
Fusion-Based Neutron Generator Production of Tc-99m and Tc-101: A Prospective Avenue to Technetium Theranostics

 , , ,
, , ,
Abstract
1. Introduction
2. Results
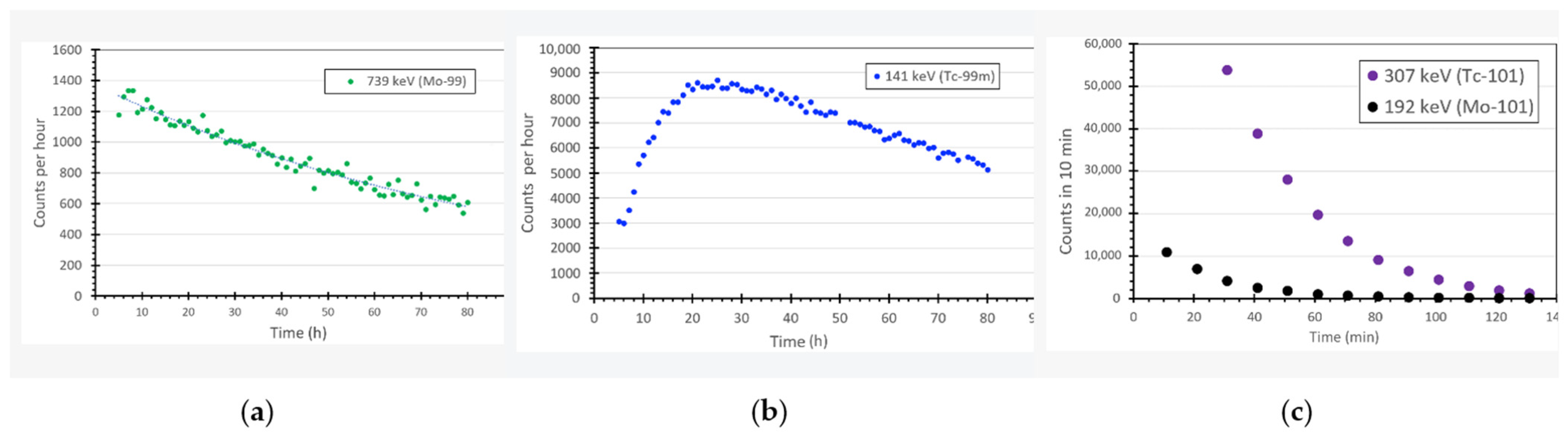
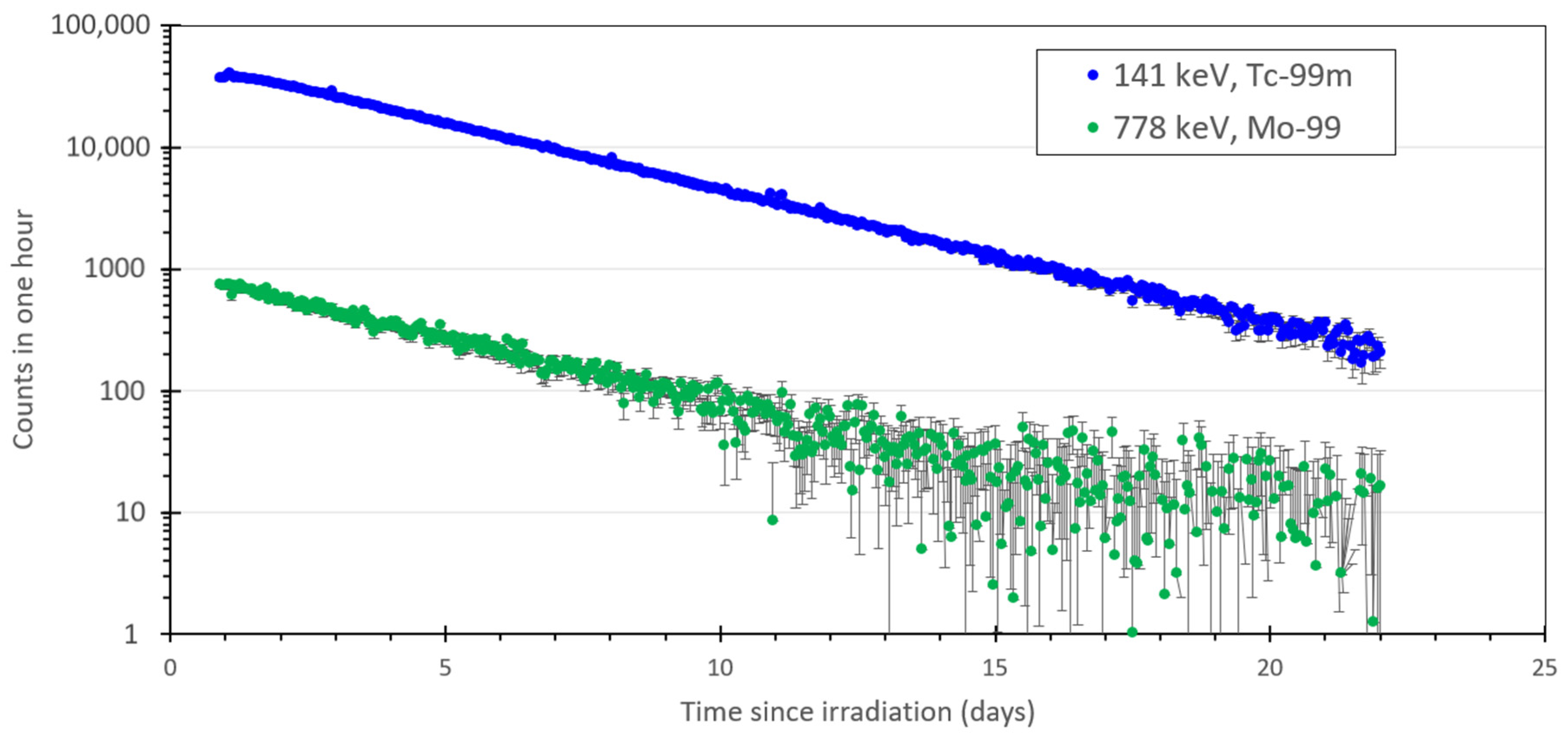
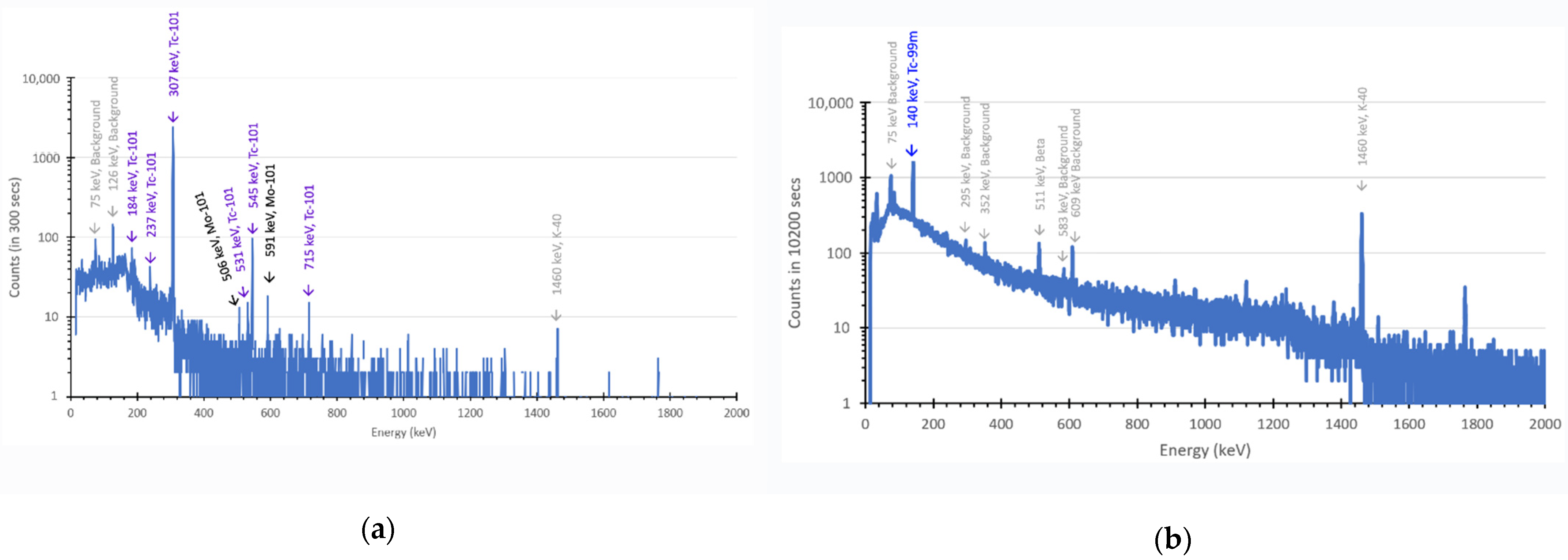
2.1. Production of 99Mo, 101Mo, 99mTc and 101Tc with a Neutron Generator
2.2. Isolation of 101Tc and 99mTc from Irradiated AHM Solution
3. Discussion
3.1. Production of 99Mo/99mTc and 101Mo/101Tc Using a Neutron Generator and Mo Targetry
3.2. Inventory of Neutron Generators Required for Worldwide Annual Production
3.3. Financial Considerations for Neutron Generator Production
3.4. Separation of Tc from LSA Mo Using AC
3.5. Technetium-101 as a Potential Ephemeral Diagnostic, Therapeutic, and/or Theranostic Agent
4. Materials and Methods
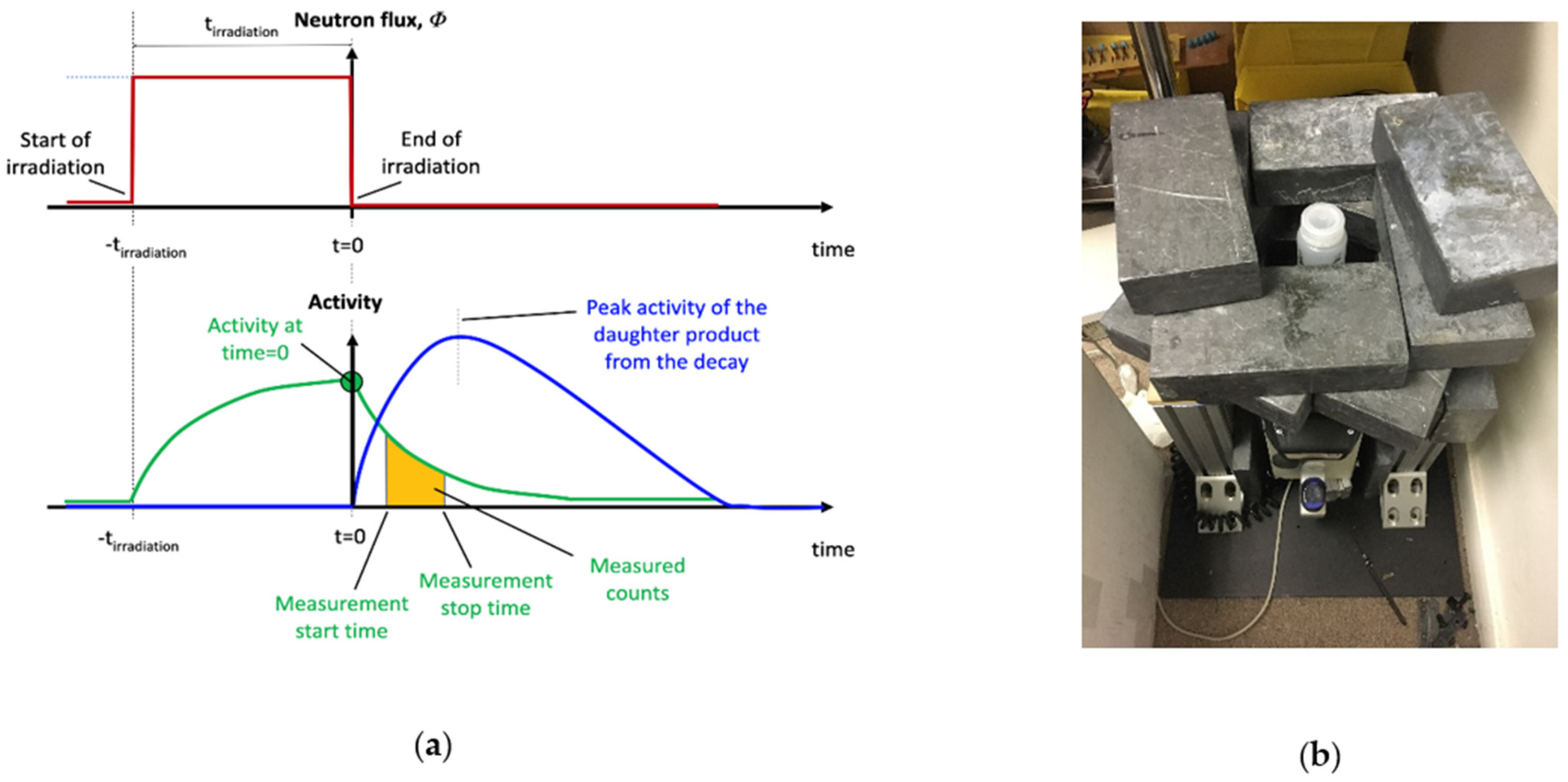
4.1. Neutron Irradiation
4.2. Gamma Spectroscopy
4.3. Measurement of the Neutron Flux (Φ) Based on Gold (Au) Reference
4.4. Extraction of Tc from Mo in a Bulk Sample of Irradiated AHM Solution
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD/NEA. The Supply of Medical Isotopes: An Economic Diagnosis and Possible Solutions; OECD Publishing: Paris, France, 2019. [Google Scholar] [CrossRef]
- Wolterbeek, B.; Kloosterman, J.L.; Lathouwers, D.; Rohde, M.; Winkelman, A.; Frima, L.; Wols, F. What is wise in the production of 99Mo? A comparison of eight possible production routes. J. Radioanal. Nucl. Chem. 2014, 302, 773–779. [Google Scholar] [CrossRef]
- Gumiela, M. Cyclotron production of 99mTc: Comparison of known separation technologies for isolation of 99mTc from molybdenum targets. Nucl. Med. Biol. 2018, 58, 33–41. [Google Scholar] [CrossRef]
- Mayordomo, N.; Rodríguez, D.M.; Schild, D.; Molodtsov, K.; Johnstone, E.V.; Hübner, R.; Shams Azzam, S.A.; Brendler, V.; Müller, K. Technetium Retention by Gamma Alumina Nanoparticles and the Effect of Sorbed Fe2+. J. Hazard. 2020, 388, 122066. [Google Scholar] [CrossRef]
- World Nuclear News. Belgium to Recycle Residues from Isotope Production. 3 January 2019. Available online: https://www.world-nuclear-news.org/Articles/Belgium-to-recycle-residues-from-isotope-productio (accessed on 5 March 2020).
- Brown, M.A. The Mo-99 Story. ANS Nuclear Café: All Things Nuclear. 24 April 2017. Available online: https://www.ans.org/news/article-1950/the-mo-99-story/ (accessed on 5 March 2020).
- Sadri, K.; Dabbagh, V.R.; Forghani, M.N.; Asadi, M.; Sadeghi, R. Lymphoscintigraphy in the Time of COVID-19: Effect of Molybdenum-99 Shortage on Feasibility of Sentinel Node Mapping. Lymphat. Res. Biol. 2021, 19, 134–140. [Google Scholar] [CrossRef]
- Ponsard, B. Mo-99 supply issues: Report and lessons learned. ENS RRFM 2010 Transactions. In Proceedings of the 14th International Topical Meeting on the Research Reactor Fuel Management (RRFM 2010), Marrakech, Morocco, 21–25 March 2010. [Google Scholar]
- Sharma, A.; Kumar, R. Medical radioisotopes availability: Time to look beyond the pandemic. Nucl. Med. Commun. 2021, 42, 711–712. [Google Scholar] [CrossRef] [PubMed]
- World Nuclear News. HFR Shuts as Maria Joins Isotope Supply Chain. 22 February 2010. Available online: https://world-nuclear-news.org/Articles/HFR-shuts-as-Maria-joins-isotope-supply-chain (accessed on 5 March 2020).
- McGowan, M. Two Workers Exposed to Unsafe Radiation Dose at Lucas Heights Nuclear Facility. The Guardian, 24 June 2019. Available online: https://www.theguardian.com/australia-news/2019/jun/24/two-workers-exposed-to-unsafe-radiation-dose-at-lucas-heights-nuclear-facility (accessed on 5 March 2020).
- Fleming, R.M.; Fleming, M.R.; Chaudhuri, T.K. Holding Big Pharma Accountable. Open J. Pharmacol. Pharmacother. 2019, 4, 001–003. [Google Scholar] [CrossRef]
- Freeman, L.M. And Then There Was One: Comments on the Price Rise for MAA and DTPA. J. Nucl. Med. 2014, 55, 9N–11N. [Google Scholar] [PubMed]
- Green, J. Reactors, Radioisotopes, and the HIFAR Controversy. Ph.D. Thesis, University of Wollongong, Wollongong, Australia, 1997. [Google Scholar]
- Jones, A.G.; Abrams, M.; Davidson, A.; Brodack, J.W.; Toothaker, A.K. Biological studies of a new class of technetium complexes: The hexakis(alkylisonitrile) technetium (I) cations. Int. Nucl. Med. Biol. 1984, 11, 225–234. [Google Scholar] [CrossRef]
- IAEA Live Chart of Nuclides. Available online: https://www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html (accessed on 11 March 2020).
- Nakano, T.; Sakai, M.; Torikai, K.; Suzuki, Y.; Noda, S.E.; Yamaguchi, M.; Takeda, S.; Nagao, Y.; Kikuchi, M.; Odaka, H.; et al. Imaging of 99mTc-DMSA and 18F-FDG in humans using a Si/CdTe Compton camera. Phys. Med. Biol. 2019, 65, 5. [Google Scholar] [CrossRef] [PubMed]
- Sharir, T.; Slomka, P. Dual-isotope myocardial perfusion SPECT imaging: Past, present, and future. J. Nucl. Cardiol. 2018, 25, 2024–2028. [Google Scholar] [CrossRef]
- Welch, M.J.; Kilbourn, M.R. A remote system for the routine production of oxygen-15 radiopharmaceuticals. J. Label. Compd. Radiopharm. 1985, 22, 1193–1200. [Google Scholar] [CrossRef]
- Gedcke, D.A. How Counting Statistics Controls Detection Limits and Peak Precision, Ortec® Application Note AN59. 2009. Available online: https://www.ortec-online.com/service-and-support/library/application-notes (accessed on 5 August 2021).
- Saptiama, I.; Lestari, E.; Sarmini, E.; Lubis, H.; Marlina, M.; Mutalib, A. Development of 99Mo/99mTc generator system for production of medical radionuclide 99mTc using a neutron-activated 99Mo and zirconium based material (ZBM) as its adsorbent. Atom. Indones. 2016, 42, 115–121. [Google Scholar] [CrossRef]
- Mausolf, E.; Johnstone, E. Direct, Continuous Transmutation of Molybdenum (Mo) for the Production and Recovery of Technetium (Tc) and Ruthenium (Ru) Using a Neutron Source. US/International Patent 2018. under view. [Google Scholar]
- Johnstone, E.V.; Czerwinski, K.R.; Hartmann, T.; Poineau, F.; Bailey, D.J.; Hyatt, N.C.; Mayordomo, N.; Nuñez, A.; Tsang, F.Y.; Sattelberger, A.P.; et al. Calcium molybdate, CaMoO4: A promising target material for 99mTc and its potential applications in nuclear medicine and nuclear waste disposition. In Proceedings of the 10th International Symposium on Technetium and Rhenium—Science and Utilization, Moscow, Russia, 3–6 October 2018. [Google Scholar] [CrossRef]
- Gerasimov, A.S.; Kiselev, G.V. Production of Mo-99 in research reactors with the use of uranium targets and Mo-98 targets. In Proceedings of the Second International Conference on Isotopes, Sydney, Australia, 12–16 October 1997. [Google Scholar]
- Toth, J.J.; Pierson, B.D.; Painter, C.L.; Greenwood, L.R.; Burns, K.A.; Love, E.F.; Soderquist, C.Z.; Lavender, C.A.; Wall, D.E.; Wittman, R.S. Production of Molybdenum-99 Using Neutron Capture Methods; Technical report; PNNL-19895, RPT-59331-01 Rev 0; Pacific Northwest National Laboratory (PNNL): Richmond, WA, USA, 2011. [Google Scholar]
- Lamson, M.; Hotte, C.E.; Ice, R.D. Practical Generator Kinetics. J. Nucl. Med. Technol. 1976, 4, 21–27. [Google Scholar]
- IAEA. Cyclotrons—What They Are and Where You Can Find Them. 27 January 2021. Available online: https://www.iaea.org/newscenter/news/cyclotrons-what-are-they-and-where-can-you-find-them (accessed on 6 August 2021).
- Piestrup, M.A.; Brown, C.M.; Cremer, J.T., Jr.; Gary, C.K.; Williams, D.L.; Chen, A.X.; Jones, G.E., Jr.; Guan, Y.Z.; Urdahl, R.S.; Amoroso, A.N. Neutron Source with Beam Shaping Apparatus for Radiography. US Patent 10,955,365, 23 March 2021. [Google Scholar]
- Piestrup, M.A.; Brown, C.M.; Chen, A.X.; Gary, C.K.; Guan, Y.Z.; Horii, Y.; Jones, E.J., Jr.; Kokubo, Y.; Yamada, N.; Komura, S. Neutron Source for Neutron Capture Therapy. US Patent 10,874,881 B2, 29 December 2020. [Google Scholar]
- Watterson, J.I.W. A Review of Accelerator Based Neutron Sources and Their Applications; IAEA-TECDOC-1153, IAEA Technical Report; International Atomic Energy Agency: Vienna, Austria, 2000. [Google Scholar]
- Chowdhury, P.; Benmerrouche, M. Radiological safety considerations of a 40 kW 35 MeV electron linear accelerator at the Canadian Light Source. In Proceedings of the RadSynch 13, Upton, NY, USA, 8–10 May 2013; pp. 1–16. [Google Scholar]
- Borio di Tigliole, A.; Cammi, A.; Chiesa, D.; Clemenza, M.; Manera, S.; Nastasi, M.; Pattavina, L.; Ponciroli, R.; Pozzi, S.; Prata Previtali, M.; et al. TRIGA reactor absolute neutron flux measurement using activated isotopes. Prog. Nucl. Energy 2014, 70, 249–255. [Google Scholar] [CrossRef]
- NTI Article, NTI (Nuclear Threat Initiative), 1776 Eye Street, NW, Suite 600, Washington, DC 20006). 1 August 2011. Available online: https://www.nti.org/learn/facilities/8/ (accessed on 7 August 2021).
- CPI Inflation Calculator. Available online: https://www.in2013dollars.com/us/inflation/1972?amount=270000 (accessed on 7 August 2021).
- Ross, C.; Galea, R.; Saull, P.; Davidson, W.; Brown, P.; Brown, D.; Harvey, J.; Messina, G.; Wassenaar, R.; de Jong, M. Using the 100Mo photoneutron reaction to meet Canada’s requirement of 99mTc. Phys. Can. 2010, 66, 19–24. [Google Scholar]
- CBC News. FAQ: Cyclotrons. 2009. Available online: https://www.cbc.ca/news/science/faq-cyclotrons-1.789206 (accessed on 7 August 2021).
- Otake, Y. RIKEN accelerator-driven compact neutron source, RANS and neutron application. In Proceedings of the 26th International Seminar on Interaction Neutrons Nuclei, Xi’an, China, 28 May–1 June 2018. [Google Scholar]
- Ito, K.; Yachidate, A.; Akiba, K. Sorption of technetium on activated carbon. CYRIC Ann. Rep. 1986, 1986, 118–120. [Google Scholar]
- Ito, K. Variation of pertechnetate ion adsorption on activated carbons from different sources. CYRIC Ann. Rep. 1991, 1991, 99–101. [Google Scholar]
- Ito, K.; Akiba, K. Adsorption of pertechnetate ion on active carbon from acids and their salt solutions. J. Radioanal. Nucl. Chem. 1991, 152, 381–390. [Google Scholar] [CrossRef]
- Holm, E.; Gäfvert, T.; Lindhal, P.; Roos, P. In situ sorption of technetium using activated carbon. Appl. Radiat. Isot. 2000, 53, 153–157. [Google Scholar] [CrossRef]
- Gu, B.; Dowlen, K.E.; Liang, L.; Clausen, J.L. Efficient separation and recovery of technetium-99 from contaminated groundwater. Sep. Technol. 1996, 6, 123–132. [Google Scholar] [CrossRef]
- Byrnes, M.E.; Mavis, J.; Rossi, A.; Tortoso, A. Potential use of activated carbon to recover Tc-99 from 200 West Area groundwater as an alternative to more expensive resins, Hanford Site, Richland, Washington. In Proceedings of the WM2010: Waste Management Symposia, Phoenix, AZ, USA, 7–11 March 2010. Hanford Report: HNF–42335–FP. [Google Scholar]
- Makarov, A.; Safonov, A.; Konevnik, Y.; Teterin, Y.; Maslakov, K.; Teterin, A.; Karaseva, Y.; German, K.; Zakharova, E. Activated carbon additives for technetium immobilization in bentonite-based engineered barriers for radioactive waste repositories. J. Hazard. Mater. 2021, 401, 123436. [Google Scholar] [CrossRef]
- Li, D.; Kaplan, D.; Knox, A.; Crapse, K.; Diprete, D. Aqueous 99Tc, 129I, and 137Cs removal from contaminated groundwater and sediments using highly-effective low-cost sorbents. J. Environ. Radioact. 2014, 136, 56–63. [Google Scholar] [CrossRef]
- Jang, J.; Uesaka, M.; Tatenuma, K.; Tsuguchi, A.; Seikimoto, S.; Ohtsuki, T. Photonuclear production of Mo-99 / Tc-99m using molybdenum trioxide and activated carbon. In Proceedings of the Mo-99 Topical Meeting 2017, Montreal, QC, Canada, 10–13 September 2017. S9–P3. [Google Scholar]
- Petrovic, D.; Dukic, A.; Kumric, K.; Babic, B.; Momcilovic, M.; Ivanovic, N.; Matovic, L. Mechanism of Sorption of Pertechnetate onto ordered mesoporous carbon. J. Radioanal. Nucl. Chem. 2014, 302, 217–224. [Google Scholar] [CrossRef]
- Ravi, A.; Oshchepkov, A.S.; German, K.E.; Kirakosyan, G.A.; Safonov, A.V.; Khrustalev, V.N.; Kataev, E.A. Finding a receptor design for selective recognition of perrhenate and pertechnetate: Hydrogen vs. halogen bonding. Chem. Commun. 2018, 54, 4826–4829. [Google Scholar] [CrossRef] [PubMed]
- Galamboš, M.; Dano, M.; Vigašlová, E.; Krivosudsky, L.; Rosskopfová, O.; Novák, I.; Berek, D.; Rajec, P. Effect of competing anions on pertechnetate adsorption by activated carbon. J. Radioanal. Nucl. Chem. 2015, 304, 1219–1224. [Google Scholar] [CrossRef]
- Daňo, M.; Viglašová, E.; Galamboš, M.; Rajec, P.; Novák, I. Sorption behaviour of pertechnetate on oxidized and reduced surface of activated carbon. J. Radioanal. Nucl. Chem. 2017, 314, 2219–2227. [Google Scholar] [CrossRef]
- Rajec, P.; Galambos, M.; Dano, M.; Rosskopfova, O.; Caplovicova, M.; Hudec, P.; Hornaek, M.; Novak, I.; Berek, D.; Caplovic, L. Preparation and characterization of adsorbent based on carbon for pertechnetate adsorption. J. Radioanal. Nucl. Chem. 2015, 303, 277–286. [Google Scholar] [CrossRef]
- Mahmudov, R.; Huang, C.P. Selective adsorption of oxyanions on activated carbon exemplified by Filtrasorb 400 (F400). Sep. Purif. Tech. 2011, 77, 294–300. [Google Scholar] [CrossRef]
- Cruywagen, J.J. Protonation, oligomerization, and condensation reactions of vanadate (V), molybdate (VI), and tungstate (VI). Adv. Inorg. Chem. 1999, 49, 127–182. [Google Scholar]
- Bettinardi, D.J.; Paulenova, A.; Tkac, P. Speciation of molybdenum(VI) in chloride media at elevated Mo concentrations. ACS Omega. 2020, 5, 23786–23792. [Google Scholar] [CrossRef] [PubMed]
- Muruchi, L.; Schaeffer, N.; Passos, H.; Mendonça, C.M.N.; Coutinho, J.A.P.; Jimenez, Y.P. Sustainable extraction and separation of rhenium and molybdenum from model copper mining effluents using a polymeric aqueous two-phase system. ACS Sustain. Chem. Eng. 2019, 7, 1778–1785. [Google Scholar] [CrossRef]
- Śmiechowski, M.; Persson, I. Hydradtion of oxometallate ions in aqueous solution. Inorg. Chem. 2020, 59, 8231–8239. [Google Scholar] [CrossRef]
- Pagnanelli, F.; Ferella, F.; De Michelis, I.; Veligo, M. Adsorption onto activated carbon for molybdenum recovery from leach liquors of exhausted hydrotreating catalysts. Hydrometallurgy 2011, 110, 67–72. [Google Scholar] [CrossRef]
- Kimura, M.; Gotou, C.; Tani, M. Separation and preconcentration of molybdenum(VI) ions in aqueous solution using activated carbon as a collector; Determination of molybdenum(VI) in tap-water, river-water and seawater. Bunseki Kagaku 1989, 38, 529. [Google Scholar] [CrossRef][Green Version]
- Hamed, M.; Rizik, H.; Ahmed, I. Adsorption behaviour of zirconium and molybdenum from nitric acid medium using low-cost adsorbent. J. Mol. Liq. 2018, 249, 361–370. [Google Scholar] [CrossRef]
- Seo, Y.S.; Choi, S.W.; Yang, T.J.; Kim, M.J.; Tran, T. Recovery of rhenium and molybdenum from a roaster fume scrubbing liquor by adsorption using activated carbon. Hydrometallurgy 2012, 129–130, 145–150. [Google Scholar] [CrossRef]
- El-Bayoumy, S.; El-Kolaly, M. Some radiochemical studies on the adsorption behaviour of molybdenum-99 on silver-coasted carbon granules and activated carbon. J. Radioanal. Chem. 1982, 68, 7–13. [Google Scholar] [CrossRef]
- Viglasova, E.; Dano, M.; Galambos, M.; Rosskopfova, O.; Rajec, P.; Novak, I. Column studies for the separation of 99mTc using activated carbon. J. Radioanal. Nucl. Chem. 2016, 307, 591–597. [Google Scholar] [CrossRef]
- Tatenuma, K.; Ishikawa, K.; Tsugugchi, A.; Komatsuzaki, Y.; Suzuki, Y.; Tanaka, A.; Kurosawa, K.; Uehara, T.; Higaki, Y.; Hanaoka, H.; et al. A mass-production process for highly-pure medical use 99mTc from natural isotopic Mo(n, γ)99Mo without using uranium. Radioisotopes 2014, 63, 501–513. [Google Scholar] [CrossRef]
- Tatenuma, K.; Tsuguchi, A.; Suzuki, Y.; Ishikawa, K. Generator of highly concentrated pure 99mTc from low specific activity 99Mo produced by reactor and/or electron accelerator. In Proceedings of the Mo-99 2016 Topical Meetings on Molybdenum-99 Technological Development, St. Louis, MO, USA, 11–14 September 2016. [Google Scholar]
- Tatenuma, K.; Ueda, T.; Kurosawa, K.; Ishikawa, K.; Tanaka, A.; Noguchi, T.; Arano, Y. Method of Recovering Enriched Radioactive Technetium and System Therefor. US Patent 9236153–B2, 12 January 2016. [Google Scholar]
- Yang, G.; Hu, J. An automated 99mTc generator device using low specific activity 99Mo. J. Nucl. Med. 2018, 59 (Suppl. 1), 1039. [Google Scholar]
- Song, Z.; Zhang, Y.; Wang, N.; Guo, H.; Zhao, H.; Zhao, H.; Gao, X. Extracting technetium from neutral molybdenum solution with activated carbon fiber. J. Isot. China 2021, 34, 133–140. [Google Scholar]
- Zhang, Y.; Song, Z.; Wang, N.; Guo, H.; Zhao, H.; Gao, X. Activated carbon fiber as matrix for three-column selectivity inversion 99Mo–99mTc generator. J. Isot. China 2021, 34, 120–126. [Google Scholar]
- Notni, J.; Wester, H.J. Re-thinking the role of radiometal isotopes: Towards a future concept for theranostic radiopharmaceuticals. J. Label. Compd. Radiopharm. 2018, 61, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M.; Scholten, B.; Neumaier, B. New developments in the production of theranostic pairs of radionuclides. J. Radioanal. Nucl. Chem. 2018, 318, 1493–1509. [Google Scholar] [CrossRef]
- Johnstone, E.V.; Yates, M.A.; Poineau, F.P.; Sattelberger, A.P.; Czerwinski, K.R. Technetium: The first radioelement on the Periodic Table. J. Chem. Educ. 2017, 94, 320–326. [Google Scholar] [CrossRef]
- Tavares, A.A.S.; Tavares, J.M.R.S. 99mTc Auger electrons for target tumor therapy: A review. Int. J. Radiat. Biol. 2010, 86, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Bigott, H.M.; Mccarthy, D.W.; Wüst, F.R.; Dahlheimer, J.L.; Piwnica-Worms, D.R.; Welch, M.J. Production, processing and uses of 94mTc. J. Label. Compd. Radiopharm. 2001, 44, S119–S121. [Google Scholar] [CrossRef]
- Hayakawa, T.; Hatsukawa, Y.; Tanimori, T. 95gTc and 96gTc as alternatives to medical radioisotope 99mTc. Heliyon 2018, 4, e00497. [Google Scholar] [CrossRef]
- Gott, M.D.; Hayes, C.R.; Wycoff, D.E.; Balkin, E.R.; Smith, B.E.; Pauzauskie, P.J.; Fassbender, M.E.; Cutler, C.S.; Ketring, A.R.; Wilbur, D.S.; et al. Accelerator-based production of the 99mTc-186Re diagnostic-therapeutic pair using metal disulfide targets (MoS2, WS2, OsS2). Appl. Radiat. Isot. 2016, 114, 159–166. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yoshihara, K. Rhenium complexes labeled with 186,188Re for nuclear medicine. In Technetium and Rhenium Their Chemistry and Its Applications; Yoshihara, K., Omori, T., Eds.; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 1996; Volume 176. [Google Scholar] [CrossRef]
- Alberto, R.; Braband, H.; Nadeem, Q. Bioorganometallic technetium and rhenium chemistry: Fundamentals for applications. Chimia 2020, 74, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Chotkowski, M.; Czerwiński, A. Electrochemistry of Technetium; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Poineau, F.; Johnstone, E.V.; Czerwinski, K.R.; Sattelberger, A.P. Recent advances in technetium halide chemistry. Acc. Chem. Res. 2014, 47, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M. Therapeutic nuclides and nuclear data. Radiochim. Acta. 2001, 89, 297–302. [Google Scholar] [CrossRef]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvygin, A.V. Target radionuclide therapy for tumors. Int. J. Mol. Sci. 2016, 17, 33. [Google Scholar] [CrossRef]
- Morgenstern, A.; Lebeda, O.; Stursa, J.; Bruchertseifer, F.; Capote, R.; McGinley, J.; Rasmussen, G.; Sin, M.; Zielinska, B.; Apostolidis, C. Production of 230U/226Th for targeted alpha therapy via proton irradiation of 231Pa. Anal. Chem. 2008, 80, 8763–8770. [Google Scholar] [CrossRef]
- Metebi, A.; Nayback, C.; Fan, J.; Johnson, N.; Diemer, J.; Grimm, T.; Zamiara, M.; Zinn, K. Therapeutic Efficacy of Pb-214-labeled Trastuzumab in a Preclinical Model of Ovarian Cancer. J. Nucl. Med. 2021, 62 (Suppl. 1), 93. [Google Scholar]
- Ferrier, M.G.; Li, Y.; Chyan, M.; Wong, R.; Li, L.; Spreckelmeyer, S.; Hamlin, D.K.; Mastren, T.; Fassbender, M.E.; Orvig, C.; et al. Thorium chelators for targeted alpha therapy: Rapid chelation of thorium-226. J. Label. Compd. Radiopharm. 2020, 63, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Pillai, M.R.A.; Dash, A.; Knapp, F.F. Sustained Availability of 99mTc: Possible Paths Forward. J. Nucl. Med. 2013, 54, 313–323. [Google Scholar] [CrossRef]
- Nagai, Y. Production scheme for diagnostic-therapeutic radioisotopes by accelerator neutrons. Proc. Jpn. Acad. Ser. B. 2021, 97, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Lapereur, N.; Lacoeuille, F.; Bouvry, C.; Hindre, F.; Garcion, E.; Cherel, M.; Noiret, N.; Garin, E.; Russ Knapp, F.F., Jr. Rhenium-188 labeled radiopharmaceuticals: Current clinical applications in oncology and promising prospectives. Front. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Ceccarelli, C.; Bianchi, F.; Trippi, D.; Di Martino, F.; Satini, P.; Elisei, R.; Pinchera, A. Location of functioning metastases from differentiated thyroid carcinoma by simultaneous double isotope acquisition of I-131 whole body scan and bone scan. J. Endocrinol. Investig. 2004, 27, 866–869. [Google Scholar] [CrossRef]
- Kotb, M.H.; Omar, W.; El-Maghraby, T.; El-Bedwihy, M.; El-Tawdy, M.; Mustafa, H.; Al-Nahhas, A. The value of simultaneous co-registration of 99mTc-MDP and 131Iodine in metastatic differentiated thyroid carcinoma. Nucl. Med. Rev. 2007, 10, 98–105. [Google Scholar]
- Vogel, W.V.; van der Marck, S.C.; Versleijen, M.W.J. Challenges and future options for the production of lutetium-177. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef] [PubMed]
- Abram, U.; Alberto, R. Technetium and rhenium: Coordination chemistry and nuclear medical applications. J. Braz. Chem. Soc. 2006, 17, 1486–1500. [Google Scholar] [CrossRef]
- Badria, F.A. Radiopharmaceuticals: On-Going Research for Better Diagnosis, Therapy, Environmental, and Pharmaceutical Applications. IntechOpen 2021. Online First. [Google Scholar] [CrossRef]
- Xu, T.; Vorobyeva, A.; Schulga, A.; Konovalova, E.; Vorontsova, O.; Ding, H.; Gräslund, T.; Tashireva, L.A.; Orlova, A.; Tolmachev, V.; et al. Imaging-Guided Therapy Simultaneously Targeting HER2 and EpCAM with Trastuzumab and EpCAM-Directed Toxin Provides Additive Effect in Ovarian Cancer Model. Cancers 2021, 13, 3939. [Google Scholar] [CrossRef]
- Adelphi Technologies Inc. Available online: https://www.adelphitech.com/ (accessed on 20 June 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Activity (Bq) Mo-99 | Activity (Bq) Tc-99m |
|---|---|---|
| 1 | 2.09 × 108 | 2.02 × 107 |
| 24 | 4.46 × 109 | 3.70 × 109 |
| 168 | 1.66 × 1010 | 1.47 × 1010 |
| 330 | 1.94 × 1010 | 1.72 × 1010 |
| Time (Days) | Commercial Generator (Doses) | Continuous Generator (Doses) | Efficiency Gain (%) |
|---|---|---|---|
| 1 | 15 | 22 | 44 |
| 5 | 52 | 110 | 112 |
| 7 | 60 | 154 | 157 |
| 14 | 91 | 308 | 238 |
| Generator Flux | 2 × 1010 n/s | 2 × 1012 n/s | ||||||
| Tc Isotope Produced | 99mTc | 101Tc | 99mTc | 101Tc | ||||
| Mo Target | Nat. Mo | 98Mo | Nat. Mo | 100Mo | Nat. Mo | 98Mo | Nat. Mo | 100Mo |
| Doses Generated per day | 5 | 22 | 53 | 220 | 534 | 2200 | 5344 | 22,000 |
| Generators Required | 21,918 | 4981 | 2068 | 298 | 205 | 50 | 21 | 5 |
| Type | Estimate of Beam Energy (MeV) | Approximate Yield Range (n/s) | Approximate System Cost, Order of Magnitude ($M) |
|---|---|---|---|
| Reactor * | Not applicable | >1017 | ~1000 |
| Electron Accelerator † with Photoneutron Converter | 30–40 | 5 × 1013 to 1 × 1014 | 10 |
| Cyclotron ‡ | 10–18.0 | 5.7 × 1012 to 2.1 × 1014 | 1–10 |
| RFQ Linac § | 1.5–3.0 | 1 × 1011 to 1.3 × 1012 | 1 |
| D-D Neutron Generator | 0.1–0.2 | 1 × 108 to 1 × 1011 | 0.1–1 |
| Isotope | Half-Life | Egamma (keV) | Ebeta (keV) | Tissue Penetration Range (mm) | Uses |
|---|---|---|---|---|---|
| 89Sr | 50.5 d | N/A | 587.10 (99.9%) | 8 | TRT 1-osseous metastases |
| 90Y | 64.00 h | N/A | 933.7 (99.9%) | 12 | TRT-hepatic malignancies, lymphoma |
| 131I | 8.02 d | 284.3 (6.1%), 364.48 (81.5%), 636.98 (7.2%) | 191.58 (89.6%), 96.62 (7.2%) | 2.4 | Diagnostic imaging (SPECT); TRT: thyroid ablation, neuroendocrine tumors, prostate seeds |
| 177Lu | 6.647 d | 208.36 (10.4%) | 149.35 (79.4%), 47.66 (11.6%) | 2.2 | Diagnostic imaging (SPECT); TRT: PRRT 2, bone pain palliation, synovectomy, neuroendocrine, metastatic prostate, etc. |
| 186Re | 3.72 d | 137.16 (9.5%) | 359.2 (71.0%), 306.1 (21.5%) | 4.5 | Bone pain palliation, synovectomy, endovascular irrad. |
| 188Re | 17.01 h | 155.04 (15.5%) | 795.41 (70.7%), 728.88 (25.8%) | 11 | Bone pain palliation, synovectomy, endovascular irrad. |
| 223Ra * | 11.43 d | ~82.0 (<2.0%) | 492.5 (99.7%), 471.3 (91.3%), 172.9 (0.3%) | - | Bone metastasises |
| 225Ac * | 9.920 d | 218.0 (11.4%), 440.45 (25.9%), 1567.1 (99.7%) | 660.34 (97.4%), 492.2 (65.9%), 197.4 (100%), 93.4 (68.8%) | - | Metastatic castration-resistant prostate cancer |
| 101Tc | 14.22 min | 306.8 (89%), 545 (5.9%) | 487 (90%), 385 (6%), 127.2 (2.64%) | N/A | N/A |
| Isotope | Energy of Key Line (keV) | Half-Life | Detector Efficiency (%) |
|---|---|---|---|
| 101Mo | 192 | 14.61 m | 9.4 ± 1 |
| 101Tc | 307 | 14.22 m | 6.6 ± 0.7 |
| 99Mo | 778 | 65.924 h | 3.3 ± 0.3 |
| 99mTc | 141 | 6.001 h | 11.8 ± 1.5 |
| 198Au | 411 | 2.697 d | 5.4 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mausolf, E.J.; Johnstone, E.V.; Mayordomo, N.; Williams, D.L.; Guan, E.Y.Z.; Gary, C.K. Fusion-Based Neutron Generator Production of Tc-99m and Tc-101: A Prospective Avenue to Technetium Theranostics. Pharmaceuticals 2021, 14, 875. https://doi.org/10.3390/ph14090875
Mausolf EJ, Johnstone EV, Mayordomo N, Williams DL, Guan EYZ, Gary CK. Fusion-Based Neutron Generator Production of Tc-99m and Tc-101: A Prospective Avenue to Technetium Theranostics. Pharmaceuticals. 2021; 14(9):875. https://doi.org/10.3390/ph14090875
Chicago/Turabian StyleMausolf, Edward J., Erik V. Johnstone, Natalia Mayordomo, David L. Williams, Eugene Yao Z. Guan, and Charles K. Gary. 2021. "Fusion-Based Neutron Generator Production of Tc-99m and Tc-101: A Prospective Avenue to Technetium Theranostics" Pharmaceuticals 14, no. 9: 875. https://doi.org/10.3390/ph14090875
APA StyleMausolf, E. J., Johnstone, E. V., Mayordomo, N., Williams, D. L., Guan, E. Y. Z., & Gary, C. K. (2021). Fusion-Based Neutron Generator Production of Tc-99m and Tc-101: A Prospective Avenue to Technetium Theranostics. Pharmaceuticals, 14(9), 875. https://doi.org/10.3390/ph14090875