
New Strategies for the Treatment of Atrial Fibrillation


Abstract
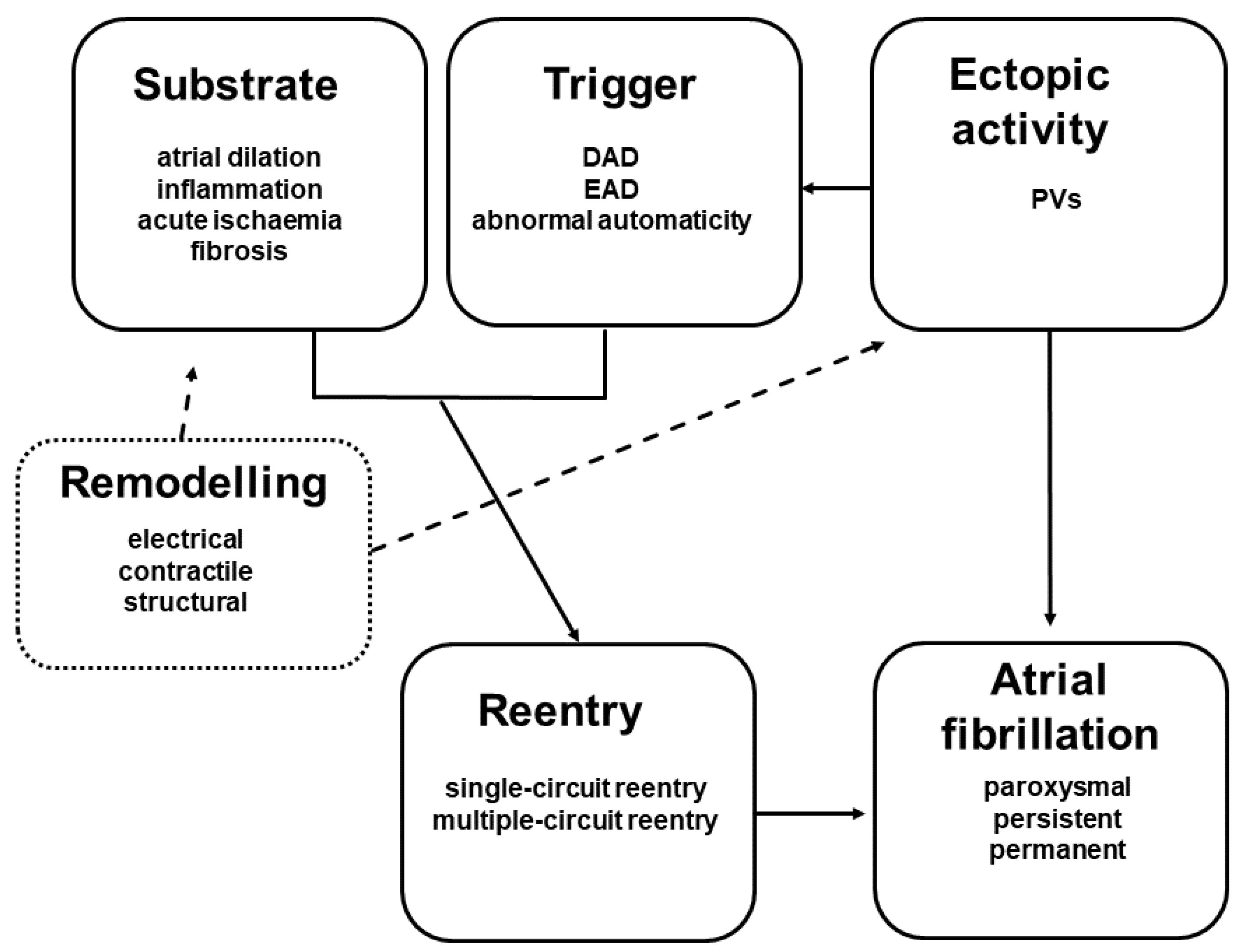
:1. Introduction
2. Mechanisms of Atrial Remodelling
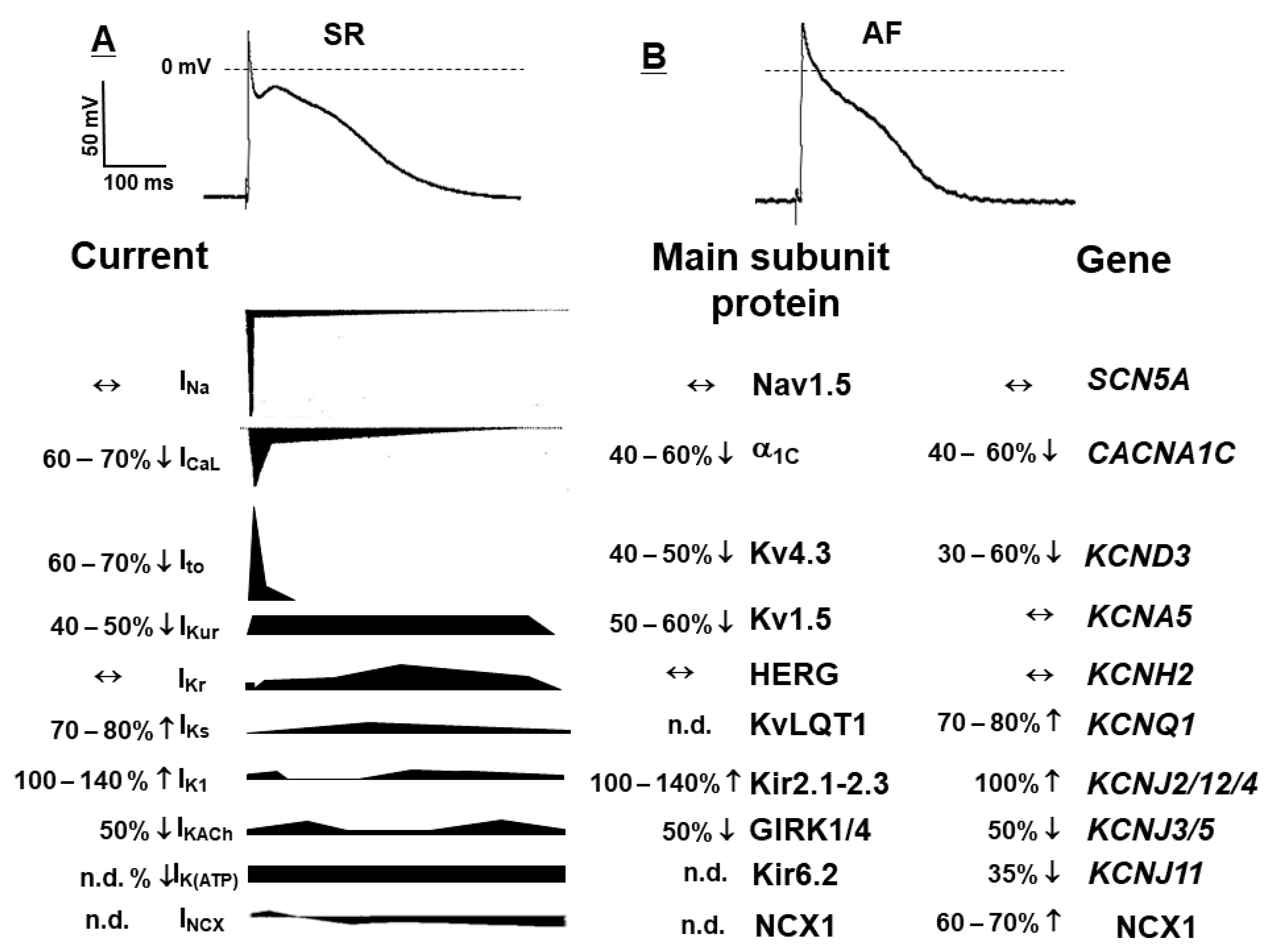
2.1. Electrical Remodelling
2.2. Contractile Remodelling
2.3. Structural Remodelling
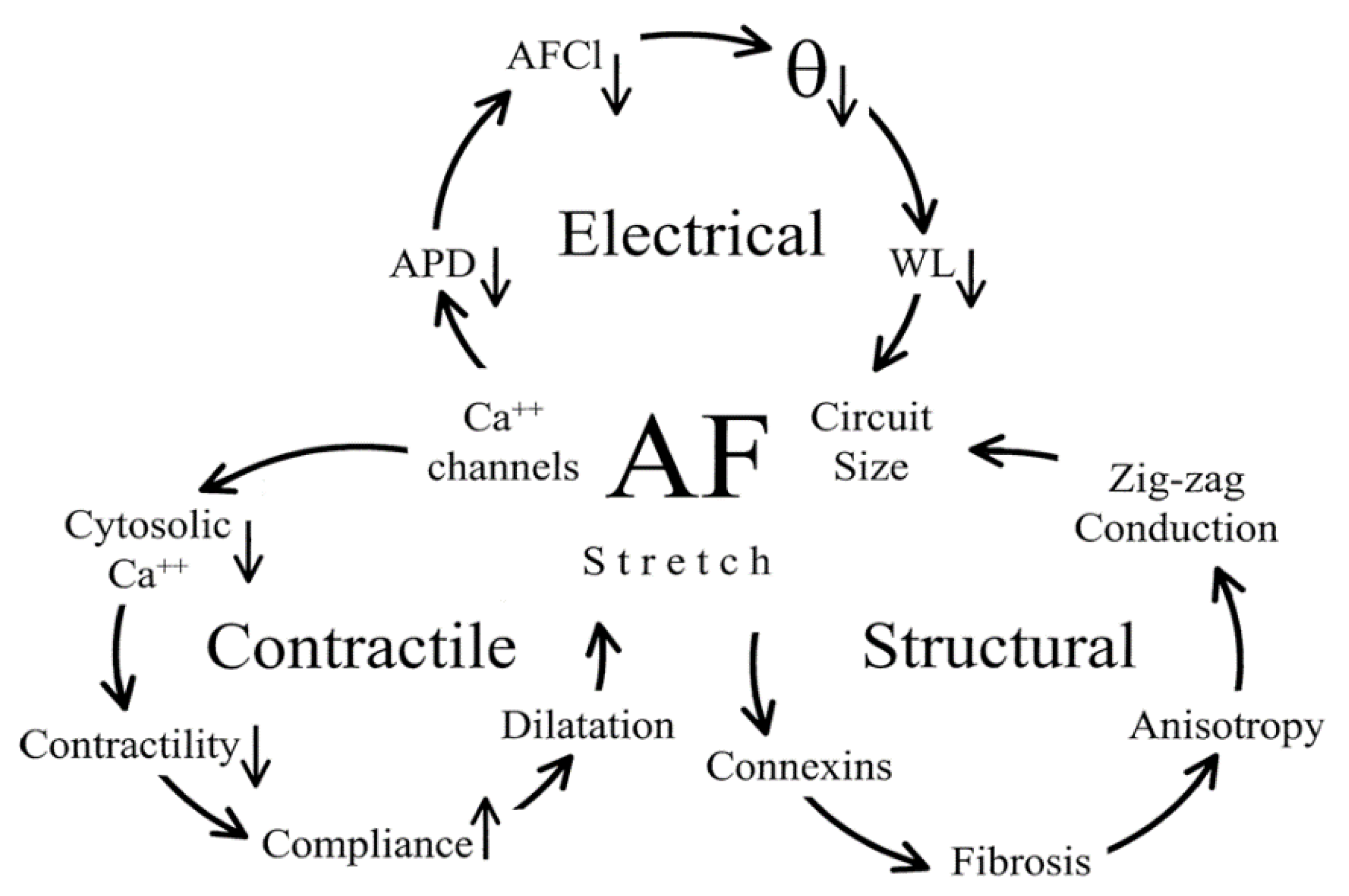
2.4. The Connection between Electrical, Contractile, and Structural Remodelling
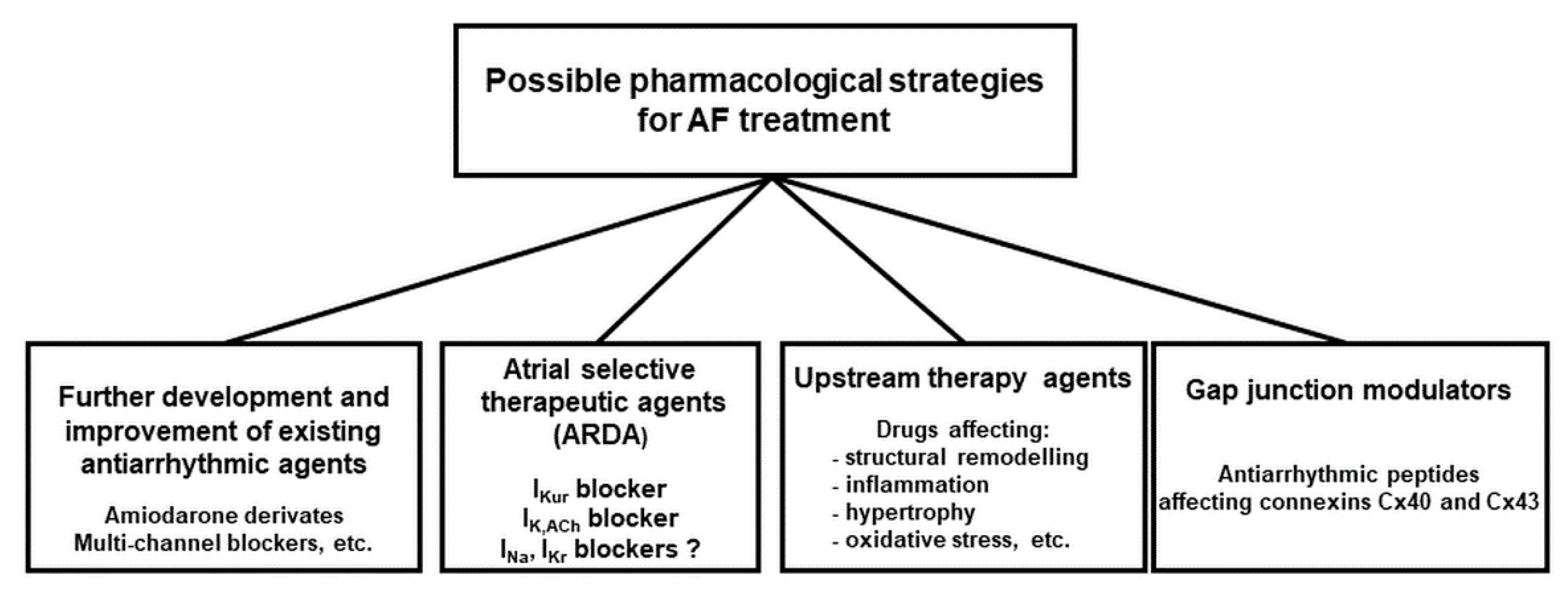
3. Therapeutic Options to Prevent and Stop Atrial Remodelling
3.1. Back to Sinus Rhythm or Just Control Ventricular Rate
3.2. New Approved Drugs and Investigational Compounds to Convert AF
3.2.1. Blockers of Specific Ion Channels vs. Multiple Ion Channel Blocker to Stop AF
Selective IKs Blockers
3.2.2. Amiodarone-Like Multichannel Blockers
3.2.3. Can Atrial Selective Ion Channel Block Stop AF?
IKur Blockers
Sodium Channel Blockers
Atrial Acetylcholine-Sensitive Potassium Current (IK,ACh) Blockers
Constitutively Active IK,ACh Channels (IK,Ach_Const)
3.2.4. NCX Modulators
3.2.5. Gap Junctions Modulators
3.3. Other Possible Ion Channel Targets for Novel Antiarrhythmic Drugs
3.4. Non Ion-Channel Blockers—Upstream Therapy of AF
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | atrial fibrillation |
| AFlu | atrial flutter |
| AERP | atrial effective refractory period |
| APD | action potential duration |
| ARDA | atrial repolarizing delayed agents |
| ATR | experimentally induced tachypaced dog model of permanent AF |
| CICR | Ca2+-induced Ca2+-release |
| CVPT | catecholaminergic polymorphic ventricular tachycardia |
| DAD | delayed afterdepolarization |
| EAD | early afterdepolarization |
| FDA | Food and Drug Administration |
| IK,Ach | acetylcholine sensitive potassium current |
| IK,Ach_Const | constitutively active acetylcholine sensitive potassium current (IK,Ach) |
| ICa,L | L-type calcium current |
| IK1 | inward rectifier potassium current |
| IKATP | ATP-sensitive potassium current |
| IKr | rapid component of the delayed rectifier potassium current |
| IKs | slow component of the delayed rectifier potassium current |
| IKur | ultra-rapid component of the delayed rectifier potassium current |
| Ito | transient outward potassium current |
| INa | fast inward sodium current |
| INaL | late sodium current |
| INCX | sodium-calcium (Na+/Ca2+) exchanger current (NCX) |
| miRNA | small single-stranded non-coding RNA) |
| NSAID | non-steroidal anti-inflammatory drugs |
| PVs | left upper pulmonary vein |
| RAAS | renin-angiotensin-aldosterone system |
| SMAD | Mothers Against Decapentaplegic is a protein from the SMAD family |
| SR | sinus rhythm |
| TGF | transforming growth factor |
References
- Beyerbach, D.M.; Zipes, D.P. Mortality as an endpoint in atrial fibrillation. Heart Rhythm. 2004, 1, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Opie, L.H. Controversies in atrial fibrillation. Lancet 2006, 367, 262–272. [Google Scholar] [CrossRef]
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed AF in adults: National implications for rhythm management and stroke prevention: The anTicoagulation and Risk Factors in AF (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- A Wolf, P.; Abbott, R.D.; Kannel, W.B. Atrial fibrillation: A major contributor to stroke in the elderly. The Framingham Study. Arch. Intern. Med. 1987, 147, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.-H. Arrhythmogenic Ion-Channel Remodeling in the Heart: Heart Failure, Myocardial Infarction, and Atrial Fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef]
- Camm, A.J.; Lip, G.Y.; De Caterina, R.; Savelieva, I.; Atar, D. Focused update of ESC Guidelines for the management of atrial fibrillation: An update of the 2010 guidelines for the management of atrial fibrillation—Developed with the special contribution of the European Heart Rhythm Association. Europace 2012, 14, 1385–1413. [Google Scholar]
- Allessie, M.A.; I Bonke, F.; Schopman, F.J. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: A new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ. Res. 1977, 41, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Ravens, U.; Poulet, C.; Wettwer, E.; Knaut, M. Atrial selectivity of antiarrhythmic drugs. J. Physiol. 2013, 591, 4087–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, S.V.; Jalife, J. Rotors and the Dynamics of Cardiac Fibrillation. Circ. Res. 2013, 112, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Dobrev, D.; Ravens, U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res. Cardiol. 2003, 98, 137–148. [Google Scholar] [CrossRef]
- Nattel, S.; Burstein, B.; Dobrev, D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ. Arrhythmia Electrophysiol. 2008, 1, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Jost, N.; Kohajda, Z.; Kristof, A.; Kovacs, P.; Juhasz, V.; Kiss, L.; Varro, A.; Virag, L.; Baczko, I. Atrial remodeling and novel pharmacological strategies for an-tiarrhythmic therapy in atrial fibrillation. Curr. Med. Chem. 2011, 18, 3675–3694. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Dobrev, D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur. Heart J. 2012, 33, 1870–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healey, J.S.; Israel, C.W.; Connolly, S.J.; Hohnloser, S.H.; Nair, G.M.; Divakaramenon, S.; Capucci, A.; Van Gelder, I.C.; Lau, C.P.; Gold, M.R.; et al. Relevance of electrical remodeling in human atrial fibrillation: Results of the asymptomatic atrial fibrillation and stroke evaluation in pacemaker patients and the atrial fibrillation reduction atrial pacing trial mechanisms of atrial fibrillation study. Circ. Arrhythm Electrophysiol. 2012, 5, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbar, D. Genetics of atrial fibrillation: Rare mutations, common polymorphisms, and clinical relevance. Heart Rhythm 2008, 5, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinor, P.T.; Yi, B.A.; MacRae, C.A. Genetics of atrial fibrillation. Med. Clin. N. Am. 2008, 92, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L. Familial Auricular Fibrillation. N. Engl. J. Med. 1943, 229, 396–398. [Google Scholar] [CrossRef]
- Fox, C.S.; Parise, H.; D’Agostino, R.B.S. Parental AF as a risk factor for AF in offspring. JAMA 2004, 291, 2851–2855. [Google Scholar] [CrossRef]
- Pastori, D.; Menichelli, D.; Lip, G.Y.H.; Sciacqua, A.; Violi, F.; Pignatelli, P.; ATHERO-AF Study Group (2020). Family history of atrial fibrillation and risk of cardiovascular events: A multicenter prospective cohort study. Circ. Arrhythm Electrophysiol. 2004, 13, e008477. [Google Scholar]
- Hucker, W.J.; Hanley, A.; Ellinor, P.T. Improving atrial fibrillation therapy: Is there a gene for that? J. Am. Coll. Cardiol. 2017, 69, 2088–2095. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J. Calcium sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiser, M.; Lederer, W.J.; Schotten, U. Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc. Res. 2010, 89, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Greiser, M.; Schotten, U. Dynamic remodeling of intracellular Ca2+ signaling during atrial fibrillation. J. Mol. Cell. Cardiol. 2013, 58, 134–142. [Google Scholar] [CrossRef]
- Voigt, N.; Dobrev, D. Cellular and molecular correlates of ectopic activity in patients with atrial fibrillation. Europace 2012, 14, v97–v105. [Google Scholar] [CrossRef] [PubMed]
- Christ, T. Atrial-selective Antiarrhythmic Activity by Vernakalant Fact or Fiction? J. Cardiovasc. Pharmacol. 2014, 63, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Berk, E.; Christ, T.; Schwarz, S.; Ravens, U.; Knaut, M.; Kaumann, A.J. In permanent atrial fibrillation, PDE3 reduces force responses to 5-HT, but PDE3 and PDE4 do not cause the blunting of atrial arrhythmias. Br. J. Pharmacol. 2016, 173, 2478–2489. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Feng, J.; Gaspo, R.; Li, G.-R.; Wang, Z.; Nattel, S. Ionic Remodeling Underlying Action Potential Changes in a Canine Model of Atrial Fibrillation. Circ. Res. 1997, 81, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gaspo, R.; Leblanc, N.; Nattel, S. Cellular mechanisms of atrial contractile dysfunction caused by sustained atrial tachycardia. Circulation 1998, 98, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schotten, U.; Duytschaever, M.; Ausma, J.; Eijsbouts, S.; Neuberger, H.-R.; Allessie, M. Electrical and Contractile Remodeling During the First Days of Atrial Fibrillation Go Hand in Hand. Circulation 2003, 107, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schotten, U.; Neuberger, H.-R.; Allessie, M.A. The role of atrial dilatation in the domestication of atrial fibrillation. Prog. Biophys. Mol. Biol. 2003, 82, 151–162. [Google Scholar] [CrossRef]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Burstein, B.; Nattel, S. Atrial Fibrosis: Mechanisms and Clinical Relevance in Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Smit, M.D.; Moes, M.L.; Maass, A.H.; Achekar, I.D.; Van Geel, P.P.; Hillege, H.L.; Van Veldhuisen, D.J.; Van Gelder, I.C. The importance of whether atrial fibrillation or heart failure develops first. Eur. J. Heart Fail. 2012, 14, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Pellman, J.; Lyon, R.C.; Sheikh, F. Extracellular matrix remodeling in atrial fibrosis: Mechanisms and implications in atrial fibrillation. J. Mol. Cell. Cardiol. 2010, 48, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Christiansen, C.; Mehnert, F.; Rothman, K.; Sørensen, H.T. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: Population based case-control study. BMJ 2011, 343, d3450. [Google Scholar] [CrossRef] [Green Version]
- Cardin, S.; Guasch, E.; Luo, X.; Naud, P.; Le Quang, K.; Shi, Y.; Tardif, J.-C.; Comtois, P.; Nattel, S. Role for MicroRNA-21 in Atrial Profibrillatory Fibrotic Remodeling Associated with Experimental Postinfarction Heart Failure. Circ. Arrhythmia Electrophysiol. 2012, 5, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lu, Y.; Yang, B. MicroRNAs and atrial fibrillation: New fundamentals. Cardiovasc. Res. 2010, 89, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, H.; Zhang, Y.; Lu, Y.; Pan, Z.; Cai, B.; Wang, N.; Xuelian, L.; Feng, T.; Hong, Y.; Yang, B. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc. Res. 2009, 83, 465–472. [Google Scholar] [CrossRef]
- Ling, T.-Y.; Wang, X.-L.; Chai, Q.; Lau, T.-W.; Koestler, C.M.; Park, S.J.; Daly, R.C.; Greason, K.L.; Jen, J.; Wu, L.-Q.; et al. Regulation of the SK3 channel by microRNA-499—Potential role in atrial fibrillation. Heart Rhythm 2013, 10, 1001–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lip, G.Y.; Fauchier, L.; Freedman, S.B.; Van Gelder, I.; Natale, A.; Gianni, C.; Nattel, S.; Potpara, T.; Rienstra, M.; Tse, H.F.; et al. Atrial fibrillation. Nat Rev Dis Primers 2016, 2, 16016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkiewicz, N.K.; Sanguinetti, M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993, 72, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salata, J.J.; Brooks, R.R. Pharmacology of Azimilide Dihydrochloride (NE-10064), A Class III Antiarrhythmic Agent. Cardiovasc. Drug Rev. 1997, 15, 137–156. [Google Scholar] [CrossRef]
- Light, P. Azimilide (Procter & Gamble). Drugs Investig. Drugs J. 2000, 3, 1534–1544. [Google Scholar]
- Karam, R.; Marcello, S.; Brooks, R.R.; E Corey, A.; Moore, A. Azimilide Dihydrochloride, a Novel Antiarrhythmic Agent. Am. J. Cardiol. 1998, 81, 40D–46D. [Google Scholar] [CrossRef]
- Connolly, S.J.; Schnell, D.J.; Page, R.L.; E Wilkinson, W.; Marcello, S.R.; Pritchett, E.L. Dose-response relations of azimilide in the management of symptomatic, recurrent, atrial fibrillation. Am. J. Cardiol. 2001, 88, 974–979. [Google Scholar] [CrossRef]
- Camm, A.J.; Pratt, C.M.; Schwartz, P.J.; Al-Khalidi, H.R.; Spyt, M.; Holroyde, M.J.; Karam, R.; Sonnenblick, E.H.; Brum, J.M. Azimilide Post Infarct Survival Evaluation (ALIVE): Azimilide does not affect mortality in post-myocardial infarction patients. Circulation 2001, 104, 104. [Google Scholar]
- Camm, A.J.; Pratt, C.M.; Schwartz, P.J.; Al-Khalidi, H.R.; Spyt, M.J.; Holroyde, M.J.; Karam, R.; Sonnenblick, E.H.; Brum, J.M. Azimilide post Infarct surVival Evaluation (ALIVE) Investigators. Mortality in patients after a recent myocardial infarction: A random-ized, placebo-controlled trial of azimilide using heart rate variability for risk stratification. Circulation 2004, 109, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, C.M.; Singh, S.N.; Al-Khalidi, H.R.; Brum, J.M.; Holroyde, M.J.; Marcello, S.R.; Schwartz, P.J.; Camm, A. The efficacy of azimilide in the treatment of atrial fibrillation in the presence of left ventricular systolic dysfunction: Results from the Azimilide Postinfarct Survival Evaluation (ALIVE) trial. J. Am. Coll. Cardiol. 2004, 43, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- So, P.P.-S.; Backx, P.H.; Dorian, P. Slow delayed rectifier K+ current block by HMR 1556 increases dispersion of repolarization and promotes Torsades de Pointes in rabbit ventricles. Br. J. Pharmacol. 2008, 155, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Gerlach, U.; Schmidt, D.; Nattel, S. In vivo electrophysiological effects of a selective slow delayed-rectifier potassium channel blocker in anesthetized dogs: Potential insights into class III actions. Cardiovasc. Res. 2004, 61, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.N.; Khrestian, C.; Carlsson, L.; Waldo, A.L. AZD7009: A new antiarrhythmic drug with predominant effects on the atria effectively terminates and prevents reinduction of atrial fibrillation and flutter in the sterile pericarditis model. J. Cardiovasc. Electrophysiol. 2004, 15, 1444–1450. [Google Scholar] [CrossRef]
- Persson, F.; Carlsson, L.; Duker, G.; Jacobson, I. Blocking Characteristics of hKv1.5 and hKv4.3/hKChIP2.2 After Administration of the Novel Antiarrhythmic Compound AZD7009. J. Cardiovasc. Pharmacol. 2005, 46, 7–17. [Google Scholar] [CrossRef]
- Wu, Y.; Carlsson, L.; Liu, T.; Kowey, P.R.; Yan, G.-X. Assessment of the Proarrhythmic Potential of the Novel Antiarrhythmic Agent AZD7009 and Dofetilide in Experimental Models of Torsades De Pointes. J. Cardiovasc. Electrophysiol. 2005, 16, 898–904. [Google Scholar] [CrossRef]
- Crijns, H.J.; Van Gelder, I.C.; Walfridsson, H.; Kulakowski, P.; Rónaszéki, A.; Dedek, V.; Malm, A.; Almgren, O. Safe and effective conversion of persistent atrial fibrillation to sinus rhythm by intravenous AZD7009. Heart Rhythm 2006, 3, 1321–1331. [Google Scholar] [CrossRef]
- Aunes-Jansson, M.; Edvardsson, N.; Stridh, M.; Sörnmo, L.; Frison, L.; Berggren, A. Decrease of the atrial fibrillatory rate, in-creased organization of the atrial rhythm and termination of atrial fibrillation by AZD7009. J. Electrocardiol. 2013, 46, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Aunes, M.; Egstrup, K.; Frison, L.; Berggren, A.; Stridh, M.; Sörnmo, L.; Edvardsson, N. Rapid slowing of the atrial fibrillatory rate after administration of AZD7009 predicts conversion of atrial fibrillation. J. Electrocardiol. 2014, 47, 316–323. [Google Scholar] [CrossRef]
- Hondeghem, L.M.; Snyders, D. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 1990, 81, 686–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondeghem, L.M.; Katzung, B.G. Antiarrhythmic agents: The modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu. Rev. Pharmacol. Toxicol. 1984, 24, 387–423. [Google Scholar] [CrossRef] [PubMed]
- Aimond, F.; Beck, L.; Gautier, P.; Chérif, O.K.; Davy, J.M.; Lorente, P.; Nisato, D.; Vassort, G. Cellular and in vivo electrophysio-logical effects of dronedarone in normal and postmyocardial infarcted rats. J. Pharmacol. Exp. Ther. 2000, 292, 415–424. [Google Scholar]
- Varró, A.; Takács, J.; Németh, M.; Hála, O.; Virág, L.; Iost, N.; Baláti, B.; Ágoston, M.; Vereckei, A.; Pastor, G.; et al. Electrophysiological effects of dronedarone (SR 33589), a noniodinated amiodarone derivative in the canine heart: Comparison with amiodarone. Br. J. Pharmacol. 2001, 133, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Connolly, S.J.; Crijns, H.J.; Roy, D.; Kowey, P.R.; Capucci, A.; Radzik, D.; Aliot, E.M.; Hohnloser, S.H. Dronedarone for Maintenance of Sinus Rhythm in Atrial Fibrillation or Flutter. N. Engl. J. Med. 2007, 357, 987–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, S.M.; Kean, S.J. Dronedarone. Drugs 2009, 69, 1647–1663. [Google Scholar] [CrossRef] [PubMed]
- Torp-Pedersen, C.; Crijns, H.J.; Gaudin, C.; Page, R.L.; Connolly, S.J.; Hohnloser, S.H.; ATHENA Investigators. Impact of dronedarone on hospitalization burden in patients with atrial fibrillation: Results from the ATHENA study. Europace 2011, 13, 1118–1126. [Google Scholar] [CrossRef]
- Burashnikov, A.; Belardinelli, L.; Antzelevitch, C. Acute dronedarone is inferior to amiodarone in terminating and preventing atrial fibrillation in canine atria. Heart Rhythm 2010, 7, 1273–1279. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.R.; Singh, S.N. Amiodarone and dronedarone: The worker bee and the drone? Heart Rhythm 2010, 7, 1280–1281. [Google Scholar] [CrossRef]
- Joghetaei, N.; Weirich, G.; Huber, W.; Büchler, P.; Estner, H. Acute Liver Failure Associated with Dronedarone. Circ. Arrhythmia Electrophysiol. 2011, 4, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Boriani, G.; Blomström-Lundqvist, C.; Hohnloser, S.H.; Bergfeldt, L.; Botto, G.L.; Capucci, A.; Lozano, I.F.; Goette, A.; Israel, C.W.; Merino, J.L.; et al. Safety and efficacy of dronedarone from clinical trials to real-world evidence: Implications for its use in atrial fibrillation. Europace 2019, 21, 1764–1775. [Google Scholar] [CrossRef]
- Jost, N.; Virag, L.; Hala, O.; Varro, A.; Thormahlen, D.; Papp, J.G. Effect of the Antifibrillatory Compound Tedisamil (KC-8857) on Transmembrane Currents in Mammalian Ventricular Myocytes. Curr. Med. Chem. 2004, 11, 3219–3228. [Google Scholar] [CrossRef]
- Beatch, G.N.; Abraham, S.; MacLeod, B.A.; Yoshida, N.R.; Walker, M. Antiarrhythmic properties of tedisamil (KC8857), a putative transient outward K+ current blocker. Br. J. Pharmacol. 1991, 102, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Flores, N.A. Tedisamil (Solvay). Curr. Opin. Investig. Drugs 2001, 2, 97–103. [Google Scholar] [PubMed]
- Barrett, T.D.; Hennan, J.K.; Fischbach, P.S.; O’Neill, B.P.; Driscoll, E.M.; Lucchesi, B.R. Tedisamil and dofetilide-induced torsades de pointes, rate and potassium dependence. Br. J. Pharmacol. 2001, 132, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Fischbach, P.S.; Johnston, P.V.; Friedrichs, G.S.; Lucchesi, B.R. Tedisamil in a Chronic Canine Model of Atrial Flutter. J. Cardiovasc. Pharmacol. 1999, 34, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, P.S.; Barrett, T.D.; Goyal, R.; Tran, B.C.; Syed, Z.A.; Hennan, J.K.; Lucchesi, B.R. Conversion of atrial fibrillation by the experi-mental antiarrhythmic drug tedisamil in two canine models. J. Cardiovasc. Electrophysiol. 2001, 12, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Hohnloser, S.H.; Dorian, P.; Straub, M.; Beckmann, K.; Kowey, P. Safety and efficacy of intravenously administered tedisamil for rapid conversion of recent-onset atrial fibrillation or atrial flutter. J. Am. Coll. Cardiol. 2004, 44, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Drugs.com. 2007. Available online: https://www.drugs.com/history/pulzium.html (accessed on 25 May 2021).
- Roden, D. Cardiology Today. FDA Panel Votes against Recommendation for Tedisamil. 2008. Available online: https://www.healio.com/news/cardiology/20120225/fda-panel-votes-against-recommendation-for-tedisamil (accessed on 25 May 2021).
- Wang, Z.; Fermini, B.; Nattel, S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ. Res. 1993, 73, 1061–1076. [Google Scholar] [CrossRef] [Green Version]
- Amos, G.J.; Wettwer, E.; Metzger, F.; Li, Q.; Himmel, H.M.; Ravens, U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J. Physiol. 1996, 491 Pt 1, 31–50. [Google Scholar] [CrossRef] [Green Version]
- Wettwer, E.; Hála, O.; Christ, T.; Heubach, J.F.; Dobrev, D.; Knaut, M.; Varró, A.; Ravens, U. Role of IKur in in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004, 110, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Wirth, K.J.; Paehler, T.; Rosenstein, B.; Knobloch, K.; Maier, T.; Frenzel, J.; Brendel, J.; Busch, A.E.; Bleich, M. Atrial effects of the novel K+-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc. Res. 2003, 60, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Christ, T.; Wettwer, E.; Voigt, N.; Hála, O.; Radicke, S.; Matschke, K.; Varró, A.; Dobrev, D.; Ravens, U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br. J. Pharmacol. 2008, 154, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Oros, A.; Volders, P.G.; Beekman, J.D.; van der Nagel, T.; Vos, M.A. Atrial-specific drug AVE0118 is free of torsades de pointes in anesthetized dogs with chronic complete atrioventricular block. Heart Rhythm 2006, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- De Haan, S.; Greiser, M.; Harks, E.; Blaauw, Y.; van Hunnik, A.; Verheule, S.; Allessie, M.; Schotten, U. AVE0118, blocker of the transient outward current (I(to)) and ultrarapid delayed rectifier current (I(Kur)), fully restores atrial contractility after cardio-version of atrial fibrillation in the goat. Circulation 2006, 114, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Blaauw, Y.; Schotten, U.; van Hunnik, A.; Neuberger, H.R.; Allessie, M.A. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc. Res. 2007, 75, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, K.J.; Steinmeyer, K.; Ruetten, H. Sensitization of upper airway mechanoreceptors as a new pharmacologic principle to treat obstructive sleep apnea: Investigations with AVE0118 in anesthetized pigs. Sleep 2013, 36, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Rivard, L.; Shiroshita-Takeshita, A.; Maltais, C.; Ford, J.; Pinnock, R.; Madge, D.; Nattel, S. Electrophysiological and atrial anti-arrhythmic effects of a novel IKur/Kv1.5 blocker in dogs. Heart Rhythm 2005, 2, S180. [Google Scholar] [CrossRef]
- Shiroshita-Takeshita, A.; Ford, J.; Madge, D.; Pinnock, R.; Nattel, S. Electrophysiological and atrial antiarrhythmic effects of a novel IKur/Kv1.5 blocker in dogs with atrial tachycardia remodeling. Heart Rhythm 2006, 3, S183. [Google Scholar] [CrossRef]
- Ford, J.; Milnes, J.; Wettwer, E.; Christ, T.; Rogers, M.; Sutton, K.; Madge, D.; Virág, L.; Jost, N.; Horváth, Z.; et al. Human electrophysiological and pharmacological properties of XEN-D0101: A novel atrial selective Kv1.5/IKur inhibitor. J. Cardiovasc. Pharmacol. 2013, 61, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, J.; Milnes, J.; El Haou, S.; Wettwer, E.; Loose, S.; Matschke, K.; Tyl, B.; Round, P.; Ravens, U. The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm 2016, 13, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Shunmugam, S.R.; Sugihara, C.; Freemantle, N.; Round, P.; Furniss, S.; Sulke, N. A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers al-lowing continuous beat-to-beat monitoring of drug efficacy. J. Interv. Card. Electrophysiol. 2018, 51, 191–197. [Google Scholar] [CrossRef]
- Stump, G.L.; Wallace, A.A.; Regan, C.P.; Lynch, J.J., Jr. In vivo antiarrhythmic and cardiac electrophysiologic effects of a novel diphenylphosphine oxide IKur blocker (2-isopropyl-5-methylcyclohexyl) diphenylphosphine oxide. J. Pharmacol. Exp. Ther. 2005, 315, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- Lagrutta, A.; Wang, J.; Fermini, B.; Salata, J.J. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, W.F.; Healey, J.S.; Bhatnagar, A.K.; Wang, P.; Gordon, J.A.; Baranchuk, A.; Deif, B.; Whitlock, R.P.; Belley-Côté, É.P. Vernakalant for cardioversion of recent-onset atrial fibrillation: A systematic review and meta-analysis. Europace 2019, 21, 1159–1166. [Google Scholar] [CrossRef]
- Fedida, D.; Orth, P.M.; Chen, J.Y.; Lin, S.; Plouvier, B.; Jung, G.; Ezrin, A.M.; Beatch, G.N. The mechanism of atrial antiarrhythmic action of RSD1235. J. Cardiovasc. Electrophysiol. 2005, 16, 1227–1238. [Google Scholar] [CrossRef]
- Wettwer, E.; Christ, T.; Endig, S.; Rozmaritsa, N.; Matschke, K.; Lynch, J.J.; Pourrier, M.; Gibson, J.K.; Fedida, D.; Knaut, M.; et al. The new antiarrhythmic drug vernakalant: Ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc. Res. 2013, 98, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burashnikov, A.; Pourrier, M.; Gibson, J.K.; Lynch, J.J.; Antzelevitch, C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel: Comparison with ranolazine and dl-sotalol. Circ. Arrhythm Electrophysiol. 2012, 5, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrev, D.; Hamad, B.; Kirkpatrick, P. Vernakalant. Nat. Rev. Drug Discov. 2010, 9, 915–916. [Google Scholar] [CrossRef]
- Van Middendorp, L.B.; Strik, M.; Houthuizen, P.; Kuiper, M.; Maessen, J.G.; Auricchio, A.; Prinzen, F.W. Electrophysiological and haemodynamic effects of vernakalant and flecainide in dyssynchronous canine hearts. Europace 2014, 16, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Camm, A.J.; Capucci, A.; Hohnloser, S.H.; Torp-Pedersen, C.; Van Gelder, I.C.; Mangal, B.; Beatch, G.; AVRO Investigators. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fi-brillation. J. Am. Coll. Cardiol. 2011, 57, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Cialdella, P.; Pedicino, D.; Santangeli, P. Novel Agents for the Acute Conversion of Atrial Fibrillation: Focus on Vernakalant. Recent Pat. Cardiovasc. Drug Discov. 2011, 6, 1–8. [Google Scholar] [CrossRef]
- Simon, A.; Niederdoeckl, J.; Janata, K.; Spiel, A.O.; Schuetz, N.; Schnaubelt, S.; Herkner, H.; Cacioppo, F.; Laggner, A.N.; Domanovits, H. Vernakalant and electrical cardioversion for AF—Safe and effective. IJC Heart Vasc. 2019, 24, 100398. [Google Scholar] [CrossRef]
- Hall, A.J.; Mitchell, A. Introducing Vernakalant into Clinical Practice. Arrhythmia Electrophysiol. Rev. 2019, 8, 70–74. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Burashnikov, A. Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J. Electrocardiol. 2009, 42, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Horváth, A.; Lemoine, M.D.; Löser, A.; Mannhardt, I.; Flenner, F.; Uzun, A.U.; Neuber, C.; Breckwoldt, K.; Hansen, A.; Girdauskas, E.; et al. Low resting membrane potential and low inward rectifier potassium currents are not inherent features of hiPSC-derived cardiomyocytes. Stem Cell Rep. 2018, 10, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Ravens, U.; Christ, T. Atrial-selective drugs for treatment of atrial fibrillation. Herzschrittmachertherapie Elektrophysiologie 2010, 21, 217–221. [Google Scholar] [CrossRef]
- Burashnikov, A.; Belardinelli, L.; Antzelevitch, C. Atrial-selective sodium channel block strategy to suppress atrial fibril-lation: Ranolazine versus propafenone. J. Pharmacol. Exp. Ther. 2012, 340, 161–168. [Google Scholar] [CrossRef]
- Szél, T.; Koncz, I.; Jost, N.; Baczkó, I.; Husti, Z.; Virág, L.; Bussek, A.; Wettwer, E.; Ravens, U.; Papp, J.G.; et al. Class I/B an-tiarrhythmic property of ranolazine, a novel antianginal agent, in dog and human cardiac preparations. Eur. J. Pharmacol. 2011, 668, 419–426. [Google Scholar]
- Flenner, F.; Friedrich, F.W.; Ungeheuer, N.; Christ, T.; Geertz, B.; Reischmann, S.; Wagner, S.; Stathopoulou, K.; Söhren, K.D.; Weinberger, F.; et al. Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc. Res. 2016, 109, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Sag, C.M.; Mallwitz, A.; Wagner, S.; Hartmann, N.; Schotola, H.; Fischer, T.H.; Ungeheuer, N.; Herting, J.; Shah, A.M.; Maier, L.S.; et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014, 76, 94–105. [Google Scholar] [CrossRef]
- Poulet, C.; Wettwer, E.; Grunnet, M.; Jespersen, T.; Fabritz, L.; Matschke, K.; Knaut, M.; Ravens, U. Late Sodium Current in Human Atrial Cardiomyocytes from Patients in Sinus Rhythm and Atrial Fibrillation. PLoS ONE 2015, 10, e0131432. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Rajamani, S.; Li, H.; January, C.T.; Shryock, J.C.; Belardinelli, L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na+ current in the heart. Am. J. Physiol. Circ. Physiol. 2009, 297, H1048–H1057. [Google Scholar] [CrossRef] [Green Version]
- Ravens, U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol. Ther. 2010, 128, 129–145. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Maier, L.S.; Mont, L.; Schwartz, P.J.; Simonis, G.; Leschke, M.; Gronda, E.; Boriani, G.; Darius, H.; Torán, L.G.; et al. Ranolazine in the treatment of atrial fibrillation: Results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following an ELectricaL CardiOversion) study. Heart Rhythm 2015, 2, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Reiffel, J.A.; Camm, A.J.; Belardinelli, L.; Zeng, D.; Karwatowska-Prokopczuk, E.; Olmsted, A.; Zareba, W.; Rosero, S.; Kowey, P. HARMONY Investigators. The HARMONY Trial: Combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: Mechanistic and therapeutic synergism. Circ. Arrhythm Electrophysiol. 2015, 8, 1048–1056. [Google Scholar] [CrossRef]
- Dobrev, D.; Friedrich, A.; Voigt, N.; Jost, N.; Wettwer, E.; Christ, T.; Knaut, M.; Ravens, U. The G Protein–Gated Potassium Current I K,ACh Is Constitutively Active in Patients With Chronic Atrial Fibrillation. Circulation 2005, 112, 3697–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettoni, M.; Zimmermann, M. Autonomic Tone Variations before the Onset of Paroxysmal Atrial Fibrillation. Circulation 2002, 105, 2753–2759. [Google Scholar] [CrossRef] [Green Version]
- Kovoor, P.; Wickman, K.; Maguire, C.T.; Pu, W.; Gehrmann, J.; Berul, C.I.; Clapham, D.E. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 2001, 37, 2136–2143. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Rozmaritsa, N.; Trausch, A.; Zimniak, T.; Christ, T.; Wettwer, E.; Matschke, K.; Dobrev, D.; Ravens, U. Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 381, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hashimoto, N. A Multiple Ion Channel Blocker, NIP-142, for the Treatment of Atrial Fibrillation. Cardiovasc. Drug Rev. 2007, 25, 342–356. [Google Scholar] [CrossRef]
- Hashimoto, N.; Yamashita, T.; Tsuruzoe, N. Characterization of in vivo and in vitro electrophysiological and anti-arrhythmic effects of a novel IK,ACh blocker, NIP-151: A comparison with an IKr-blocker dofetilide. J. Cardiovasc. Pharmacol. 2008, 51, 162–169. [Google Scholar] [CrossRef]
- Makary, S.; Voigt, N.; Maguy, A.; Wakili, R.; Nishida, K.; Harada, M.; Dobrev, D.; Nattel, S. Differential Protein Kinase C Isoform Regulation and Increased Constitutive Activity of Acetylcholine-Regulated Potassium Channels in Atrial Remodeling. Circ. Res. 2011, 109, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Kohajda, Z.; Kristóf, A.; Kovács, P.P.; Virág, L.; Varró, A.; Jost, N. The properties of the transient outward and ultra-rapid delayed rectifier potassium currents in canine atrial myocytes (FCVB 2010 Meeting-2010, abstract). Cardiovasc. Res. 2010, 87, S51. [Google Scholar]
- Juhász, V.; Hornyik, T.; Benák, A.; Nagy, N.; Husti, Z.; Pap, R.; Sághy, L.; Virág, L.; Varró, A.; Baczkó, I. Comparison of the effects of IK,ACh, IKr, and INa block in conscious dogs with atrial fibrillation and on action potentials in remodeled atrial trabeculae. Can. J. Physiol. Pharmacol. 2018, 96, 18–25. [Google Scholar] [CrossRef]
- Walfridsson, H.; Anfinsen, O.-G.; Berggren, A.; Frison, L.; Jensen, S.; Linhardt, G.; Nordkam, A.-C.; Sundqvist, M.; Carlsson, L. Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace 2015, 17, 473–482. [Google Scholar] [CrossRef]
- Podd, S.J.; Freemantle, N.; Furniss, S.S.; Sulke, N. First clinical trial of specific IK, ACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: Pacemaker assessment of BMS-914392 in patients with par-oxysmal atrial fibrillation. Europace 2016, 18, 340–346. [Google Scholar] [CrossRef]
- Pogwizd, S.M. Clinical potential of sodium-calcium exchanger inhibitors as antiarrhythmic agents. Drugs 2003, 63, 439–452. [Google Scholar] [CrossRef]
- Tóth, A.; Kiss, L.; Varró, A.; Nánási, P.P. Potential therapeutic effects of Na+/Ca2+ exchanger inhibition in cardiac diseases. Curr. Med. Chem. 2009, 16, 3294–3321. [Google Scholar] [CrossRef]
- Kovács, P.P.; Simon, J.; Christ, T.; Wettwer, E.; Varró, A.; Ravens, U. NCX current is increased in human chronic atrial fibril-lation: A possible explanation for contractile dysfunction? (FCVB 2010 Meeting-2010, abstract). Cardiovasc. Res. 2010, 87, S50. [Google Scholar]
- Elias, C.L.; Lukas, A.; Shurraw, S.; Scott, J.; Omelchenko, A.; Gross, G.J.; Hnatowich, M.; Hryshko, L.V. Inhibition of Na+/Ca2+ exchange by KB-R7943: Transport mode selectivity and antiarrhythmic consequences. Am. J. Physiol. Circ. Physiol. 2001, 281, H1334–H1345. [Google Scholar] [CrossRef]
- Birinyi, P.; Acsai, K.; Bányász, T.; Tóth, A.; Horváth, B.; Virág, L.; Szentandrássy, N.; Magyar, J.; Varró, A.; Fülöp, F.; et al. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyo-cytes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2005, 372, 63–70. [Google Scholar] [CrossRef]
- Schotten, U.; Greiser, M.; Benke, D.; Buerkel, K.; Ehrenteidt, B.; Stellbrink, C.; Vazquez-Jimenez, J.F.; Schoendube, F.; Hanrath, P.; Allessie, M. Atrial fibrillation-induced atrial contractile dysfunction: A tachycardiomyopathy of a different sort. Cardiovasc. Res. 2002, 53, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Kohajda, Z.; Farkas-Morvay, N.; Jost, N.; Nagy, N.; Geramipour, A.; Horváth, A.; Varga, R.S.; Hornyik, T.; Corici, C.; Acsai, K.; et al. The Effect of a Novel Highly Selective Inhibitor of the Sodium/Calcium Exchanger (NCX) on Cardiac Arrhythmias in In Vitro and In Vivo Experiments. PLoS ONE 2016, 11, e0166041. [Google Scholar] [CrossRef]
- Wolfes, J.; Ellermann, C.; Broer, N.; Rath, B.; Willy, K.; Leitz, P.R.; Lange, P.S.; Eckardt, L.; Frommeyer, G. Antiarrhythmic Effect of Ranolazine in Combination with Selective NCX-Inhibition in an Experimental Model of Atrial Fibrillation. Pharmaceuticals 2020, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Kanter, H.L.E.; Saffitz, J.; Beyer, E.C. Cardiac myocytes express multiple gap junction proteins. Circ. Res. 1992, 70, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.M.; Rodefeld, M.E.; Green, K.; Beyer, E.C.; Saffitz, J.E. Gap junction protein phenotypes of the human heart and con-duction system. J. Cardiovasc. Electrophysiol. 1995, 6, 813–822. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [Green Version]
- De Mazière, A.M.; Scheuermann, D.W. Structural changes in cardiac gap junctions after hypoxia and reoxygenation: A quantitative freeze-fracture analysis. Cell Tissue Res. 1990, 261, 183–194. [Google Scholar] [CrossRef]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 Associates with Nav1.5 in the Cardiomyocyte Perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; Krusemann, K.; Schaefer, T. Effects of the gap junction uncoupler palmitoleic acid on the activation and re-polarization wavefronts in isolated rabbit hearts. Br. J. Pharmacol. 1999, 128, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, E. Cardiac Ionic Currents and Acute Ischemia: From Channels to Arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef]
- Coronel, R.; Fiolet, J.W.; Wilms-Schopman, J.G.; Opthof, T.; Schaapherder, A.F.; Janse, M.J. Distribution of extracellular potassium and electrophysiologic changes during two-stage coronary ligation in the isolated, perfused canine heart. Circulation 1989, 80, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesh, M.D.; Pring, M.; Spear, J.F. Cellular uncoupling can unmask dispersion of action potential duration in ventricular myocardium. A computer modeling study. Circ. Res. 1989, 65, 1426–1440. [Google Scholar] [CrossRef] [Green Version]
- Varró, A.; Baczkó, I. Cardiac ventricular repolarization reserve: A principle for understanding drug-related proarrhythmic risk. Br. J. Pharmacol. 2011, 164, 14–36. [Google Scholar] [CrossRef] [Green Version]
- Zaniboni, M.; Pollard, A.E.; Yang, L.; Spitzer, K.W. Beat-to-beat repolarization variability in ventricular myocytes and its suppression by electrical coupling. Am. J. Physiol. Circ. Physiol. 2000, 278, H677–H687. [Google Scholar] [CrossRef] [Green Version]
- Spach, M.S.; Heidlage, J.F.; Barr, R.C.; Dolber, P.C. Cell size and communication: Role in structural and electrical development and remodeling of the heart. Heart Rhythm 2004, 1, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L.; Peters, N.S. The role of gap junctions in the arrhythmias of ischemia and infarction. Heart Rhythm 2012, 9, 308–311. [Google Scholar] [CrossRef]
- Kléber, A.G.; Rudy, Y. Basic Mechanisms of Cardiac Impulse Propagation and Associated Arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [Green Version]
- Polontchouk, L.; Haefliger, J.-A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, U.; Boldt, A.; Lauschke, J.; Weigl, J.; Schirdewahn, P.; Dorszewski, A.; Doll, N.; Hindricks, G.; Dhein, S.; Kottkamp, H. Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 2005, 91, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Morley, G.E.; Vaidya, D.; Liu, F.; Chen, F.L.; Stuhlmann, H.; Fishman, G. Heterogeneous Expression of Gap Junction Channels in the Heart Leads to Conduction Defects and Ventricular Dysfunction. Circulation 2001, 104, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Papp, R.; Gönczi, M.; Kovács, M.; Seprényi, G.; Végh, Á. Gap junctional uncoupling plays a trigger role in the antiarrhythmic effect of ischaemic preconditioning. Cardiovasc. Res. 2007, 74, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.-W. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: The action of antiarrhythmic peptides. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 381, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Haugan, K.; Miyamoto, T.; Takeishi, Y.; Kubota, I.; Nakayama, J.; Shimojo, H.; Hirose, M. Rotigaptide (ZP123) improves atrial conduction slowing in chronic volume overload-induced dilated atria. Basic Clin. Pharmacol. Toxicol. 2006, 99, 71–79. [Google Scholar] [CrossRef]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Keith, J.C., Jr.; Schaub, R.G.; Smith, R.P.; Feldman, H.S.; Haugan, K.; Kantrowitz, J.; Wang, P.J.; et al. Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduced infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J. Pharmacol. Exp. Ther. 2006, 317, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, T.; Finet, J.E.; Takeuchi, A.; Fujino, Y.; Strom, M.; Greener, I.D.; Rosenbaum, D.S.; Donahue, J.K. Connexin Gene Transfer Preserves Conduction Velocity and Prevents Atrial Fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Shiroshita-Takeshita, A.; Sakabe, M.; Haugan, K.; Hennan, J.K.; Nattel, S. Model-dependent effects of the gap junction con-duction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 2007, 115, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, J.M.; Everett TH 4th Lee, K.W.; Wilson, E.; Olgin, J.E. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 2006, 114, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; Leong-Poi, H.; Mangat, I.; Moe, G.W.; Hu, X.; So, P.P.-S.; Tarulli, E.; Ramadeen, A.; Rossman, E.I.; Hennan, J.K.; et al. Effects of Chronic Gap Junction Conduction–Enhancing Antiarrhythmic Peptide GAP-134 Administration on Experimental Atrial Fibrillation in Dogs. Circ. Arrhythmia Electrophysiol. 2009, 2, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Aonuma, S.; Kohama, Y.; Akai, K.; Komiyama, Y.; Nakajima, S.; Wakabayashi, M.; Makino, T. Studies on heart. XIX. Isolation of an atrial peptide that improves the rhythmicity of cultured myocardial cell clusters. Chem. Pharm. Bull. 1980, 28, 3332–3339. [Google Scholar] [CrossRef] [Green Version]
- Grover, R.; Dhein, S. Structure-activity relationships of novel peptides related to the antiarrhythmic peptide AAP10 which reduce the dispersion of epicardial action potential duration. Peptides 2001, 22, 1011–1021. [Google Scholar] [CrossRef]
- Kjolbye, A.L.; Knudsen, C.B.; Jepsen, T.; Larsen, B.D.; Petersen, J.S. Pharmacological characterization of the new stable anti-arrhythmic peptide analog Ac-D-Tyr-D-Pro-D-Hyp-Gly-D-Ala-Gly-NH2 (ZP123): In vivo and in vitro studies. J. Pharmacol. Exp. Ther. 2003, 306, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Butera, J.A.; Larsen, B.D.; Hennan, J.K.; Kerns, E.; Di, L.; Alimardanov, A.; Swillo, R.E.; Morgan, G.A.; Liu, K.; Wang, Q.; et al. Discovery of (2S,4R)-1-(2-Aminoacetyl)-4-benzamidopyrrolidine-2-carboxylic Acid Hydrochloride (GAP-134)13, an Orally Active Small Molecule Gap-Junction Modifier for the Treatment of Atrial Fibrillation. J. Med. Chem. 2009, 52, 908–911. [Google Scholar] [CrossRef]
- Müller, A.; Gottwald, M.; Tudyka, T.; Linke, W.; Klaus, W.; Dhein, S. Increase in gap junction conductance by an antiarrhythmic peptide. Eur. J. Pharmacol. 1997, 327, 65–72. [Google Scholar] [CrossRef]
- Hagen, A.; Dietze, A.; Dhein, S. Human cardiac gap-junction coupling: Effects of antiarrhythmic peptide AAP10. Cardiovasc. Res. 2009, 83, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.; Lauven, M.; Schaefer, T.; Polontchouk, L.; Grover, R.; Dhein, S. Pharmacological modification of gap junction coupling by an antiarrhythmic peptide via protein kinase C activation. FASEB J. 2002, 16, 1114–1116. [Google Scholar] [CrossRef]
- Easton, J.A.; Petersen, J.S.; Martin, P.E. The anti-arrhythmic peptide AAP10 remodels Cx43 and Cx40 expression and function. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 11–24. [Google Scholar] [CrossRef]
- Ueda, N.; Yamamoto, M.; Honjo, H.; Kodama, I.; Kamiya, K. The role of gap junctions in stretch-induced atrial fibrillation. Cardiovasc. Res. 2014, 104, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.C.; Lin, J.C.; Hung, C.Y.; Li, C.H.; Lin, S.F.; Yeh, H.I.; Huang, J.L.; Lo, C.P.; Haugan, K.; Larsen, B.D.; et al. Gap junction modifier rotigaptide decreases the susceptibility to ventricular arrhythmia by enhancing conduction velocity and suppressing discordant alternans during therapeutic hypothermia in isolated rabbit hearts. Heart Rhythm 2016, 13, 251–261. [Google Scholar] [CrossRef]
- Kjølbye, A.L.; Haugan, K.; Hennan, J.K.; Petersen, J.S. Pharmacological Modulation of Gap Junction Function with the Novel Compound Rotigaptide: A Promising New Principle for Prevention of Arrhythmias. Basic Clin. Pharmacol. Toxicol. 2007, 101, 215–230. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kléber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and Intracellular Redistribution of Ventricular Connexin43 During Electrical Uncoupling Induced by Ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Stahlhut, M.; Petersen, J.S.; Hennan, J.K.; Ramirez, M.T. The Antiarrhythmic Peptide Rotigaptide (ZP123) Increases Connexin 43 Protein Expression in Neonatal Rat Ventricular Cardiomyocytes. Cell Commun. Adhes. 2006, 13, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, T.C.; Thomas, D.; Petersen, J.S.; Evans, W.H.; Martin, P.E.M. The antiarrhythmic peptide rotigaptide (ZP123) increases gap junction intercellular communication in cardiac myocytes and HeLa cells expressing connexin 43. Br. J. Pharmacol. 2006, 147, 486–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.-P.; Zhang, X.-S.; Xiang, B.-R. Discontinued drugs in 2010: Cardiovascular drugs. Expert Opin. Investig. Drugs 2011, 20, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Rossman, E.I.; Liu, K.; Morgan, G.A.; Swillo, R.E.; Krueger, J.A.; Gardell, S.J.; Butera, J.; Gruver, M.; Kantrowitz, J.; Feldman, H.S.; et al. The gap junction modifier, GAP-134 [(2S,4R)-1-(2-aminoacetyl)-4-benzamido-pyrrolidine-2-carboxylic acid], improves conduction and reduces atrial fibrilla-tion/flutter in the canine sterile pericarditis model. J. Pharmacol. Exp. Ther. 2009, 329, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engstrøm, T.; Nepper-Christensen, L.; Helqvist, S.; Kløvgaard, L.; Holmvang, L.; Jørgensen, E.; Pedersen, F.; Saunamaki, K.; Tilsted, H.-H.; Steensberg, A.; et al. Danegaptide for primary percutaneous coronary intervention in acute myocardial infarction patients: A phase 2 randomised clinical trial. Heart 2018, 104, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Prasain, K.; Nguyen, T.D.; Hua, D.H.; Nguyen, T.A. The effect of the PQ1 anti-breast cancer agent on normal tissues. Anti-Cancer Drugs 2012, 23, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Bigelow, K.; Nguyen, T.A. Increase of gap junction activities in SW480 human colorectal cancer cells. BMC Cancer 2014, 14, 502. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Nguyen, T.A. PQ1, a quinoline derivative, induces apoptosis in T47D breast cancer cells through activation of caspase-8 and caspase-9. Apoptosis 2013, 18, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Cheng, C.-C.; Chen, Y.-C.; Kao, Y.-H.; Chen, S.-A.; Chen, Y.-J. Gap junction modifiers regulate electrical activities of the sinoatrial node and pulmonary vein: Therapeutic implications in atrial arrhythmogenesis. Int. J. Cardiol. 2016, 221, 529–536. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Lu, Y.-Y.; Cheng, C.-C.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. Sinoatrial node electrical activity modulates pulmonary vein arrhythmogenesis. Int. J. Cardiol. 2014, 173, 447–452. [Google Scholar] [CrossRef]
- Schmidt, C.; Wiedmann, F.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Cardiac two-pore-domain potassium channels (K2P): Physiology, pharmacology, and therapeutic potential. Dtsch. Med. Wochenschr. 2012, 137, 1654–1658. [Google Scholar]
- Skibsbye, L.; Poulet, C.; Diness, J.G.; Bentzen, B.H.; Yuan, L.; Kappert, U.; Matschke, K.; Wettwer, E.; Ravens, U.; Grunnet, M.; et al. Small-conductance calcium-activated potassisium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc. Res. 2014, 103, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Deng, C.; Wu, R.; Guo, H.; Zheng, S.; Yu, X.; Shan, Z.; Kuang, S.; Lin, Q. Decreased expression of small-conductance Ca2+-activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci. 2012, 90, 219–227. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, H.-J.; Che, H.; Sun, H.-Y.; Cheng, L.-C.; Li, X.; Au, W.-K.; Tse, H.F.; Li, G.-R. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflügers Archiv 2013, 465, 1439–1449. [Google Scholar] [CrossRef]
- Ninio, D.M.; Saint, D.A. The role of stretch-activated channels in atrial fibrillation and the impact of intracellular acidosis. Prog. Biophys. Mol. Biol. 2008, 97, 401–416. [Google Scholar] [CrossRef]
- Heidbüchel, H. A paradigm shift in treatment for atrial fibrillation: From electrical to structural therapy? Eur. Heart J. 2003, 24, 2077–2078. [Google Scholar] [CrossRef]
- Goette, A.; Bukowska, A.; Lendeckel, U. Non-ion channel blockers as anti-arrhythmic drugs (reversal of structural remod-eling). Curr. Opin. Pharmacol. 2007, 7, 219–224. [Google Scholar] [CrossRef]
- Dąbrowski, R.; Szwed, H. Antiarrhythmic potential of aldosterone antagonists in atrial fibrillation. Cardiol. J. 2012, 19, 223–229. [Google Scholar] [CrossRef]
- Zhou, X.; Du, J.-L.; Yuan, J.; Chen, Y.-Q. Statins Therapy Can Reduce the Risk of Atrial Fibrillation in Patients with Acute Coronary Syndrome: A Meta-Analysis. Int. J. Med Sci. 2013, 10, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Berkowitsch, A.; Neumann, T.; Kuniss, M.; Janin, S.; Wojcik, M.; Zaltsberg, S.; Mitrovic, V.; Pitschner, H.F. Therapy with Ren-in-Angiotensin system blockers after pulmonary vein isolation in patients with atrial fibrillation: Who is a responder? Pacing Clin. Electrophysiol. 2010, 33, 1101–1111. [Google Scholar] [CrossRef]
- Veronese, G.; Montomoli, J.; Schmidt, M.; Horváth-Puhó, E.; Sørensen, H.T. Statin use and risk of atrial fibrillation or flutter: A population-based case-control study. Am. J. Ther. 2015, 22, 186–194. [Google Scholar] [CrossRef]
- Fauchier, L.; Clementy, N.; Babuty, D. Statin therapy and atrial fibrillation: Systematic review and updated meta-analysis of published randomized controlled trials. Curr. Opin. Cardiol. 2013, 28, 7–18. [Google Scholar] [CrossRef]
- Fang, W.T.; Li, H.J.; Zhang, H.; Jiang, S. The role of statin therapy in the prevention of atrial fibrillation: A meta-analysis of randomized controlled trials. Br. J. Clin. Pharmacol. 2012, 74, 744–756. [Google Scholar] [CrossRef] [Green Version]
- Alegret, J.M.; Aragonès, G.; Elosua, R.; Beltrán-Debón, R.; Hernández-Aguilera, A.; Romero-Menor, C.; Camps, J.; Joven, J. The relevance of the association between inflammation and atrial fibrillation. Eur. J. Clin. Investig. 2013, 43, 324–331. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
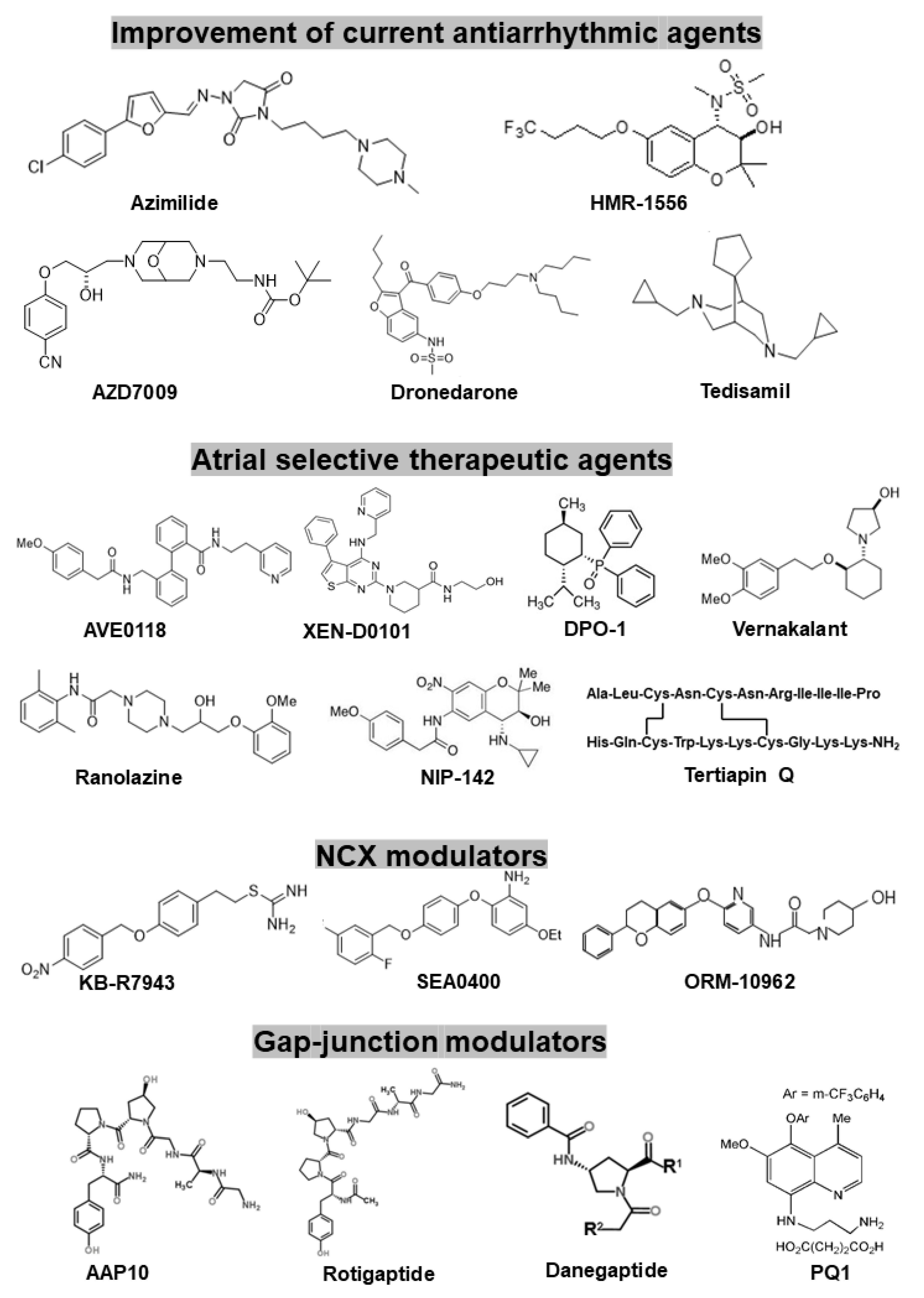
| Name of Compounds or Drugs | Major Effects | Clinical Studies | |
|---|---|---|---|
| Further development and improvement of existing antiarrhythmic drugs or compounds | Azimilide (FDA approval) | primarily IKr and IKs blocker but additionally blocks ICaL and INa (multi-channel blocker) | ALIVE, A-STAR, A-COMET I and II Studies |
| HMR-1556 | highly selective IKs blocker | not performed | |
| AZD7009 | primarily IKr and INa blocker, but additionally blocks Ito, IKur and IKs (multi-channel blocker) | small single centre clinical trial | |
| Dronedarone (FDA approval) | Amiodarone-like multichannel blocker (INa, ICa, IKr blocker) | ADONIS, ATHENA, EURIDIS etc. | |
| Tedisamil | multichannel blocker (INa, Ito, IKr, IKs, IKATP, blocker) | small single centre clinical trial | |
| Atrial selective therapeutic agents (existing drugs or investigational compounds) | AVE0118 | primarily IKur, Ito and IK,ACh blocker | not carried out |
| XEN-D0101 | highly selective IKur blocker | small single centre clinical trial | |
| DP01 | highly selective IKur blocker | not carried out | |
| Vernakalant | primarily IKr and INa blocker, but additionally blocks Ito, INa, IKr and IKs (multichannel blocker) | AVRO | |
| Ranolazine (FDA approval) | primarily INaf and INaL, and IKr blocker, but additionally blocks ICaL and IKs (multichannel blocker) | MERLIN-TIMI | |
| NIP-142, NIP-152 | highly selective IK,ACh blockers | not carried out | |
| Tertiapin Q | highly selective IK,ACh blocker | not carried out | |
| NCX modulators | KB-R7943 | initially developed as selective NCX blocker, but additionally blocks Ito, IK, IK1, INa, and ICaL | not carried out |
| SEA-0400 | selective NCX blocker, but additionally blocks ICaL | not carried out | |
| ORM-10962 | highly selective and potent NCX blocker | not carried out | |
| Gap-junction modulators | AAP10 | selective gap junction enhancer peptide | not carried out |
| Rotigaptide | selective gap junction enhancer peptide | ClinicalTrials.gov Identifier NCT00137332 | |
| Danegaptide (GAP-134) | selective gap junction enhancer peptide | ClinicalTrials.gov Identifier NCT00510029, NCT00543946, and NCT00783341 | |
| PQ1 | selective gap junction enhancer | not carried out |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jost, N.; Christ, T.; Magyar, J. New Strategies for the Treatment of Atrial Fibrillation. Pharmaceuticals 2021, 14, 926. https://doi.org/10.3390/ph14090926
Jost N, Christ T, Magyar J. New Strategies for the Treatment of Atrial Fibrillation. Pharmaceuticals. 2021; 14(9):926. https://doi.org/10.3390/ph14090926
Chicago/Turabian StyleJost, Norbert, Torsten Christ, and János Magyar. 2021. "New Strategies for the Treatment of Atrial Fibrillation" Pharmaceuticals 14, no. 9: 926. https://doi.org/10.3390/ph14090926
APA StyleJost, N., Christ, T., & Magyar, J. (2021). New Strategies for the Treatment of Atrial Fibrillation. Pharmaceuticals, 14(9), 926. https://doi.org/10.3390/ph14090926