
Adsorption and Sustained Delivery of Small Molecules from Nanosilicate Hydrogel Composites



Abstract
:
1. Introduction
2. Results
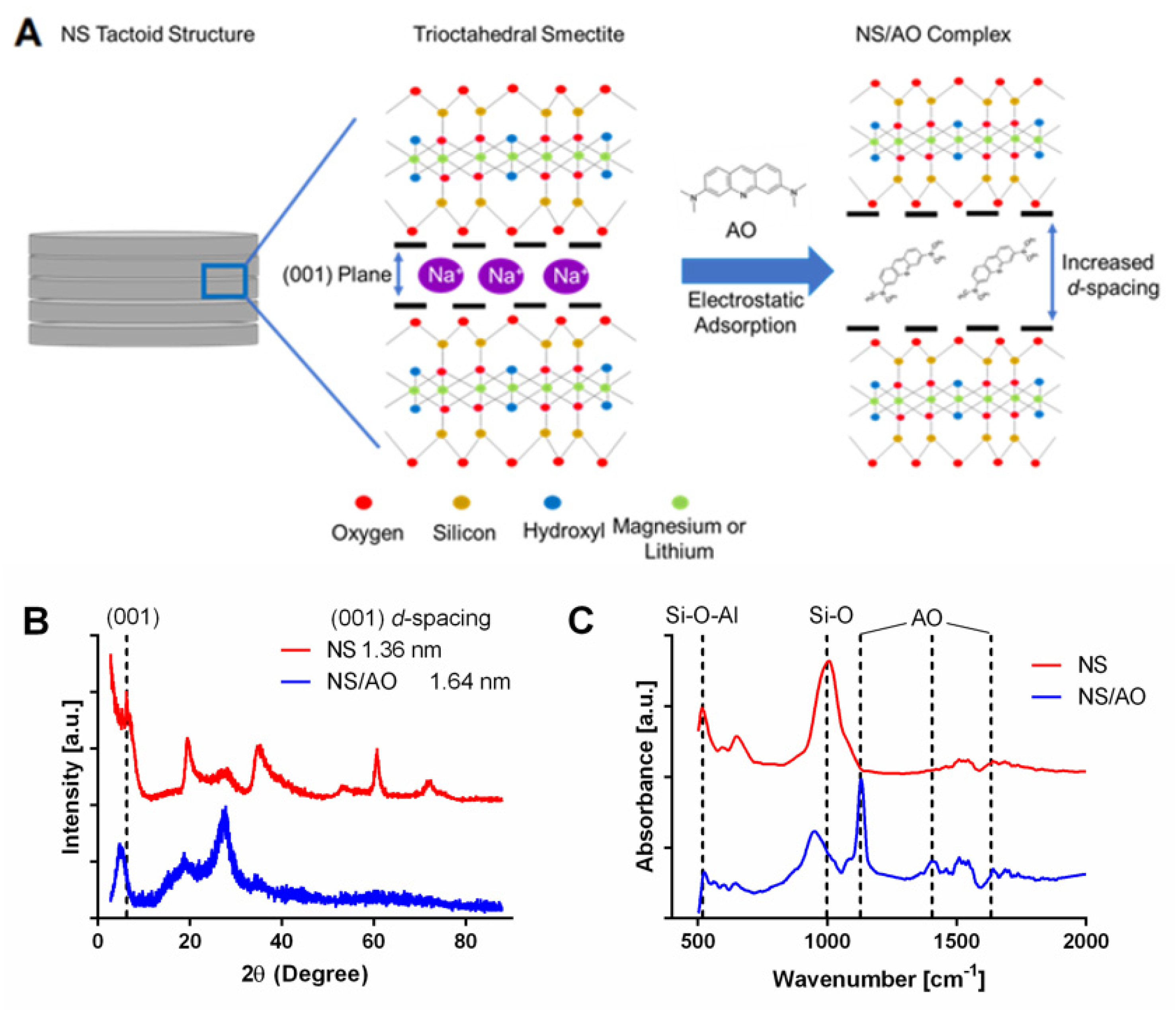
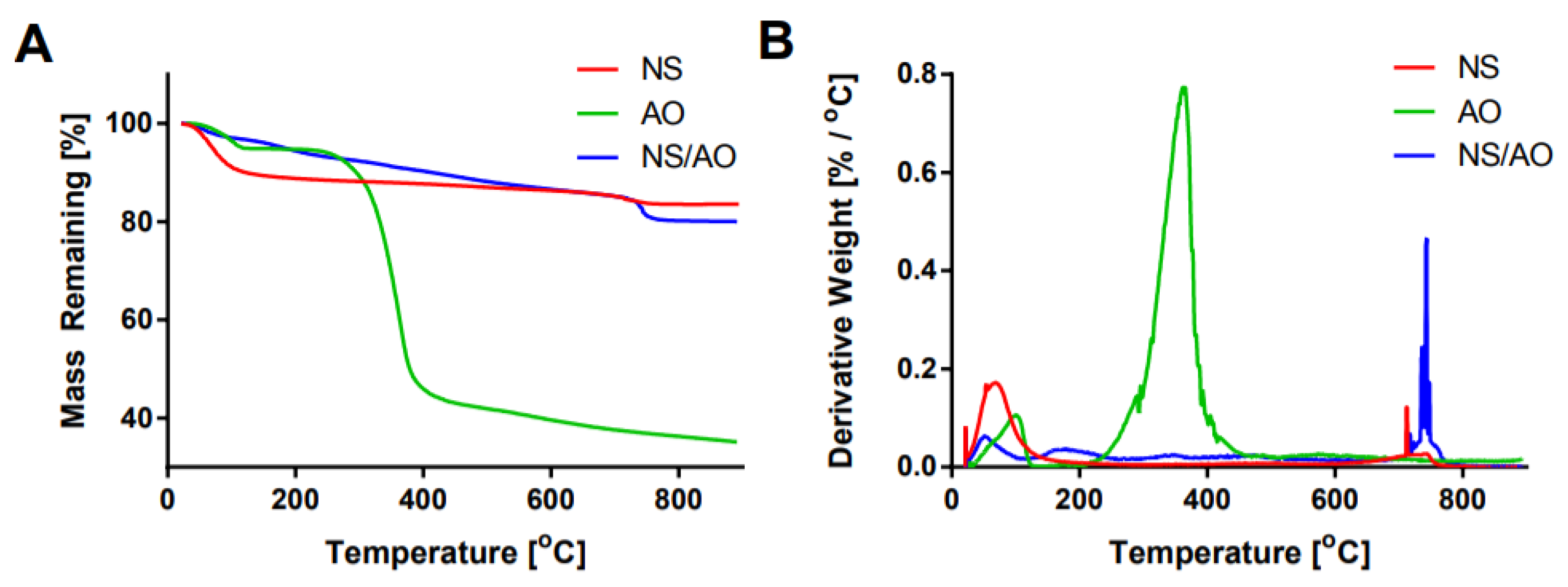
2.1. Characterization of AO Adsorption onto and Intercalation with NS to Form NS/AO Complexes
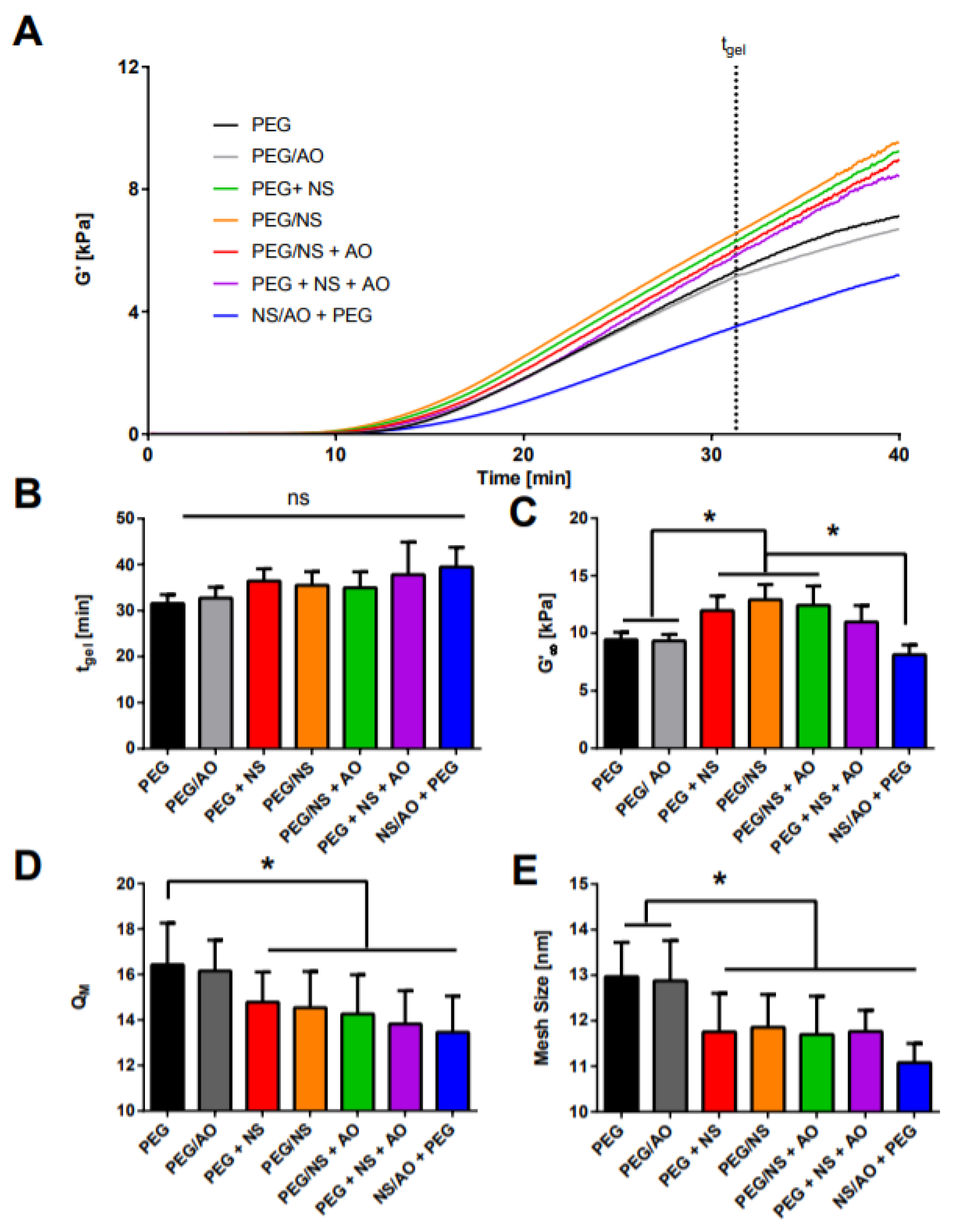
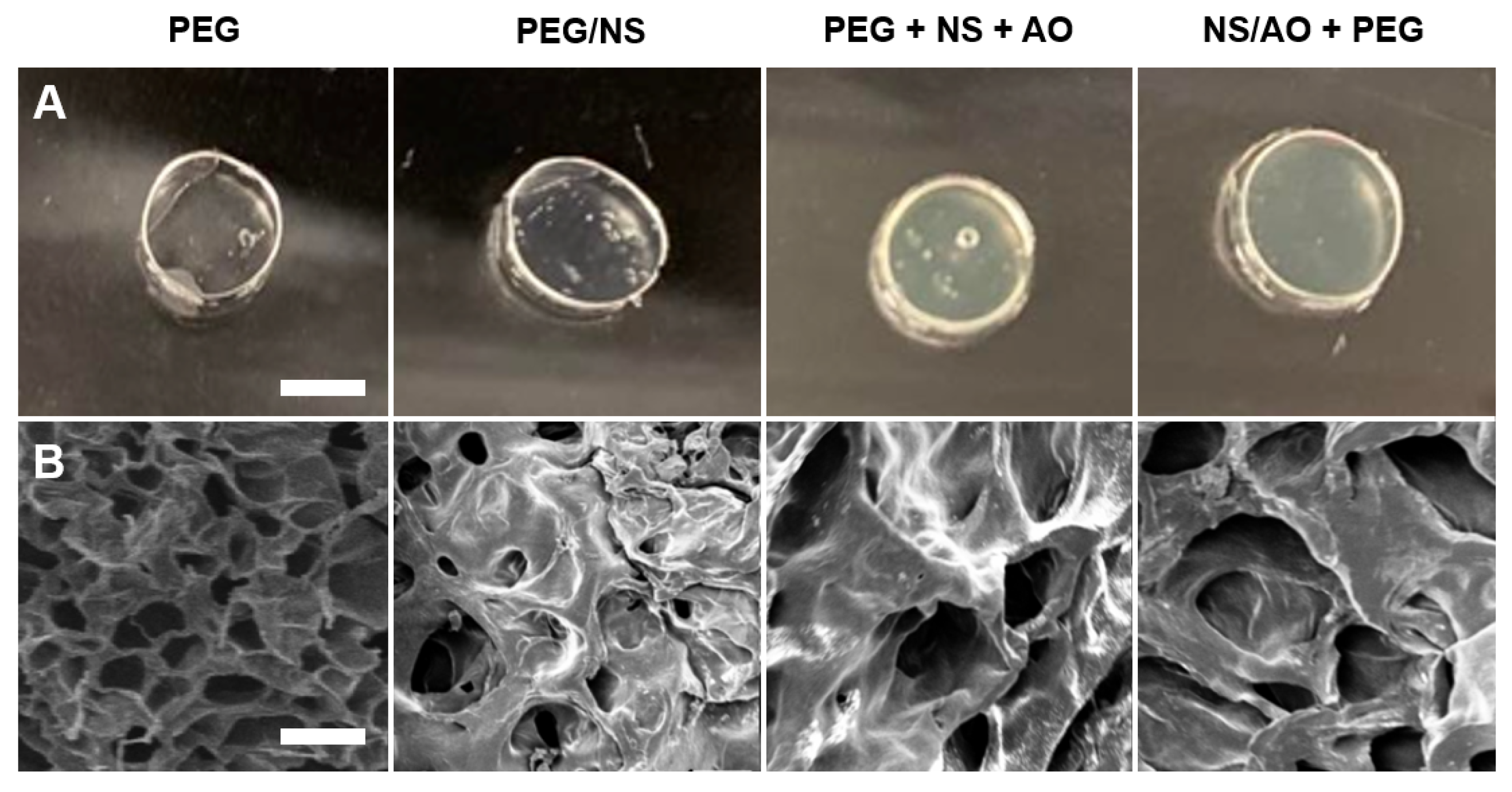
2.2. Mechanical and Physical Properties of NS-Hydrogel Composites
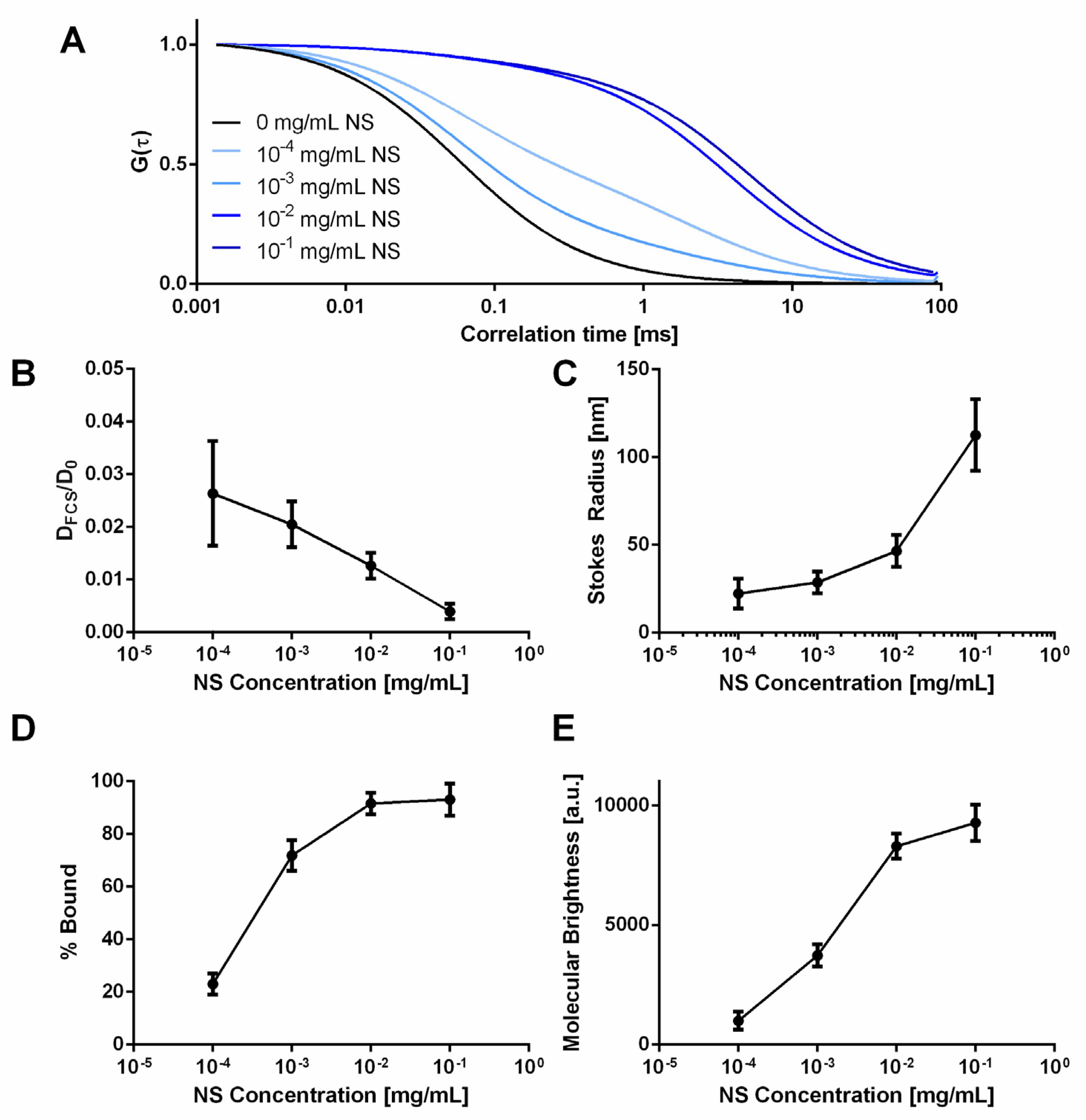
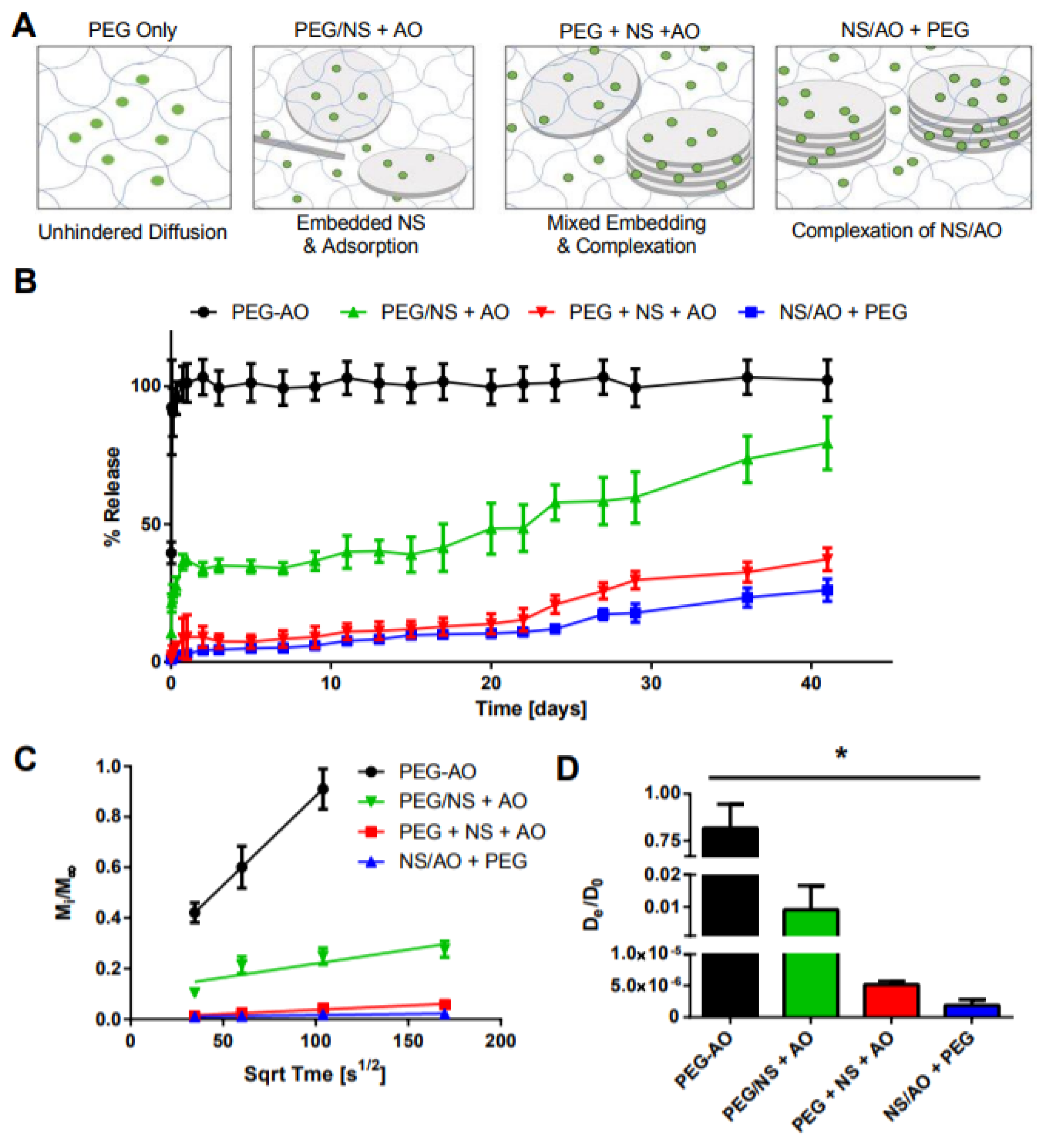
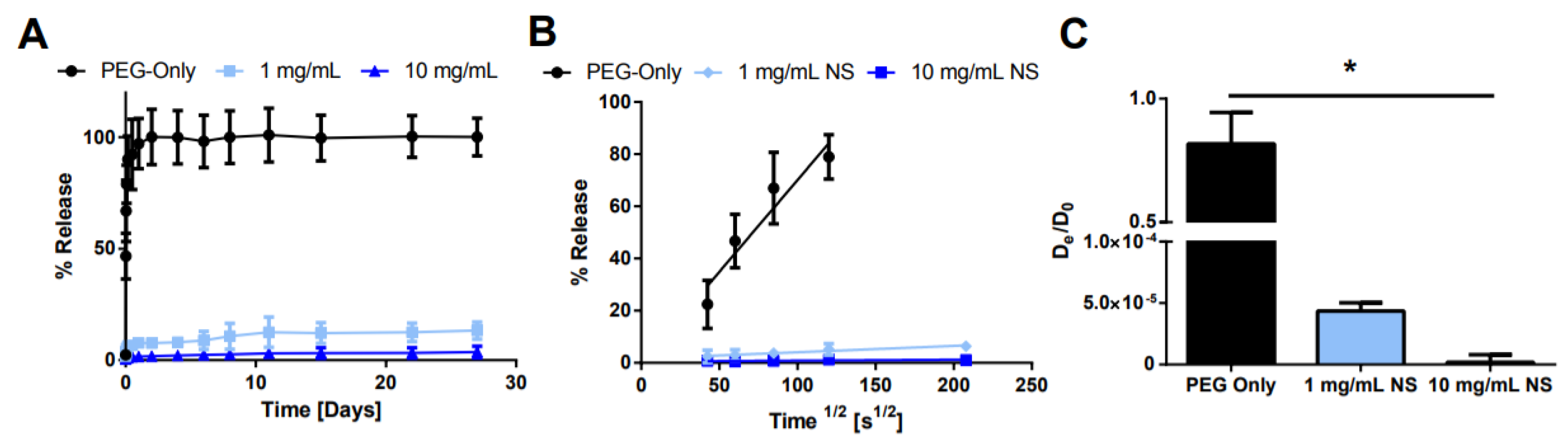
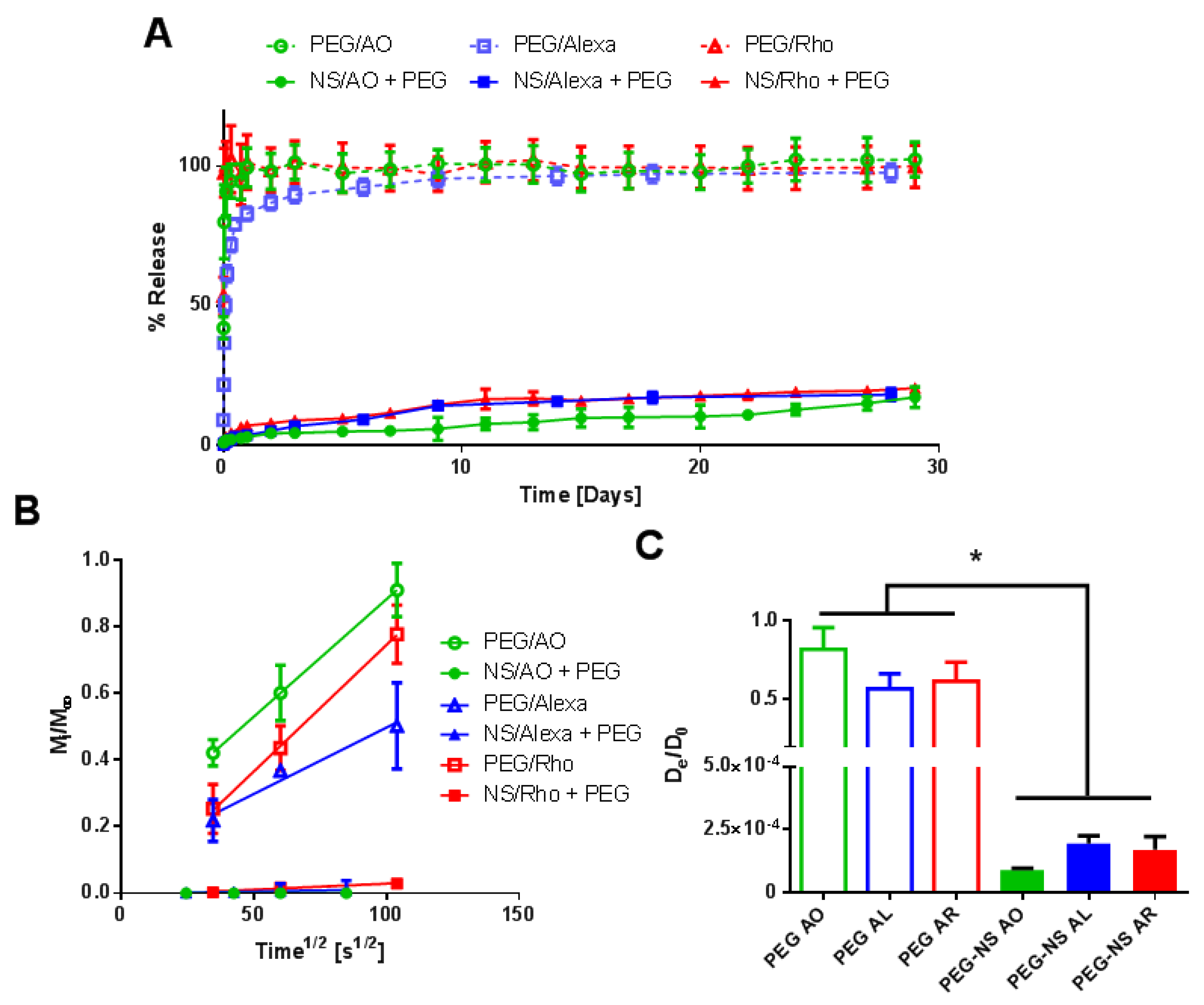
2.3. Characterization of Small Molecule Diffusivity and Release from Nanocomposite Hydrogels
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Characterization of NS–AO Interactions
4.3. Fluorescence Correlation Spectroscopy
4.4. Hydrogel Fabrication
4.5. Rheological Measurements
4.6. Hydrogel Swelling and Mesh Size
4.7. Scanning Electron Microscopy
4.8. Bulk Release Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vigata, M.; Meinert, C.; Hutmacher, D.W.; Bock, N. Hydrogels as Drug Delivery Systems: A Review of Current Characterization and Evaluation Techniques. Pharmaceutics 2020, 12, 1188. [Google Scholar] [CrossRef]
- Tong, X.; Lee, S.; Bararpour, L.; Yang, F. Long-Term Controlled Protein Release from Poly (Ethylene Glycol) Hydrogels by Modulating Mesh Size and Degradation. Macromol. Biosci. 2015, 15, 1679–1686. [Google Scholar] [CrossRef] [Green Version]
- Zustiak, S.P.; Leach, J.B. Characterization of protein release from hydrolytically degradable poly(ethylene glycol) hydrogels. Biotechnol. Bioeng. 2011, 108, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S. Understanding the burst release phenomenon: Toward designing effective nanoparticulate drug-delivery systems. Ther. Deliv. 2020, 12, 21–36. [Google Scholar] [CrossRef]
- Hezaveh, H.; Muhamad, I.I. Controlled drug release via minimization of burst release in pH-response kappa-carrageenan/polyvinyl alcohol hydrogels. Chem. Eng. Res. Des. 2013, 91, 508–519. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Ashley, G.W.; Henise, J.; Reid, R.; Santi, D.V. Hydrogel Drug Delivery System with Predictable and Tunable Drug Release and Degradation Rates. Proc. Natl. Acad. Sci. USA 2013, 110, 2318–2323. [Google Scholar] [CrossRef] [Green Version]
- DuBose, J.W.; Cutshall, C.; Metters, A.T. Controlled release of tethered molecules via engineered hydrogel degradation: Model development and validation. J. Biomed. Mater. Res. Part A 2005, 74, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S.; Stayton, P.S. Conjugates of stimuli-responsive polymers and proteins. Prog. Polym. Sci. 2007, 32, 922–932. [Google Scholar] [CrossRef]
- Mauri, E.; Negri, A.; Rebellato, E.; Masi, M.; Perale, G.; Rossi, F. Hydrogel-Nanoparticles Composite System for Controlled Drug Delivery. Gels 2018, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Sheikhi, A.; Afewerki, S.; Oklu, R.; Gaharwar, A.K.; Khademhosseini, A. Effect of ionic strength on shear-thinning nanoclay-polymer composite hydrogels. Biomater. Sci. 2018, 6, 2073–2083. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Cross, L.M.; Peak, C.W.; Gold, K.; Carrow, J.K.; Brokesh, A.; Singh, K.A. 2D Nanoclay for Biomedical Applications: Regenerative Medicine, Therapeutic Delivery, and Additive Manufacturing. Adv. Mater. 2019, 31, 1900332. [Google Scholar] [CrossRef]
- Jansson, M.; Belić, D.; Forsman, J.; Skepö, M. Nanoplatelet interactions in the presence of multivalent ions: The effect of overcharging and stability. J. Colloid Interface Sci. 2020, 579, 573–581. [Google Scholar] [CrossRef]
- Cross, L.M.; Carrow, J.K.; Ding, X.; Singh, K.A.; Gaharwar, A.K. Sustained and Prolonged Delivery of Protein Therapeutics from Two-Dimensional Nanosilicates. ACS Appl Mater. Interfaces 2019, 11, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; Lenton, S.; Plivelic, T.S.; Skepö, M. Intercalation of cationic peptides within Laponite layered clay minerals in aqueous suspensions: The effect of stoichiometry and charge distance matching. J. Colloid Interface Sci. 2019, 557, 767–776. [Google Scholar] [CrossRef]
- Jatav, S.; Joshi, Y.M. Chemical stability of Laponite in aqueous media. Appl. Clay Sci. 2014, 97, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Ghadiri, M.; Chrzanowski, W.; Lee, W.H.; Fathi, A.; Dehghani, F.; Rohanizadeh, R. Physico-chemical, mechanical and cytotoxicity characterizations of Laponite®/alginate nanocomposite. Appl. Clay Sci. 2013, 85, 64–73. [Google Scholar] [CrossRef]
- Liu, B.; Li, J.; Lei, X.; Miao, S.; Zhang, S.; Cheng, P.; Song, Y.; Wu, H.; Gao, Y.; Bi, L.; et al. Cell-loaded injectable gelatin/alginate/LAPONITE® nanocomposite hydrogel promotes bone healing in a critical-size rat calvarial defect model. RSC Adv. 2020, 10, 25652–25661. [Google Scholar] [CrossRef]
- Ghadiri, M.; Hau, H.; Chrzanowski, W.; Agus, H.; Rohanizadeh, R. Laponite clay as a carrier for in situ delivery of tetracycline. RSC Adv. 2013, 3, 20193–20201. [Google Scholar] [CrossRef]
- Ghadiri, M.; Chrzanowski, W.; Lee, W.H.; Rohanizadeh, R. Layered silicate clay functionalized with amino acids: Wound healing application. RSC Adv. 2014, 4, 35332–35343. [Google Scholar] [CrossRef]
- Gonçalves, M.; Figueira, P.; Maciel, D.; Rodrigues, J.; Qu, X.; Liu, C.; Tomás, H.; Li, Y. pH-sensitive Laponite®/doxorubicin/alginate nanohybrids with improved anticancer efficacy. Acta Biomater. 2014, 10, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Li, Z.; Jiang, W.-T.; Chang, P.-H.; Jean, J.-S.; Lin, K.-H. Mechanism of acridine orange removal from water by low-charge swelling clays. Chem. Eng. J. 2011, 174, 603–611. [Google Scholar] [CrossRef]
- Koshy, S.T.; Zhang, D.K.Y.; Grolman, J.M.; Stafford, A.G.; Mooney, D.J. Injectable nanocomposite cryogels for versatile protein drug delivery. Acta Biomater. 2018, 65, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gong, Z.; Huang, X.; Wang, J.; Xia, K.; Ying, L.; Shu, J.; Yu, C.; Zhou, X.; Li, F.; et al. An injectable heparin-Laponite hydrogel bridge FGF4 for spinal cord injury by stabilizing microtubule and improving mitochondrial function. Theranostics 2019, 9, 7016–7032. [Google Scholar] [CrossRef]
- Câmara, G.B.M.; Barbosa, R.D.M.; García-Villén, F.; Viseras, C.; Almeida Júnior, R.F.D.; Machado, P.R.L.; Câmara, C.A.; Farias, K.J.S.; de Lima e Moura, T.F.A.; Dreiss, C.A.; et al. Nanocomposite gels of poloxamine and Laponite for β-Lapachone release in anticancer therapy. Eur. J. Pharm. Sci. 2021, 163, 105861. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Yang, X.; Shi, L.; Lanham, S.A.; Hilborn, J.; Oreffo, R.O.C.; Ossipov, D.; Dawson, J.I. Bisphosphonate nanoclay edge-site interactions facilitate hydrogel self-assembly and sustained growth factor localization. Nat. Commun. 2020, 11, 1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimen, Z.; Babadag, S.; Odabas, S.; Altuntas, S.; Demirel, G.; Demirel, G.B. Injectable and Self-Healable pH-Responsive Gelatin–PEG/Laponite Hybrid Hydrogels as Long-Acting Implants for Local Cancer Treatment. ACS Appl. Polym. Mater. 2021, 3, 3504–3518. [Google Scholar] [CrossRef]
- Mignon, A.; Pezzoli, D.; Prouvé, E.; Lévesque, L.; Arslan, A.; Pien, N.; Schaubroeck, D.; Van Hoorick, J.; Mantovani, D.; Van Vlierberghe, S.; et al. Combined effect of Laponite and polymer molecular weight on the cell-interactive properties of synthetic PEO-based hydrogels. React. Funct. Polym. 2019, 136, 95–106. [Google Scholar] [CrossRef]
- Chang, C.-W.; van Spreeuwel, A.; Zhang, C.; Varghese, S. PEG/clay nanocomposite hydrogel: A mechanically robust tissue engineering scaffold. Soft Matter 2010, 6, 5157–5164. [Google Scholar] [CrossRef]
- Khelifi, S.; Mallah, B.; Ayadi, M.T.; Oueslati, M.H.; Sbihi, H.M.; Ayari, F. Performance of a Local Clay Deposit in Adsorptive and Photochemical Removal of Acridine Orange Dye and DNA Indicator from Wastewater. Desalination Water Treat. 2020, 206, 396–406. [Google Scholar] [CrossRef]
- Xiong, Z.-Q.; Li, X.-D.; Fu, F.; Li, Y.-N. Performance evaluation of laponite as a mud-making material for drilling fluids. Pet. Sci. 2019, 16, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Lagutschenkov, A.; Dopfer, O. Infrared spectrum of a protonated fluorescence dye: Acridine orange. J. Mol. Spectrosc. 2011, 268, 66–77. [Google Scholar] [CrossRef]
- Imamura, K.; Ikeda, E.; Nagayasu, T.; Sakiyama, T.; Nakanishi, K. Adsorption behavior of methylene blue and its congeners on a stainless steel surface. J. Colloid Interface Sci 2002, 245, 50–57. [Google Scholar] [CrossRef]
- Guggenheim, S.; Martin, R.T. Definition of clay and clay mineral: Joint report of the AIPEA and CMS Nomenclature Committees. Clay Miner. 1995, 30, 257–259. [Google Scholar] [CrossRef]
- Rehmann, M.S.; Skeens, K.M.; Kharkar, P.M.; Ford, E.M.; Maverakis, E.; Lee, K.H.; Kloxin, A.M. Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules 2017, 18, 3131–3142. [Google Scholar] [CrossRef]
- Shah, K.; Vasileva, D.; Karadaghy, A.; Zustiak, S.P. Development and characterization of polyethylene glycol-carbon nanotube hydrogel composite. J. Mater. Chem B 2015, 3, 7950–7962. [Google Scholar] [CrossRef]
- Stealey, S.T.; Gaharwar, A.K.; Pozzi, N.; Zustiak, S.P. Development of Nanosilicate–Hydrogel Composites for Sustained Delivery of Charged Biopharmaceutics. ACS Appl. Mater. Interfaces 2021, 13, 27880–27894. [Google Scholar] [CrossRef]
- Chen, P.; Xu, S.; Wu, R.; Wang, J.; Gu, R.; Du, J. A transparent Laponite polymer nanocomposite hydrogel synthesis via in-situ copolymerization of two ionic monomers. Appl. Clay Sci. 2013, 72, 196–200. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. In situ forming poly(ethylene glycol)-based hydrogels via thiol-maleimide Michael-type addition. J. Biomed. Mater. Res. Part A 2011, 98, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Loebus, A.; de Vicente, G.; Ren, F.; Arafeh, M.; Ouyang, Z.; Lensen, M.C. Synthesis of Poly(ethylene glycol)-based Hydrogels via Amine-Michael Type Addition with Tunable Stiffness and Postgelation Chemical Functionality. Chem. Mater. 2014, 26, 3624–3630. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Das, R.; Bhattacharjee, C. 23–Hydrogel nanocomposite for controlled drug release. In Applications of Nanocomposite Materials in Drug Delivery; Inamuddin, A.A.M., Mohammad, A., Eds.; Woodhead Publishing: Sawston, UK, 2018; pp. 575–588. [Google Scholar] [CrossRef]
- Das, S.S.; Neelam; Hussain, K.; Singh, S.; Hussain, A.; Faruk, A.; Tebyetekerwa, M. Laponite-based Nanomaterials for Biomedical Applications: A Review. Curr. Pharm. Des. 2019, 25, 424–443. [Google Scholar] [CrossRef]
- Becher, T.B.; Mendonça, M.C.P.; de Farias, M.A.; Portugal, R.V.; de Jesus, M.B.; Ornelas, C. Soft Nanohydrogels Based on Laponite Nanodiscs: A Versatile Drug Delivery Platform for Theranostics and Drug Cocktails. ACS Appl. Mater. Interfaces 2018, 10, 21891–21900. [Google Scholar] [CrossRef]
- Li, Y.; Maciel, D.; Tomás, H.; Rodrigues, J.; Ma, H.; Shi, X. pH sensitive Laponite/alginate hybrid hydrogels: Swelling behaviour and release mechanism. Soft Matter 2011, 7, 6231–6238. [Google Scholar] [CrossRef]
- Wang, S.; Wu, Y.; Guo, R.; Huang, Y.; Wen, S.; Shen, M.; Wang, J.; Shi, X. Laponite Nanodisks as an Efficient Platform for Doxorubicin Delivery to Cancer Cells. Langmuir 2013, 29, 5030–5036. [Google Scholar] [CrossRef]
- Zustiak, S.P.; Leach, J.B. Hydrolytically degradable poly(ethylene glycol) hydrogel scaffolds with tunable degradation and mechanical properties. Biomacromolecules 2010, 11, 1348–1357. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.E.; LeMay, H.E.; Bursten, B.E.; Murphy, C.; Woodward, P.; Stoltzfus, M. Chemistry: The Central Science, 14th ed.; Pearson: London, UK, 2018; p. 21739. [Google Scholar]
- Wang, B.; Zhou, M.; Rozynek, Z.; Fossum, J.O. Electrorheological properties of organically modified nanolayered laponite: Influence of intercalation, adsorption and wettability. J. Mater. Chem. 2009, 19, 1816–1828. [Google Scholar] [CrossRef]
- Martinez, A.W.; Caves, J.M.; Ravi, S.; Li, W.; Chaikof, E.L. Effects of crosslinking on the mechanical properties, drug release and cytocompatibility of protein polymers. Acta Biomater. 2014, 10, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Arno, M.C.; Inam, M.; Weems, A.C.; Li, Z.; Binch, A.L.A.; Platt, C.I.; Richardson, S.M.; Hoyland, J.A.; Dove, A.P.; O’Reilly, R.K. Exploiting the role of nanoparticle shape in enhancing hydrogel adhesive and mechanical properties. Nat. Commun. 2020, 11, 1420. [Google Scholar] [CrossRef] [Green Version]
- Gaharwar, A.K.; Rivera, C.P.; Wu, C.-J.; Schmidt, G. Transparent, elastomeric and tough hydrogels from poly(ethylene glycol) and silicate nanoparticles. Acta Biomater. 2011, 7, 4139–4148. [Google Scholar] [CrossRef] [PubMed]
- Afghah, F.; Altunbek, M.; Dikyol, C.; Koc, B. Preparation and characterization of nanoclay-hydrogel composite support-bath for bioprinting of complex structures. Sci. Rep. 2020, 10, 5257. [Google Scholar] [CrossRef]
- Boyaci, T.; Orakdogen, N. Poly(N,N-dimethylaminoethyl methacrylate-co-2-acrylamido-2-methyl-propanosulfonic acid)/Laponite nanocomposite hydrogels and cryogels with improved mechanical strength and rapid dynamic properties. Appl. Clay Sci. 2016, 121–122, 162–173. [Google Scholar] [CrossRef]
- Park, H.; Guo, X.; Temenoff, J.S.; Tabata, Y.; Caplan, A.I.; Kasper, F.K.; Mikos, A.G. Effect of swelling ratio of injectable hydrogel composites on chondrogenic differentiation of encapsulated rabbit marrow mesenchymal stem cells in vitro. Biomacromolecules 2009, 10, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Kroger, S.; Hill, L.; Jain, E.; Stock, A.; Bracher, P.; Fahu, H.; Zustiak, S.P. Design of Hydrolytically Resorbable Polyethylene Glycol Crosslinkers for Facile Control of Hydrogel Degradation. Macromol. Biosci. 2020, 20, 2000085. [Google Scholar] [CrossRef] [PubMed]
- Kaberova, Z.; Karpushkin, E.; Nevoralová, M.; Vetrík, M.; Šlouf, M.; Dušková-Smrčková, M. Microscopic Structure of Swollen Hydrogels by Scanning Electron and Light Microscopies: Artifacts and Reality. Polymers 2020, 12, 578. [Google Scholar] [CrossRef] [Green Version]
- Peppas, N.A.; Keys, K.B.; Torres-Lugo, M.; Lowman, A.M. Poly(ethylene glycol)-containing hydrogels in drug delivery. J. Control. Release 1999, 62, 81–87. [Google Scholar] [CrossRef]
- Lin, C.-C.; Anseth, K.S. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm. Res. 2009, 26, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Parlato, M.; Reichert, S.; Barney, N.; Murphy, W.L. Poly(ethylene glycol) hydrogels with adaptable mechanical and degradation properties for use in biomedical applications. Macromol. Biosci. 2014, 14, 687–698. [Google Scholar] [CrossRef]
- Suman, K.; Joshi, Y.M. Microstructure and Soft Glassy Dynamics of an Aqueous Laponite Dispersion. Langmuir 2018, 34, 13079–13103. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.W.; Butterworth, J.T. The nature of laponite and its aqueous dispersions. J. Colloid Interface Sci. 1992, 151, 236–243. [Google Scholar] [CrossRef]
- Das, K.; Rawat, K.; Bohidar, H.B. Surface patch binding induced interaction of anisotropic nanoclays with globular plasma proteins. RSC Adv. 2016, 6, 104117–104125. [Google Scholar] [CrossRef]
- Cummins, H.Z. Liquid, glass, gel: The phases of colloidal Laponite. J. Non-Cryst. Solids 2007, 353, 3891–3905. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.; Webb, W.W. Thermodynamic Fluctuations in a Reacting System—Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett. 1972, 29, 705–708. [Google Scholar] [CrossRef]
- Michelman-Ribeiro, A.; Horkay, F.; Nossal, R.; Boukari, H. Probe diffusion in aqueous poly (vinyl alcohol) solutions studied by fluorescence correlation spectroscopy. Biomacromolecules 2007, 8, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Zustiak, S.P.; Boukari, H.; Leach, J.B. Solute diffusion and interactions in cross-linked poly (ethylene glycol) hydrogels studied by fluorescence correlation spectroscopy. Soft Matter 2010, 6, 3609–3618. [Google Scholar] [CrossRef] [Green Version]
- Fouqueau, A.; Meuwly, M.; Bemish, R.J. Adsorption of acridine orange at a C8,18/water/acetonitrile interface. J. Phys. Chem. B 2007, 111, 10208–10216. [Google Scholar] [CrossRef]
- Ghassemi, Z.; Ruesing, S.; Leach, J.B.; Zustiak, S.P. Stability of proteins encapsulated in Michael-type addition polyethylene glycol hydrogels. Biotechnol. Bioeng. 2021, 118, 4840–4853. [Google Scholar] [CrossRef]
- Wilson, S.A.; Cross, L.M.; Peak, C.W.; Gaharwar, A.K. Shear-Thinning and Thermo-Reversible Nanoengineered Inks for 3D Bioprinting. ACS Appl. Mater. Interfaces 2017, 9, 43449–43458. [Google Scholar] [CrossRef]
- Bruns, J.; McBride-Gagyi, S.; Zustiak, S.P. Injectable and Cell-Adhesive Polyethylene Glycol Cryogel Scaffolds: Independent Control of Cryogel Microstructure and Composition. Macromol. Mater. Eng. 2018, 303, 1800298. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, L.; Liao, J.; Tan, Y.; Ouyang, K.; Ning, C.; Ni, G.; Tan, G. Cell-laden photocrosslinked GelMA–DexMA copolymer hydrogels with tunable mechanical properties for tissue engineering. J. Mater. Sci. Mater. Med. 2014, 25, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, M.; Castilla, C.; Maška, M.; Ederra, C.; Peláez, R.; Morales, X.; Muñoz-Arrieta, G.; Mujika, M.; Kozubek, M.; Muñoz-Barrutia, A.; et al. Characterization of three-dimensional cancer cell migration in mixed collagen-Matrigel scaffolds using microfluidics and image analysis. PLoS ONE 2017, 12, e0171417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Molecule | Acridine Orange | Alexa 647 | Atto Rho 13 |
|---|---|---|---|
| Abbreviation | AO | Alexa | Rho |
| Molecular Weight [g/mol] | 265 | 1025 | 867 |
| Diffusion Coefficient in Water at 37 °C [×10−6 cm2/s] | 8.7 | 3.3 | 4.6 |
| Ex./Em. Wavelength [nm] | 500/540 | 640/670 | 590/630 |
| Net Charge at pH 7.4 | +1 | +1 | +1 |
| Chemical Structure | 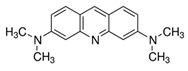 | 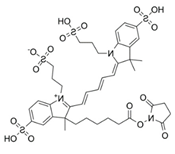 | 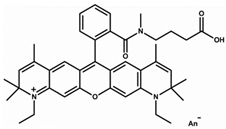 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stealey, S.; Khachani, M.; Zustiak, S.P. Adsorption and Sustained Delivery of Small Molecules from Nanosilicate Hydrogel Composites. Pharmaceuticals 2022, 15, 56. https://doi.org/10.3390/ph15010056
Stealey S, Khachani M, Zustiak SP. Adsorption and Sustained Delivery of Small Molecules from Nanosilicate Hydrogel Composites. Pharmaceuticals. 2022; 15(1):56. https://doi.org/10.3390/ph15010056
Chicago/Turabian StyleStealey, Samuel, Mariam Khachani, and Silviya Petrova Zustiak. 2022. "Adsorption and Sustained Delivery of Small Molecules from Nanosilicate Hydrogel Composites" Pharmaceuticals 15, no. 1: 56. https://doi.org/10.3390/ph15010056
APA StyleStealey, S., Khachani, M., & Zustiak, S. P. (2022). Adsorption and Sustained Delivery of Small Molecules from Nanosilicate Hydrogel Composites. Pharmaceuticals, 15(1), 56. https://doi.org/10.3390/ph15010056