Scholar] [CrossRef]
- Heiser, D.; Labi, V.; Erlacher, M.; Villunger, A. The Bcl-2 protein family and its role in the development of neoplastic disease. Exp. Gerontol. 2004, 39, 1125–1135. [Google Scholar] [CrossRef]
- Lauricella, M.; D’Anneo, A.; Giuliano, M.; Calvaruso, G.; Emanuele, S.; Vento, R.; Tesoriere, G. Induction of apoptosis in human osteosarcoma Saos-2 cells by the proteasome inhibitor MG132 and the protective effect of pRb. Cell Death Differ. 2003, 10, 930–932. [Google Scholar] [CrossRef]
- Zhao, H.; Hou, W.; Tao, J.; Zhao, Y.; Wan, G.; Ma, C.; Xu, H. Upregulation of lncRNA HNF1A-AS1 promotes cell proliferation and metastasis in osteosarcoma through activation of the Wnt/β-catenin signaling pathway. Am. J. Transl. Res. 2016, 8, 3503–3512. [Google Scholar] [PubMed]
- Liu, Z.; Wei, X.; Zhang, A.; Li, C.; Bai, J.; Dong, J. Long non-coding RNA HNF1A-AS1 functioned as an oncogene and autophagy promoter in hepatocellular carcinoma through sponging hsa-miR-30b-5p. Biochem. Biophys. Res. Commun. 2016, 473, 1268–1275. [Google Scholar] [CrossRef]
- Tao, H.; Chen, F.; Liu, H.; Hu, Y.; Wang, Y.; Li, H. Wnt/β-catenin signaling pathway activation reverses gemcitabine resistance by attenuating Beclin1-mediated autophagy in the MG63 human osteosarcoma cell line. Mol. Med. Rep. 2017, 16, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Műzes, G.; Sipos, F. Anti-tumor immunity, autophagy and chemotherapy. World J. Gastroenterol. 2012, 18, 6537–6540. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.-T.; Xu, Y.; Yi, H.-W.; Su, J.; Yu, H.-M.; Xiang, X.-Y.; Li, X.-N.; Zhang, Z.-C.; Sun, L.-K. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett. 2012, 323, 180–187. [Google Scholar] [CrossRef]
- Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramírez, Á.; Castillo-Rodríguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Luo, P.; Xu, H.; Dai, S.; Rao, W.; Peng, C.; Ma, W.; Wang, J.; Xu, H.; Zhang, L.; et al. RNF146 Inhibits Excessive Autophagy by Modulating the Wnt-β-Catenin Pathway in Glutamate Excitotoxicity Injury. Front. Cell. Neurosci. 2017, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Feng, J.; Dong, S.; Pan, R.; Zhuang, H.; Ding, Z. Regulation of autophagy of prostate cancer cells by β-catenin signaling. Cell. Physiol. Biochem. 2015, 35, 926–932. [Google Scholar] [CrossRef]
- Park, J.-M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.M.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef]
- Nàger, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Shimozaki, S.; Yamamoto, N.; Domoto, T.; Nishida, H.; Hayashi, K.; Kimura, H.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Kato, T.; et al. Efficacy of glycogen synthase kinase-3β targeting against osteosarcoma via activation of β-catenin. Oncotarget 2016, 7, 77038–77051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolick, N.L.; Geller, A.C. Epidemiology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 57–72. [Google Scholar] [CrossRef]
- Peek, G.; Olsen, C.M.; Baade, P.; Youlden, D.R.; Aitken, J.F.; Green, A.C.; Khosrotehrani, K. Survival in patients with multiple primary melanomas: Systematic review and meta-analysis. J. Am. Acad. Dermatol. 2020, 83, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Marsden, J.R.; Newton-Bishop, J.A.; Burrows, L.; Cook, M.; Corrie, P.G.; Cox, N.H.; Gore, M.E.; Lorigan, P.; MacKie, R.; Nathan, P.; et al. Revised UK guidelines for the management of cutaneous melanoma 2010. J. Plast. Reconstr. Aesthet. Surg. 2010, 63, 1401–1419. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.E. Loss of beta-catenin expression is associated with disease progression in malignant melanoma. Br. J. Dermatol. 2002, 146, 1104. [Google Scholar] [CrossRef]
- Maelandsmo, G.M.; Holm, R.; Nesland, J.M.; Fodstad, Ø.; Flørenes, V.A. Reduced beta-catenin expression in the cytoplasm of advanced-stage superficial spreading malignant melanoma. Clin. Cancer Res. 2003, 9, 3383–3388. [Google Scholar]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Kuphal, S.; Lodermeyer, S.; Bataille, F.; Schuierer, M.; Hoang, B.H.; Bosserhoff, A.K. Expression of Dickkopf genes is strongly reduced in malignant melanoma. Oncogene 2006, 25, 5027–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, L.M.; Deeds, J.; Hunter, J.; Shao, J.; Holmgren, L.M.; Woolf, E.A.; Tepper, R.I.; Shyjan, A.W. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998, 58, 1515–1520. [Google Scholar]
- Yamamoto, H.; Kishida, S.; Kishida, M.; Ikeda, S.; Takada, S.; Kikuchi, A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3beta regulates its stability. J. Biol. Chem. 1999, 274, 10681–10684. [Google Scholar] [CrossRef] [Green Version]
- Biechele, T.L.; Kulikauskas, R.M.; Toroni, R.A.; Lucero, O.M.; Swift, R.D.; James, R.G.; Robin, N.C.; Dawson, D.W.; Moon, R.T.; Chien, A.J. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci. Signal. 2012, 5, ra3. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.; Shibamoto, S.; Nusse, R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 1999, 13, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, W.H.; Swift, R.D.; Biechele, T.L.; Kulikauskas, R.M.; Moon, R.T.; Chien, A.J. Regulating the response to targeted MEK inhibition in melanoma: Enhancing apoptosis in NRAS- and BRAF-mutant melanoma cells with Wnt/β-catenin activation. Cell Cycle 2012, 11, 3724–3730. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Li, L.; Brafford, P.A.; van den Eijnden, M.; Halloran, M.B.; Sproesser, K.; Haass, N.K.; Smalley, K.S.M.; Tsai, J.; Bollag, G.; et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010, 23, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Chien, A.J.; Conrad, W.H.; Moon, R.T. A Wnt survival guide: From flies to human disease. J. Investig. Dermatol. 2009, 129, 1614–1627. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Zimmerman, Z.F.; Kulikauskas, R.M.; Bomsztyk, K.; Moon, R.T.; Chien, A.J. Activation of Wnt/β-catenin signaling increases apoptosis in melanoma cells treated with trail. PLoS ONE 2013, 8, e69593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, B.; Kim, W.; Kim, Y.K.; Hwang, B.N.; Park, S.Y.; Yoon, J.W.; Park, W.S.; Cho, J.W.; Bedford, M.T.; Jho, E. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene 2011, 30, 2379–2389. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- You, C.; Zhang, S.; Sun, Y.; Zhang, S.; Tang, G.; Tang, F.; Liu, X.; Xiao, Y.; Zhang, J.; Gong, Y.; et al. β-catenin decreases acquired TRAIL resistance in non-small-cell lung cancer cells by regulating the redistribution of death receptors. Int. J. Oncol. 2018, 53, 2258–2268. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Mollinedo, F. Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J. Biol. Chem. 2005, 280, 11641–11647. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Bischof, H.; Gilby, D.; Belloni, B.; Dummer, R.; Wellbrock, C. In melanoma, beta-catenin is a suppressor of invasion. Oncogene 2011, 30, 4531–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serini, S.; Zinzi, A.; Ottes Vasconcelos, R.; Fasano, E.; Riillo, M.G.; Celleno, L.; Trombino, S.; Cassano, R.; Calviello, G. Role of β-catenin signaling in the anti-invasive effect of the omega-3 fatty acid DHA in human melanoma cells. J. Dermatol. Sci. 2016, 84, 149–159. [Google Scholar] [CrossRef]
- Hino, S.; Tanji, C.; Nakayama, K.I.; Kikuchi, A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol. Cell. Biol. 2005, 25, 9063–9072. [Google Scholar] [CrossRef] [Green Version]
- Serini, S.; Fasano, E.; Piccioni, E.; Monego, G.; Cittadini, A.R.M.; Celleno, L.; Ranelletti, F.O.; Calviello, G. DHA induces apoptosis and differentiation in human melanoma cells in vitro: Involvement of HuR-mediated COX-2 mRNA stabilization and β-catenin nuclear translocation. Carcinogenesis 2012, 33, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Prim. 2019, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoranjan, B.; Venugopal, C.; Bakhshinyan, D.; Adile, A.A.; Richards, L.; Kameda-Smith, M.M.; Whitley, O.; Dvorkin-Gheva, A.; Subapanditha, M.; Savage, N.; et al. Wnt activation as a therapeutic strategy in medulloblastoma. Nat. Commun. 2020, 11, 4323. [Google Scholar] [CrossRef]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [Green Version]
- Rogers, H.A.; Miller, S.; Lowe, J.; Brundler, M.-A.; Coyle, B.; Grundy, R.G. An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET). Br. J. Cancer 2009, 100, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Schüller, U.; Heine, V.M.; Mao, J.; Kho, A.T.; Dillon, A.K.; Han, Y.-G.; Huillard, E.; Sun, T.; Ligon, A.H.; Qian, Y.; et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008, 14, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.; Deutschmann, M.; Ahlfeld, J.; Prix, C.; Koch, A.; Smits, R.; Fodde, R.; Kretzschmar, H.A.; Schüller, U. Severe alterations of cerebellar cortical development after constitutive activation of Wnt signaling in granule neuron precursors. Mol. Cell. Biol. 2011, 31, 3326–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöschl, J.; Bartels, M.; Ohli, J.; Bianchi, E.; Kuteykin-Teplyakov, K.; Grammel, D.; Ahlfeld, J.; Schüller, U. Wnt/β-catenin signaling inhibits the Shh pathway and impairs tumor growth in Shh-dependent medulloblastoma. Acta Neuropathol. 2014, 127, 605–607. [Google Scholar] [CrossRef]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Manoranjan, B.; Adile, A.A.; Venugopal, C.; Singh, S.K. WNT: An unexpected tumor suppressor in medulloblastoma. Mol. Cell. Oncol. 2020, 7, 1834903. [Google Scholar] [CrossRef] [PubMed]
- Manoranjan, B.; Wang, X.; Hallett, R.M.; Venugopal, C.; Mack, S.C.; McFarlane, N.; Nolte, S.M.; Scheinemann, K.; Gunnarsson, T.; Hassell, J.A.; et al. FoxG1 interacts with Bmi1 to regulate self-renewal and tumorigenicity of medulloblastoma stem cells. Stem. Cells 2013, 31, 1266–1277. [Google Scholar] [CrossRef]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Salaroli, R.; Ronchi, A.; Buttarelli, F.R.; Cortesi, F.; Marchese, V.; Della Bella, E.; Renna, C.; Baldi, C.; Giangaspero, F.; Cenacchi, G. Wnt activation affects proliferation, invasiveness and radiosensitivity in medulloblastoma. J. Neurooncol. 2015, 121, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Martin, D.C.; Castelo-Branco, P.; Zhang, C.H.; Fraser, M.; Tse, K.; Poon, R.; Shih, D.J.H.; et al. WNT activation by lithium abrogates TP53 mutation associated radiation resistance in medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 174. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.C.; Hill, R.M.; Megahed, H.; Lusher, M.E.; Schwalbe, E.C.; Cole, M.; Hogg, T.L.; Gilbertson, R.J.; Ellison, D.W.; Bailey, S.; et al. TP53 mutations in favorable-risk Wnt/Wingless-subtype medulloblastomas. J. Clin. Oncol. 2011, 29, e344–e346; author reply e347–e348. [Google Scholar] [CrossRef] [PubMed]
- Salaroli, R.; Di Tomaso, T.; Ronchi, A.; Ceccarelli, C.; Cammelli, S.; Cappellini, A.; Martinelli, G.N.; Barbieri, E.; Giangaspero, F.; Cenacchi, G. Radiobiologic response of medulloblastoma cell lines: Involvement of beta-catenin? J. Neurooncol. 2008, 90, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Damalas, A.; Kahan, S.; Shtutman, M.; Ben-Ze’ev, A.; Oren, M. Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001, 20, 4912–4922. [Google Scholar] [CrossRef] [Green Version]
- Levina, E.; Oren, M.; Ben-Ze’ev, A. Downregulation of beta-catenin by p53 involves changes in the rate of beta-catenin phosphorylation and Axin dynamics. Oncogene 2004, 23, 4444–4453. [Google Scholar] [CrossRef] [Green Version]
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-regulation of beta-catenin by activated p53. Mol. Cell. Biol. 2001, 21, 6768–6781. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Kim, W.; Khan, S.K.; Yang, Y. Interacting network of Hippo, Wnt/β-catenin and Notch signaling represses liver tumor formation. BMB Rep. 2017, 50, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.-H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.-S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, S.; Wang, R.; Chen, C.; Ma, W.; Cai, H. Anti-tumor effect of LATS2 on liver cancer death: Role of DRP1-mediated mitochondrial division and the Wnt/β-catenin pathway. Biomed. Pharmacother. 2019, 114, 108825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J. Am. Heart Assoc. 2017, 6, e005328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.-Q.; Zuo, G.-W.; Feng, Z.-Q.; Zhao, L.-C.; Luo, L.; You, Z.-M.; Li, D.-Y.; Xia, J.; Li, J.; Chen, D.-L. Effect of trichostatin A on anti HepG2 liver carcinoma cells: Inhibition of HDAC activity and activation of Wnt/β-Catenin signaling. Asian Pac. J. Cancer Prev. 2014, 15, 7849–7855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosimann, C.; Hausmann, G.; Basler, K. Beta-catenin hits chromatin: Regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. [Google Scholar] [CrossRef]
- Su, N.; Wang, P.; Li, Y. Role of Wnt/β-catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol. Lett. 2016, 12, 4623–4629. [Google Scholar] [CrossRef] [Green Version]
- Ming, M.; Wang, S.; Wu, W.; Senyuk, V.; Le Beau, M.M.; Nucifora, G.; Qian, Z. Activation of Wnt/β-catenin protein signaling induces mitochondria-mediated apoptosis in hematopoietic progenitor cells. J. Biol. Chem. 2012, 287, 22683–22690. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Shao, N.; Zou, J.; Li, J.; Chen, F.; Dai, J.; Qu, X.; Sun, X.; Ma, D.; Ji, C. Hyper-activation of WNT/β-catenin signaling pathway mediates anti-tumor effects of histone deacetylase inhibitors in acute T lymphoblastic leukemia. Leuk. Lymphoma 2012, 53, 1769–1778. [Google Scholar] [CrossRef]
- Kato, Y.; Naiki, Y.; Komatsu, T.; Takahashi, K.; Nakamura, J.; Koide, N. A Wnt pathway activator induces apoptosis and cell death in mouse monocytic leukemia cells. Oncol. Res. 2017, 25, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Yao, R.; Sun, X.; Xie, Y.; Liu, L.; Han, D.; Yao, Y.; Li, H.; Li, Z.; Xu, K. Lithium chloride inhibits cell survival, overcomes drug resistance, and triggers apoptosis in multiple myeloma via activation of the Wnt/β-catenin pathway. Am. J. Transl. Res. 2018, 10, 2610–2618. [Google Scholar] [PubMed]
- Ma, C.; Fan, L.; Wang, J.; Hao, L.; He, J. Hippo/Mst1 overexpression induces mitochondrial death in head and neck squamous cell carcinoma via activating β-catenin/Drp1 pathway. Cell Stress Chaperones 2019, 24, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.M.; Liu, W.W.; Liu, C.J.; Wen, C.; Lu, H.F.; Wan, F.S. Mst1 overexpression inhibited the growth of human non-small cell lung cancer in vitro and in vivo. Cancer Gene Ther. 2013, 20, 453–460. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, K.; Pei, Y.; Ma, J.; Tan, J.; Zhao, J. Mst1 regulates non-small cell lung cancer A549 cell apoptosis by inducing mitochondrial damage via ROCK1/F-actin pathways. Int. J. Oncol. 2018, 53, 2409–2422. [Google Scholar] [CrossRef]
- Lin, L.; Li, M.; Lin, L.; Xu, X.; Jiang, G.; Wu, L. FPPS mediates TGF-β1-induced non-small cell lung cancer cell invasion and the EMT process via the RhoA/Rock1 pathway. Biochem. Biophys. Res. Commun. 2018, 496, 536–541. [Google Scholar] [CrossRef]
- Hommura, F.; Furuuchi, K.; Yamazaki, K.; Ogura, S.; Kinoshita, I.; Shimizu, M.; Moriuchi, T.; Katoh, H.; Nishimura, M.; Dosaka-Akita, H. Increased expression of beta-catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer 2002, 94, 752–758. [Google Scholar] [CrossRef]
- Kren, L.; Hermanová, M.; Goncharuk, V.N.; Kaur, P.; Ross, J.S.; Pavlovský, Z.; Dvorák, K. Downregulation of plasma membrane expression/cytoplasmic accumulation of beta-catenin predicts shortened survival in non-small cell lung cancer. A clinicopathologic study of 100 cases. Cesk. Patol. 2003, 39, 17–20. [Google Scholar]
- Kase, S.; Sugio, K.; Yamazaki, K.; Okamoto, T.; Yano, T.; Sugimachi, K. Expression of E-cadherin and beta-catenin in human non-small cell lung cancer and the clinical significance. Clin. Cancer Res. 2000, 6, 4789–4796. [Google Scholar] [CrossRef]
- Peng, Y.; Cao, J.; Yao, X.-Y.; Wang, J.-X.; Zhong, M.-Z.; Gan, P.-P.; Li, J.-H. TUSC3 induces autophagy in human non-small cell lung cancer cells through Wnt/β-catenin signaling. Oncotarget 2017, 8, 52960–52974. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-G.; Liang, N.-X.; Qin, Y.-Z.; Ma, D.-J.; Huang, C.-J.; Liu, L.; Li, S.-Q. Effects of RNAi-mediated TUSC3 silencing on radiation-induced autophagy and radiation sensitivity of human lung adenocarcinoma cell line A549 under hypoxic condition. Tumour Biol. 2016, 37, 16357–16365. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef]
- Lowy, A.M.; Knight, J.; Groden, J. Restoration of E-cadherin/beta-catenin expression in pancreatic cancer cells inhibits growth by induction of apoptosis. Surgery 2002, 132, 141–148. [Google Scholar] [CrossRef]
- Krishnadath, K.K.; Tilanus, H.W.; van Blankenstein, M.; Hop, W.C.; Kremers, E.D.; Dinjens, W.N.; Bosman, F.T. Reduced expression of the cadherin-catenin complex in oesophageal adenocarcinoma correlates with poor prognosis. J. Pathol. 1997, 182, 331–338. [Google Scholar] [CrossRef]
- Jawhari, A.; Jordan, S.; Poole, S.; Browne, P.; Pignatelli, M.; Farthing, M.J. Abnormal immunoreactivity of the E-cadherin-catenin complex in gastric carcinoma: Relationship with patient survival. Gastroenterology 1997, 112, 46–54. [Google Scholar] [CrossRef]
- Song, Y.-H.; Jeong, S.-J.; Kwon, H.-Y.; Kim, B.; Kim, S.-H.; Yoo, D.-Y. Ursolic acid from Oldenlandia diffusa induces apoptosis via activation of caspases and phosphorylation of glycogen synthase kinase 3 beta in SK-OV-3 ovarian cancer cells. Biol. Pharm. Bull. 2012, 35, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Annavarapu, S.R.; Cialfi, S.; Dominici, C.; Kokai, G.K.; Uccini, S.; Ceccarelli, S.; McDowell, H.P.; Helliwell, T.R. Characterization of Wnt/β-catenin signaling in rhabdomyosarcoma. Lab. Investig. 2013, 93, 1090–1099. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, S.; Schwartz, V.K.; Jackson, I.A.; Clark Gamblin, T.; Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 2018, 18, 560. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Higgins, D.G.; Kolch, W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Fernandez, R.; Watters, K.; Piskareva, O.; Stallings, R.L.; Bray, I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr. Surg. Int. 2013, 29, 101–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenström, P.; Heuchel, R.; Heldin, N.-E.; ten Dijke, P.; Heldin, C.-H.; Landström, M. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlund, S.; Lee, S.Y.; Grimsby, S.; Zhang, S.; Aspenström, P.; Heldin, C.-H.; Landström, M. Interaction between Smad7 and beta-catenin: Importance for transforming growth factor beta-induced apoptosis. Mol. Cell. Biol. 2005, 25, 1475–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trejo-Solis, C.; Escamilla-Ramirez, A.; Jimenez-Farfan, D.; Castillo-Rodriguez, R.A.; Flores-Najera, A.; Cruz-Salgado, A. Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals 2021, 14, 871. https://doi.org/10.3390/ph14090871
Trejo-Solis C, Escamilla-Ramirez A, Jimenez-Farfan D, Castillo-Rodriguez RA, Flores-Najera A, Cruz-Salgado A. Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals. 2021; 14(9):871. https://doi.org/10.3390/ph14090871
Chicago/Turabian StyleTrejo-Solis, Cristina, Angel Escamilla-Ramirez, Dolores Jimenez-Farfan, Rosa Angelica Castillo-Rodriguez, Athenea Flores-Najera, and Arturo Cruz-Salgado. 2021. "Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells" Pharmaceuticals 14, no. 9: 871. https://doi.org/10.3390/ph14090871