
FDA-Approved Oximes and Their Significance in Medicinal Chemistry



Abstract
:1. Introduction
1.1. The Sources and Discovery of Oximes
1.2. The Synthesis of Oximes
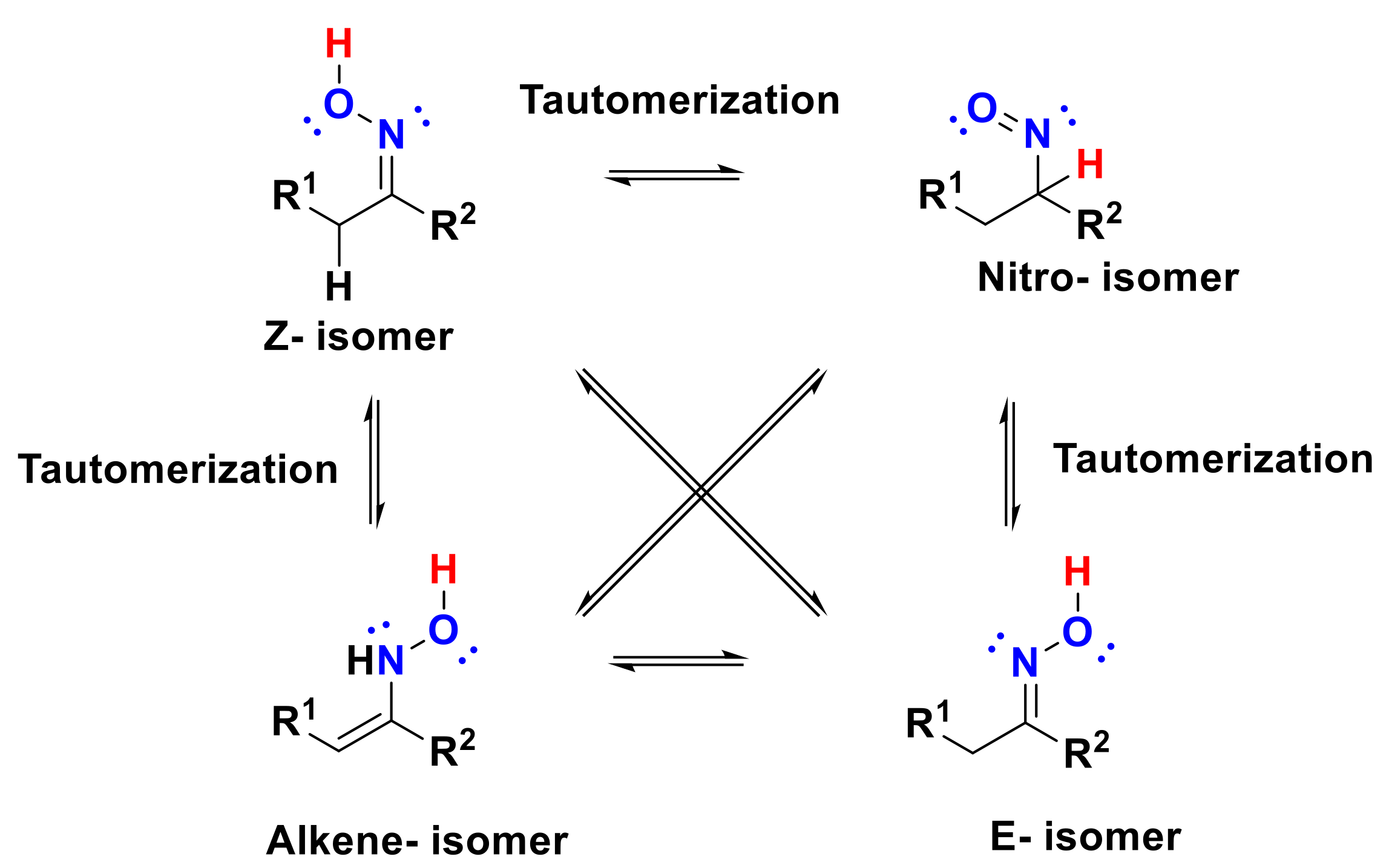
1.3. Isomerism of Oximes
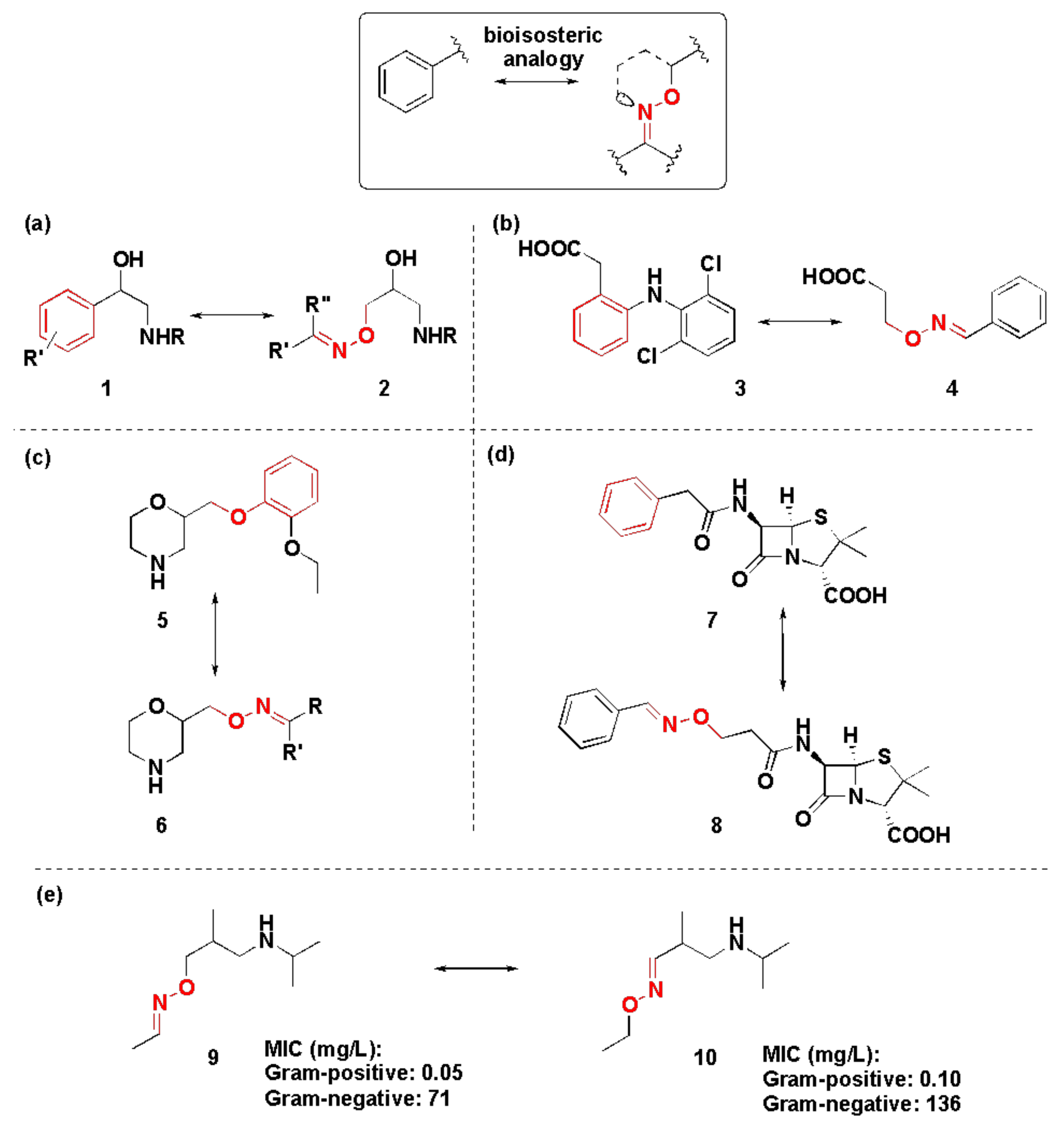
1.4. Bioisosterism and Its Pharmacological Relevance in Oximes
2. Classification of FDA-Approved Oximes
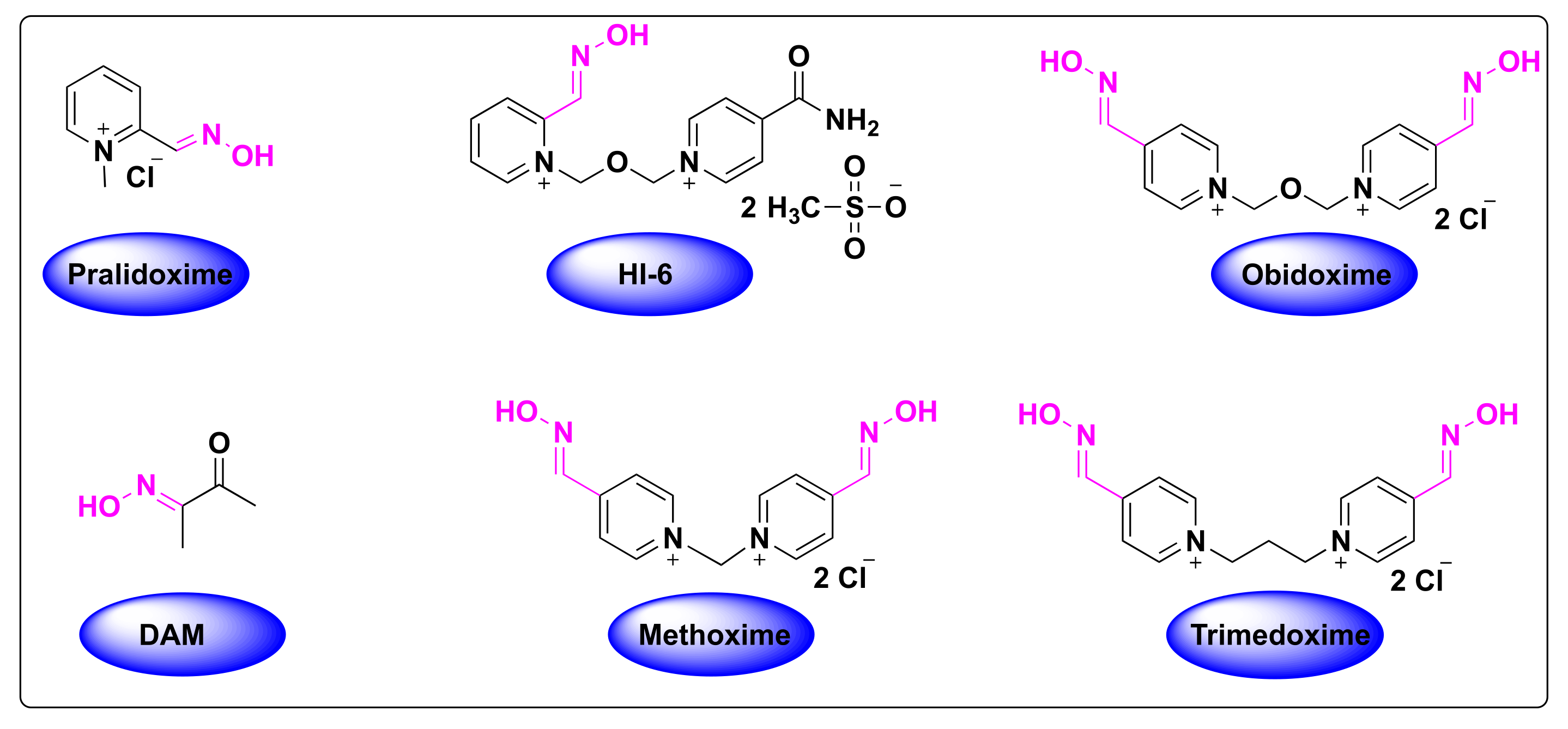
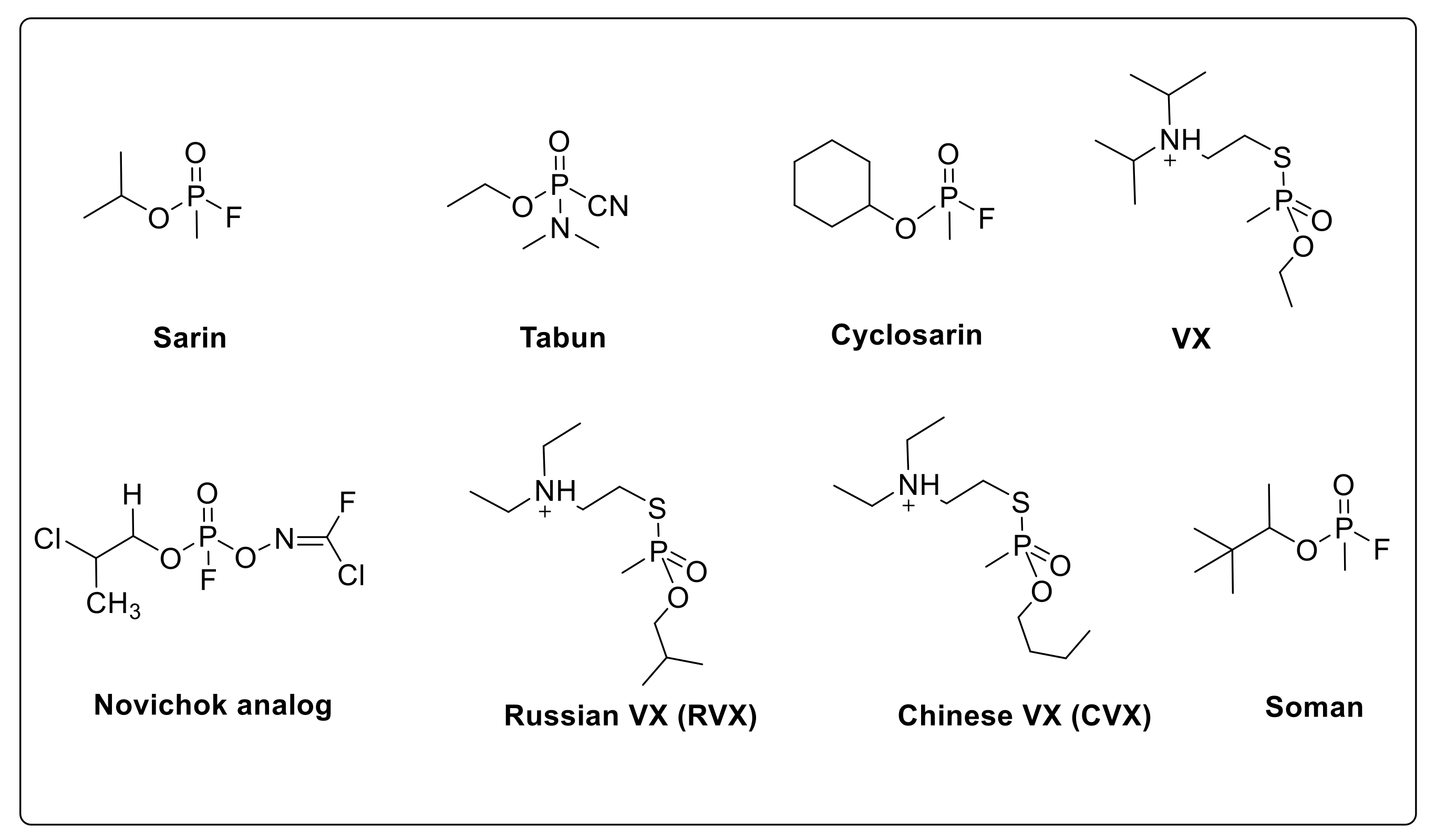
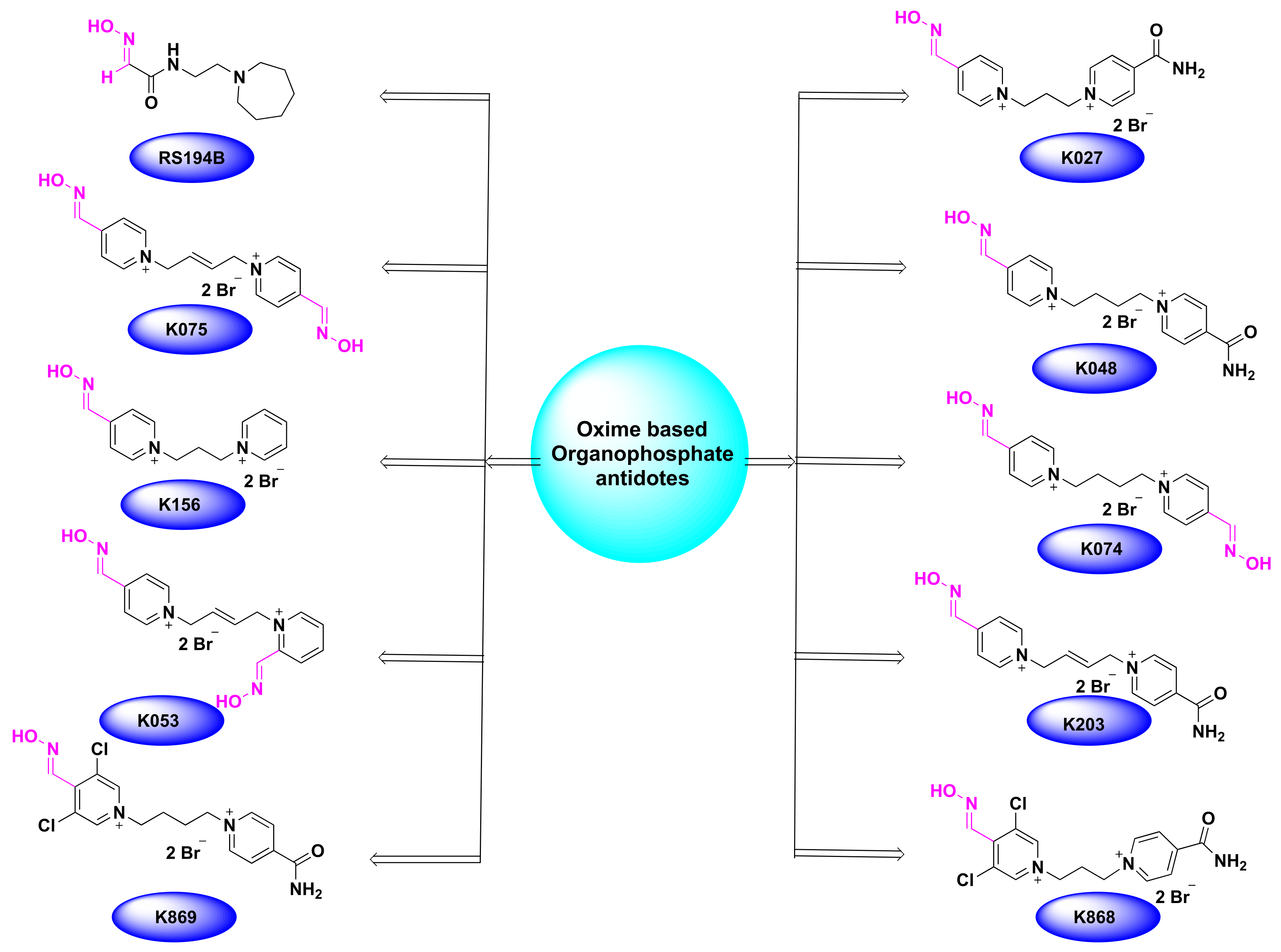
2.1. Oximes as Organophosphate (OP) Poisoning Antidotes
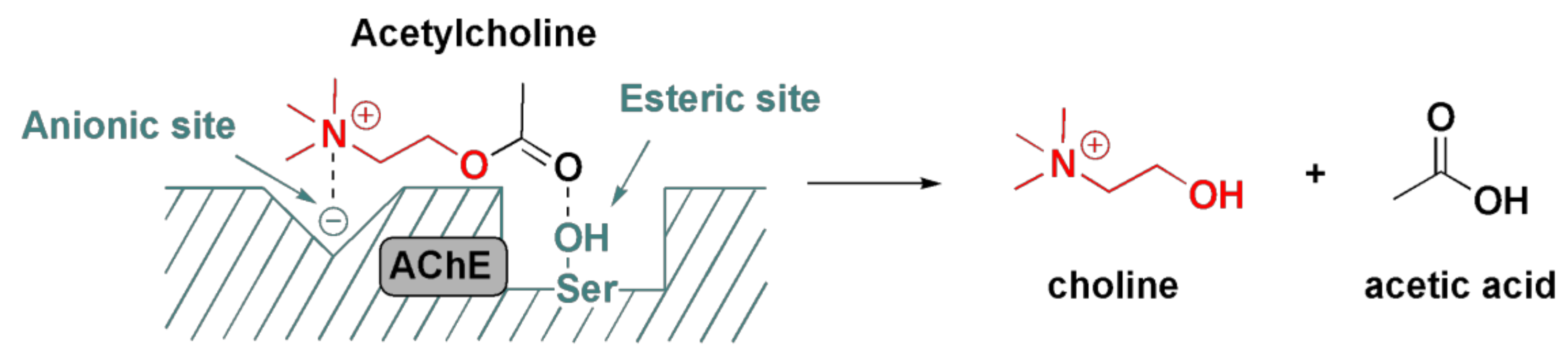
2.2. Significance of Acetylcholinesterase and Its Inhibition in OP Poisoning
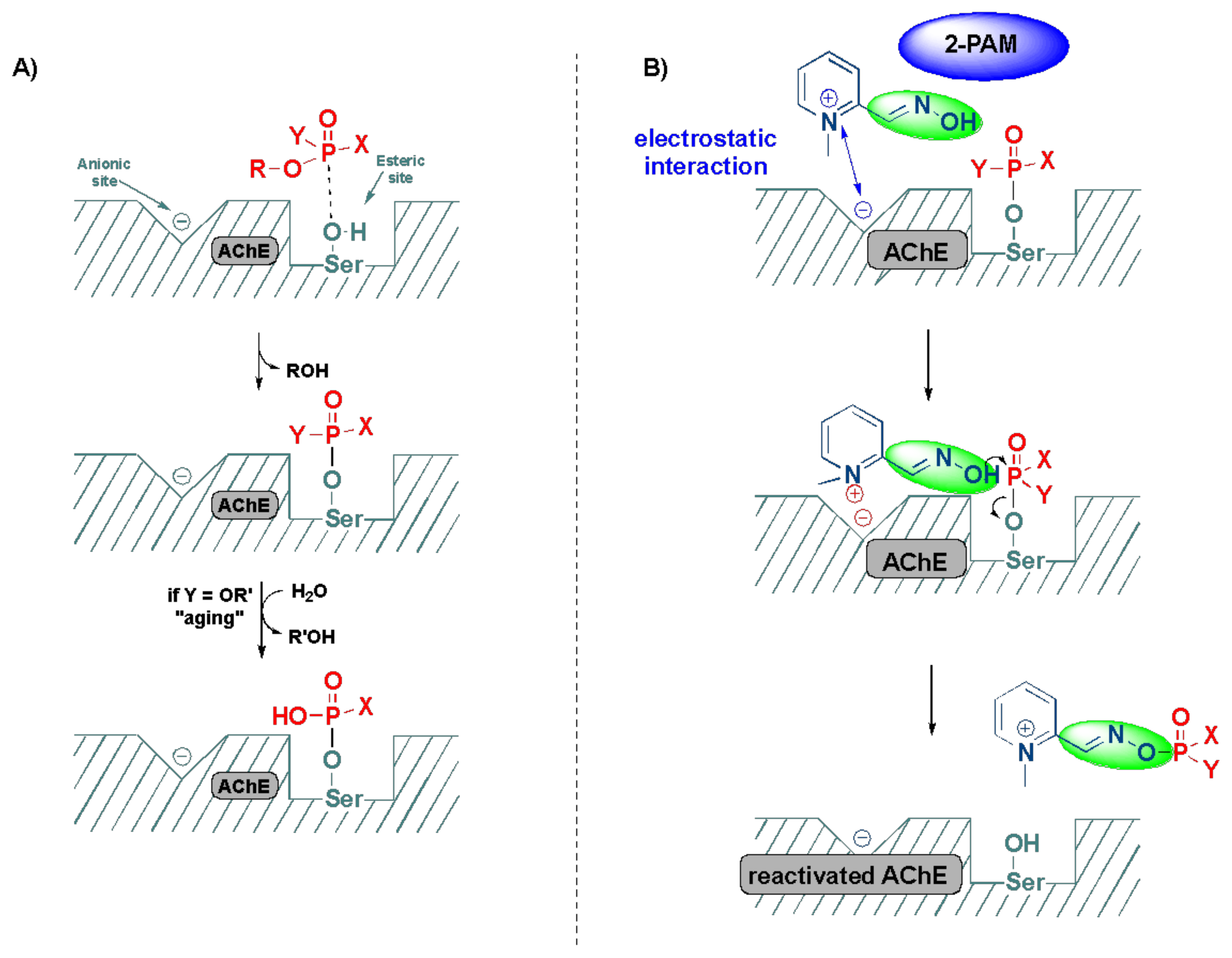
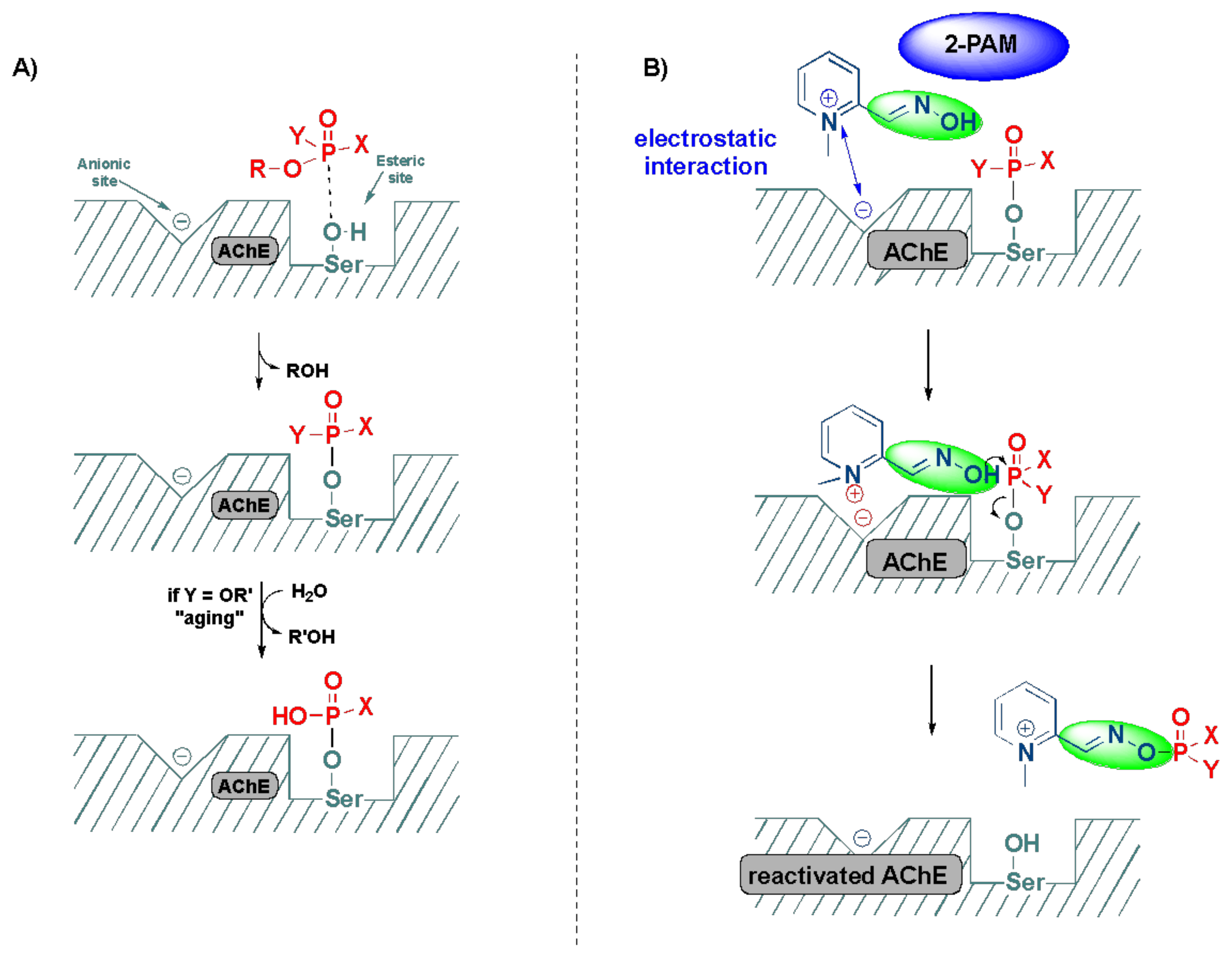
2.3. Detoxification of OP Poisoning by Pralidoxime
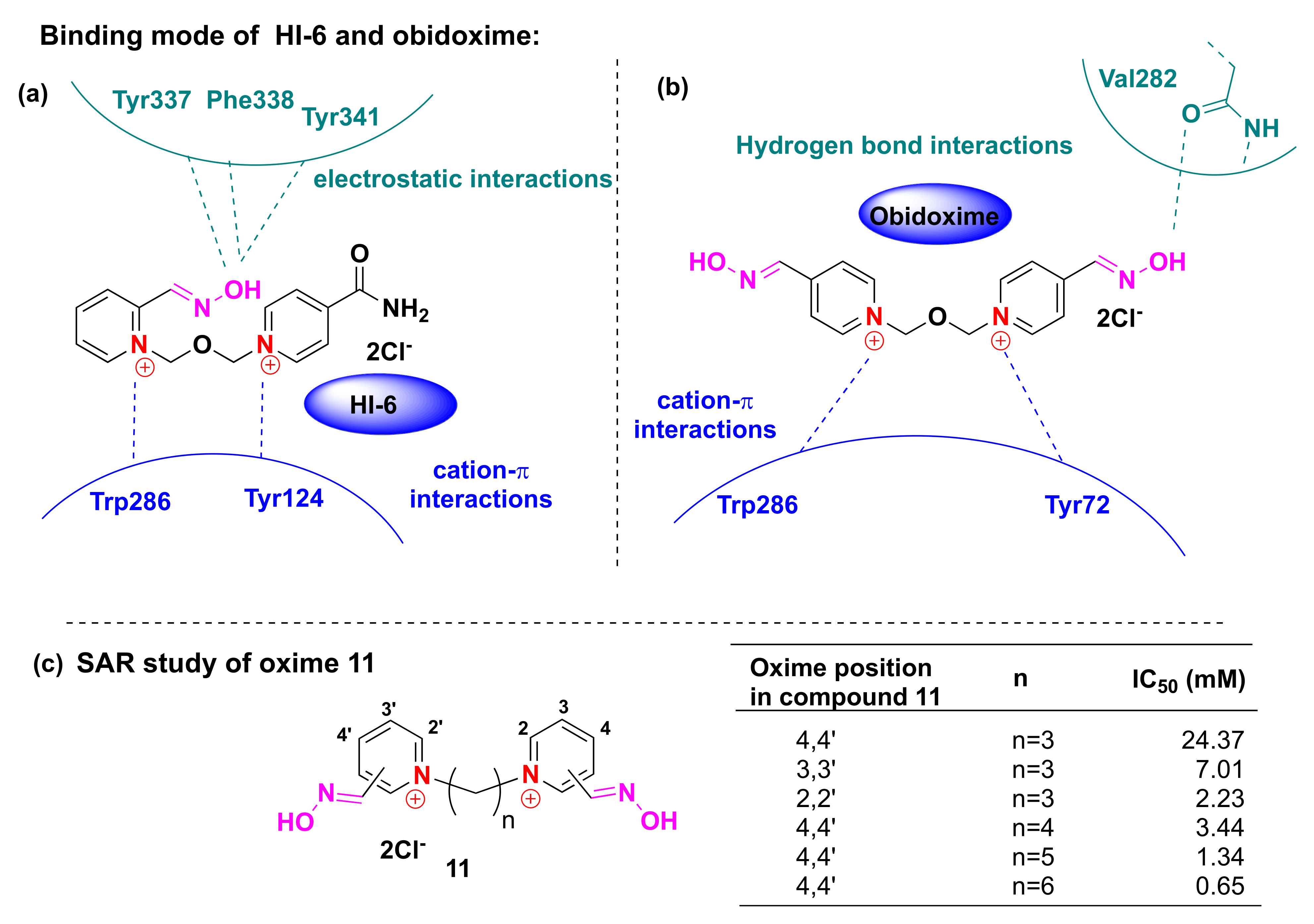
3. Computational and SAR Studies of AChE Reactivators
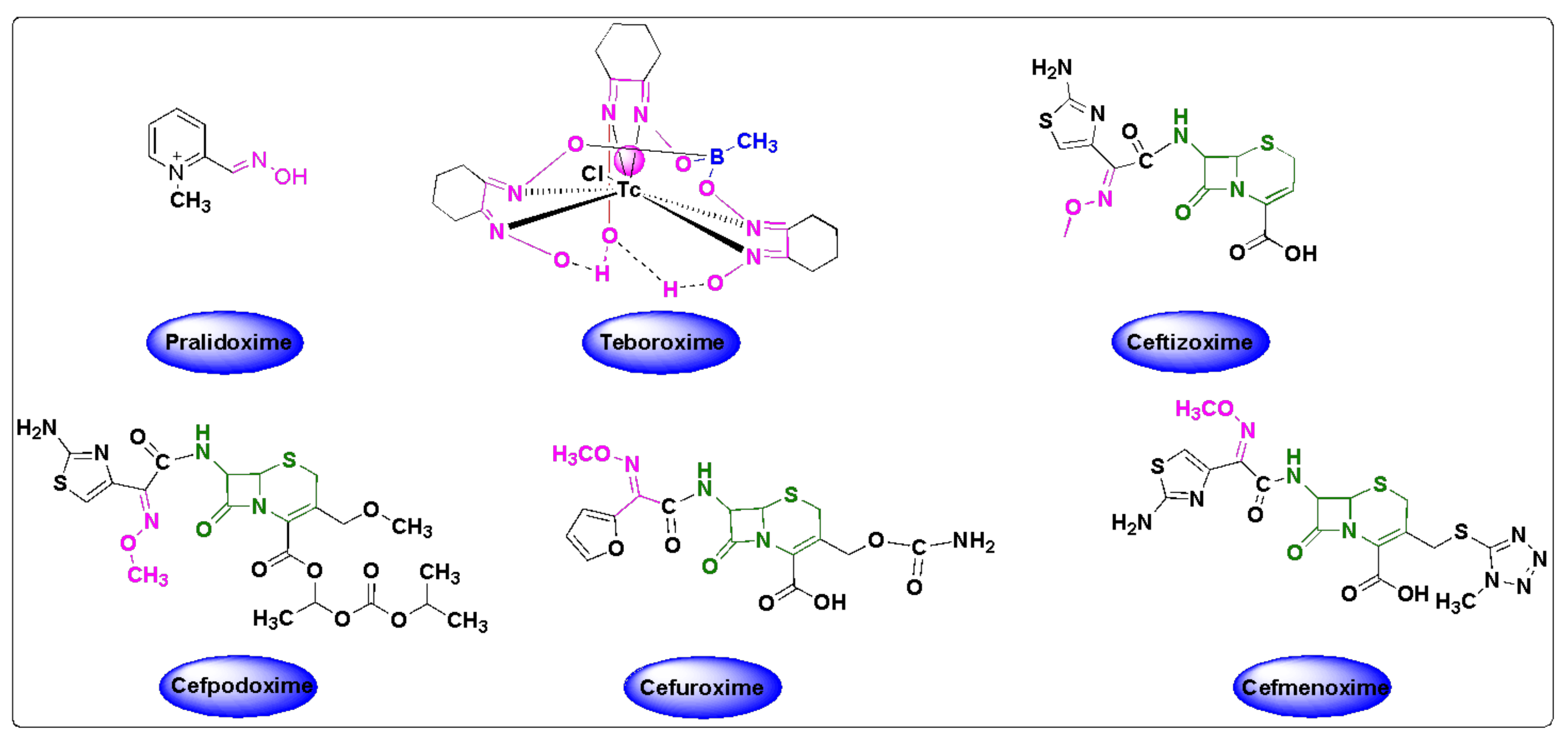
4. Oxime Based FDA Approved Drugs
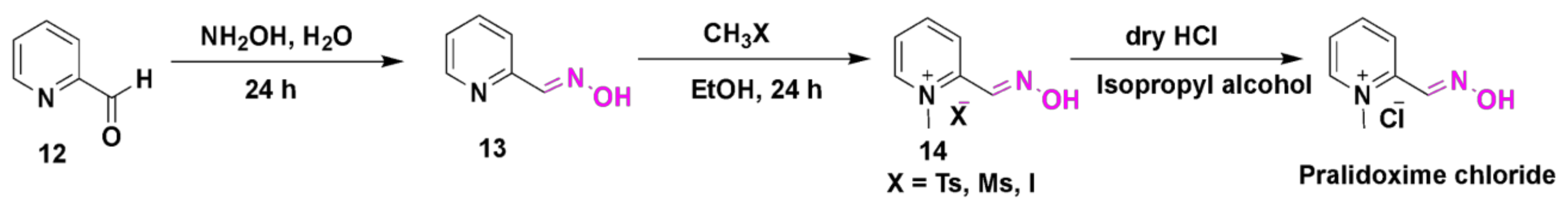
4.1. Pralidoxime

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Brand | Applications (References) ** |
|---|---|---|
 | Atnaa, Duodote (with atropine), Pralidoxime Chloride, Protopam Chloride | Organophosphate poisoning and pre-treatment [57,82]. |
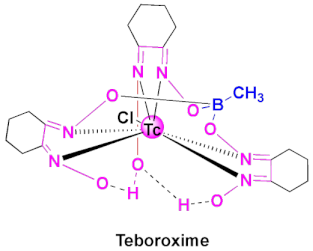 | Cardiotec | Agent for myocardial perfusion imaging [84,85]. |
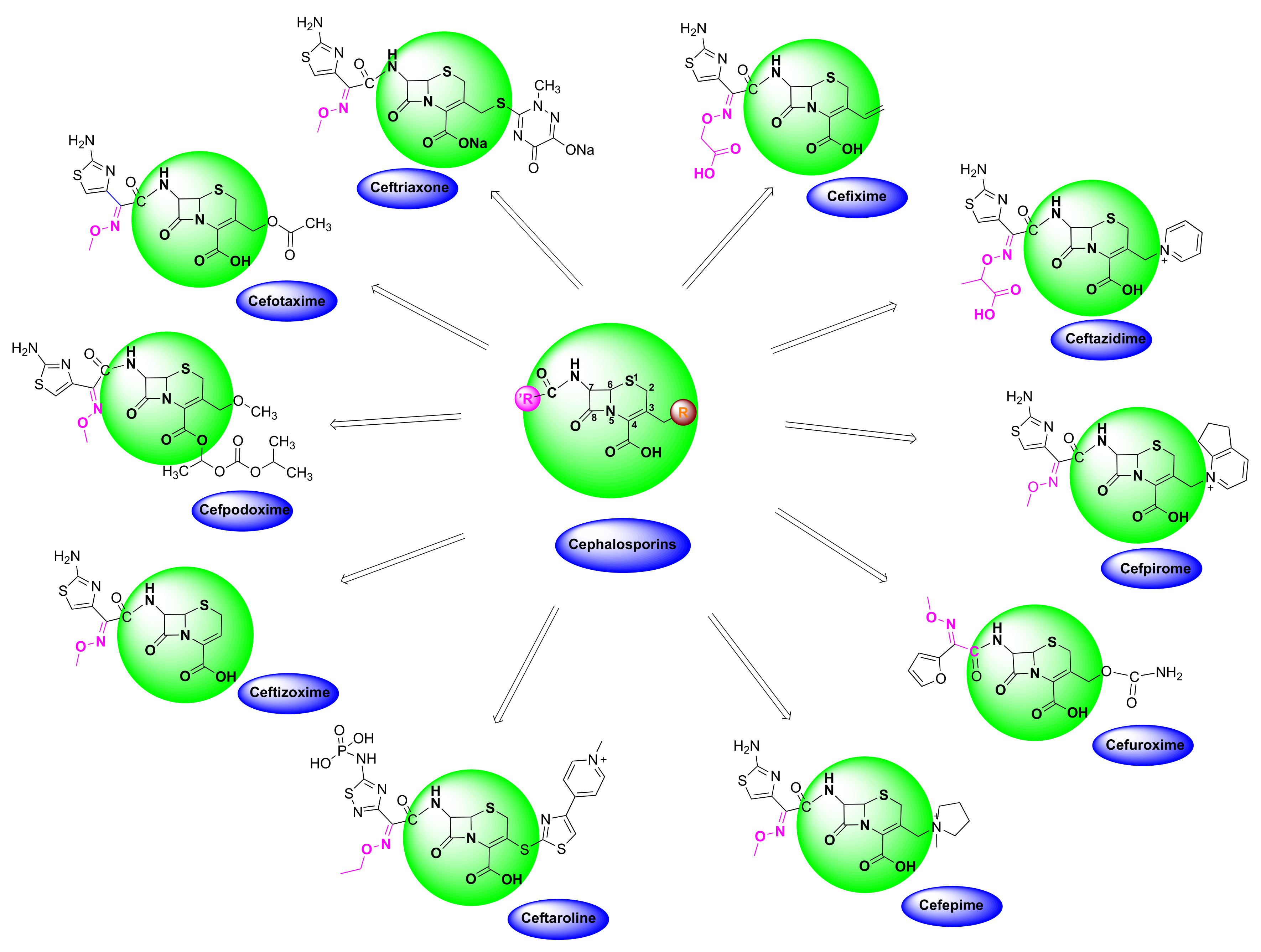
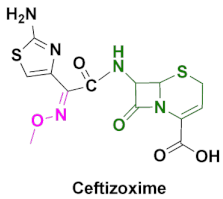 | Cefizox | Gonorrhea, pelvic inflammatory disease, urinary tract infections, cystitis, epiglottitis, meningitis, osteomyelitis, pneumonia, skin/soft tissue infection and other diseases caused by Gram(+) and Gram(-) bacteria [86]. |
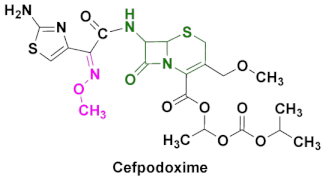 | Banan, Vantin, Cefpodoxime Proxetil | Acute bronchitis, pneumonia, pharyngitis/tonsillitis, gonorrhea, urinary tract infections, otitis and other diseases caused by Gram(+) and Gram(-) bacteria [87,88,89]. |
 | Cefmax | Treatment of female gynecologic and obstetric infection, gonorrhea, otitis, skin/soft tissue infection, sinusitis and other diseases caused by Gram(+) and Gram(-) bacteria [90,91]. |
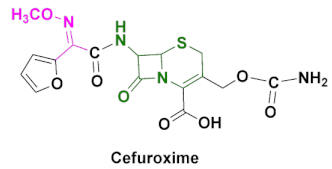 | Ceftin, Cefuroxime, Cefuroxime sodium, Kefurox, Zinacef | Skin and middle ear infections, tonsillitis, throat infections, laryngitis, bronchitis, pneumonia, urinary tract infections, gonorrhea and other diseases caused by Gram(+) and Gram(-) bacteria [92,93]. |
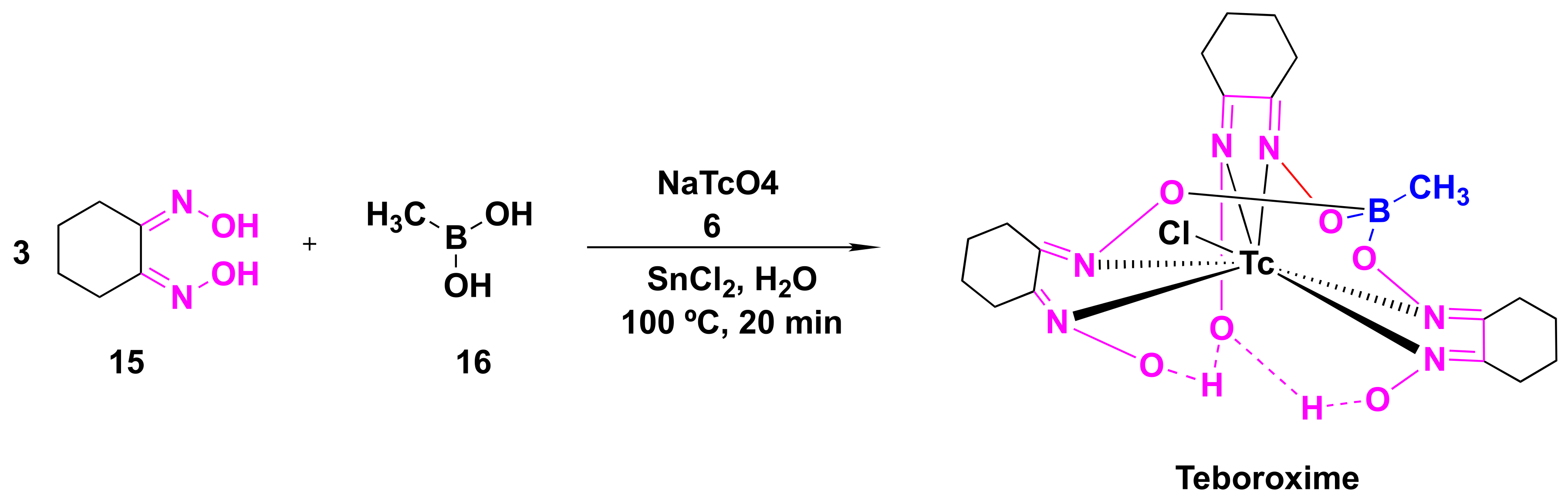
4.2. Teboroxime
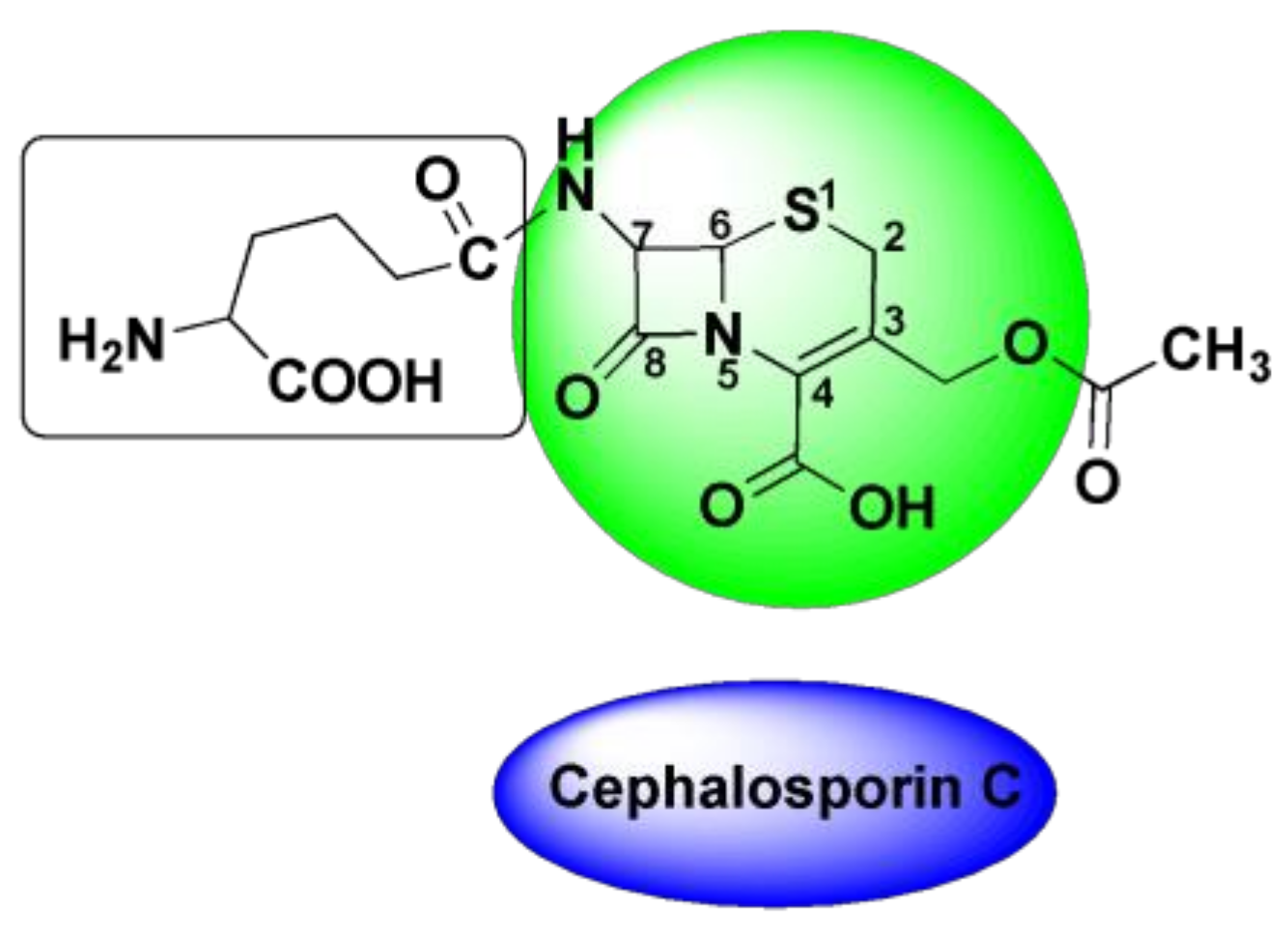
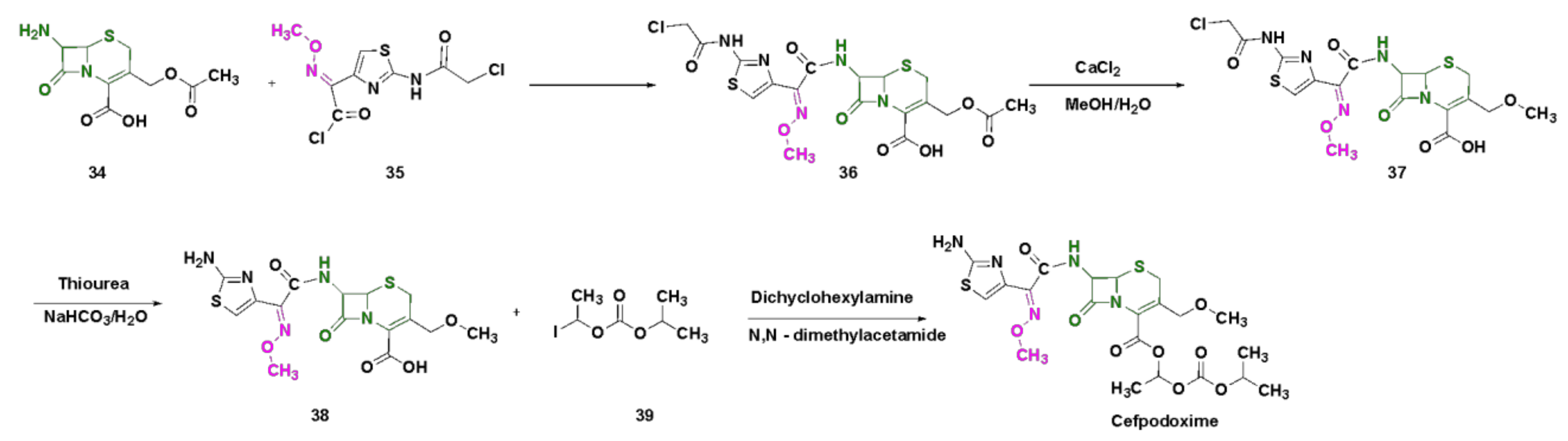
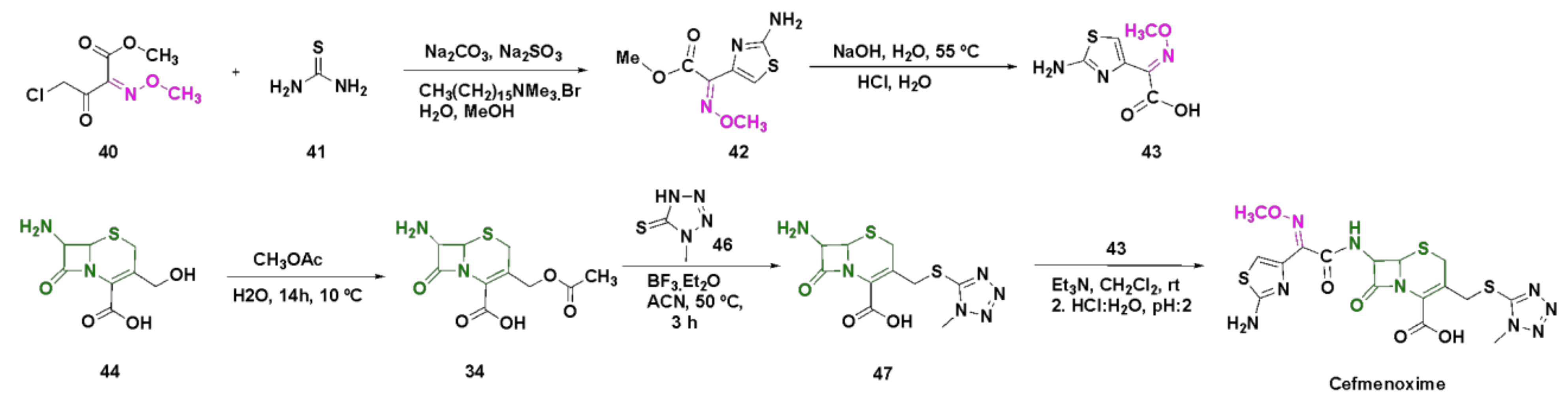
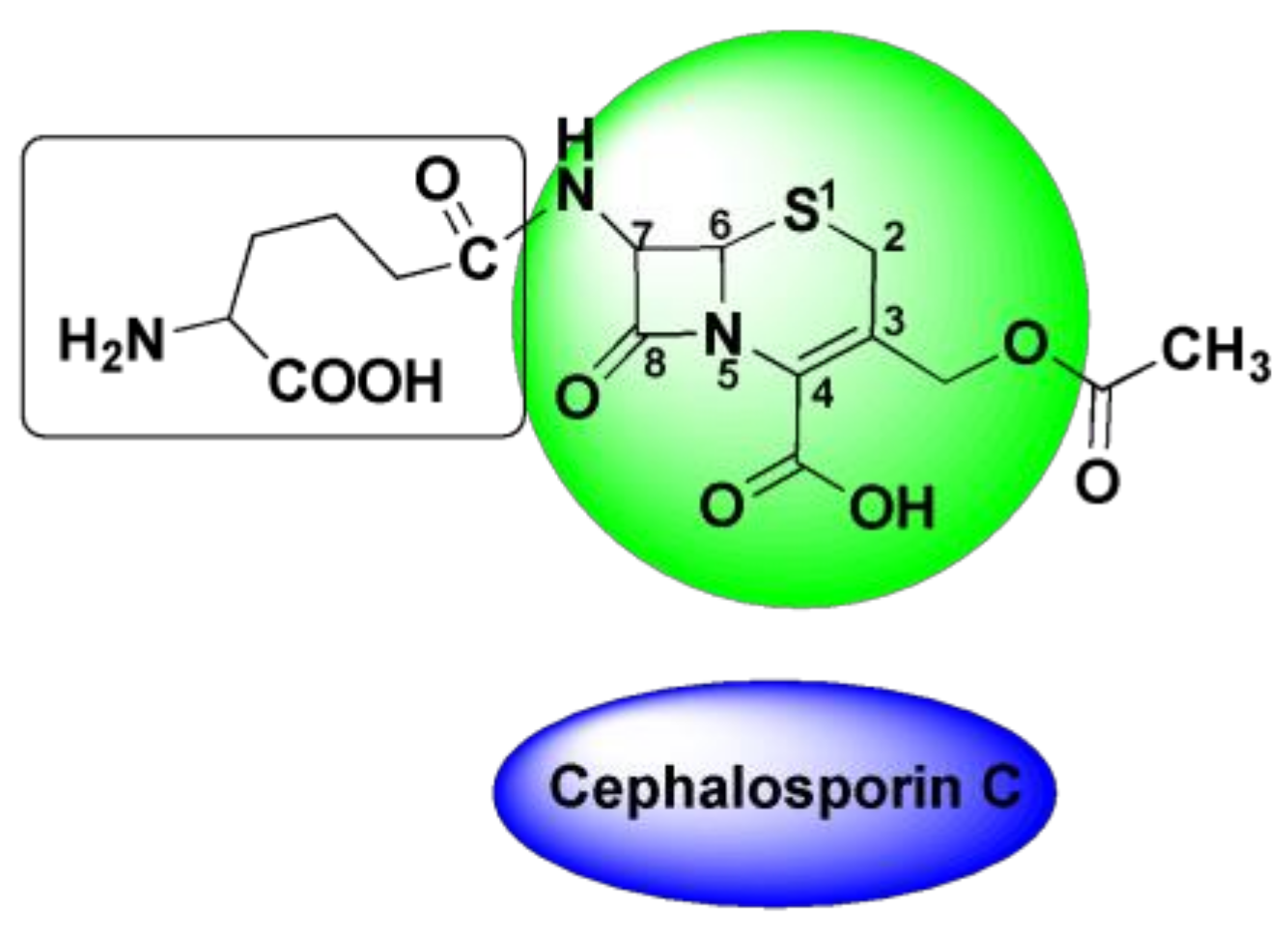
4.3. Oxime-Based Cephalosporins
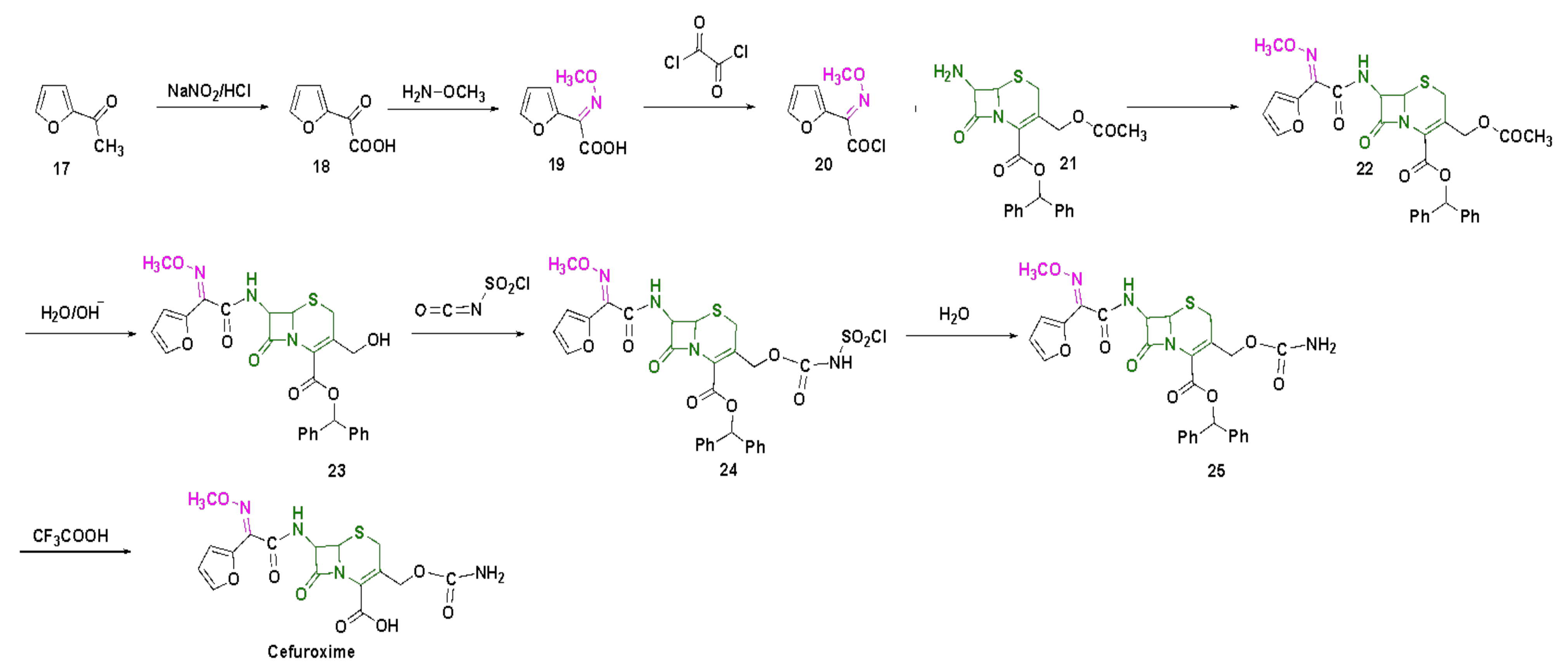
4.4. Cefuroxime
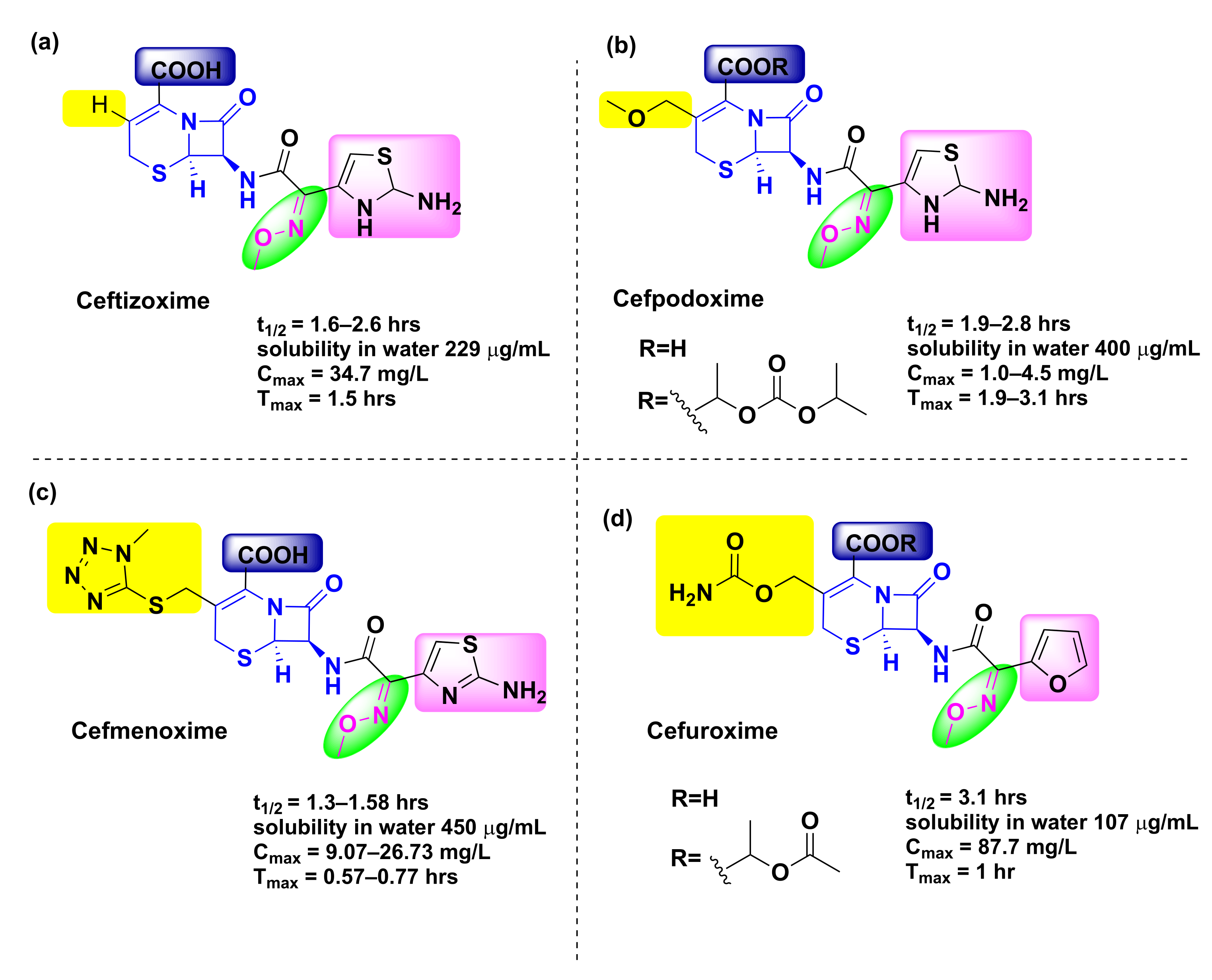
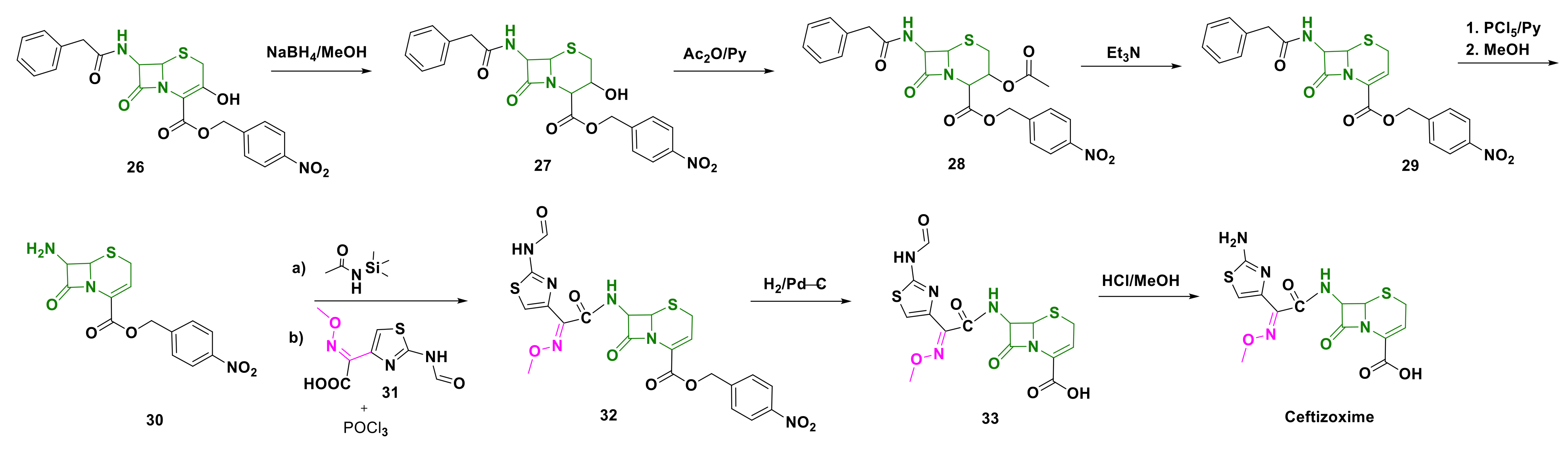
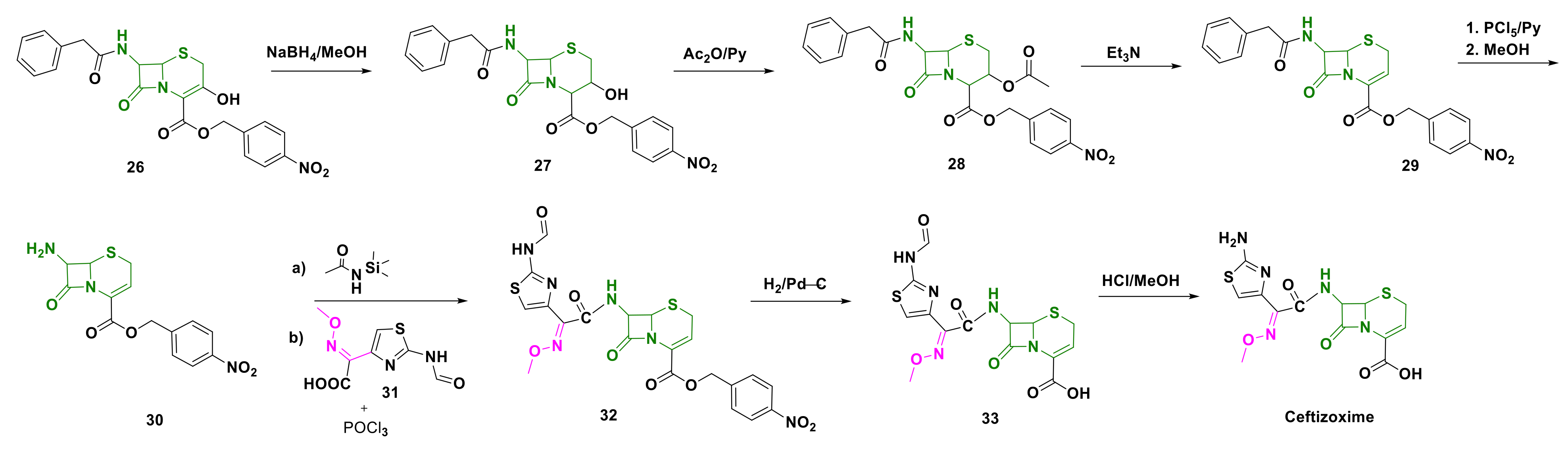
4.5. Ceftizoxime
| Compound | EC50 (mg/L) | t1/2 (h) | Solubility | Cmax (mg/L) | Tmax (h) | Target |
|---|---|---|---|---|---|---|
| Ceftizoxime [113] | B. fragilis: 202 E. cloacae: 51 | 5.7–9.4 (rabbits) [107] 1.6–2.57 (people) [114,115,116] | Water (229 mg/L) [117] | 34.7 | 1.5 [118] | S. aureus: PBP 2 [119] |
| Cefpodoxime [120] | H. influenzae: 0.04 M. catarrhalis: 0.12 S. pneumoniae: 0.27 [121] | 1.9–2.8 | Water (400 µg/mL) [122] | 1.0–4.5 | 1.9–3.1 | E. coli: Peptidoglycan synthase FtsI [123] |
| Cefmenoxime [124,125] | ND | 1.3–1.5 | Water (450 mg/L) [126] | 9.07–26.73 | 0.57–0.77 | E. coli: Peptidoglycan synthase FtsI [90] |
| Cefuroxime [127,128] | K. pneumoniae: 1.61 [124] | 1.2–2.4 [93] | Water (107 mg/L), good in acetone, sparingly soluble in chloroform, ethyl acetate, methanol | 4.1–4.8 8.6–9.0 | 2.0–2.5; 1.8–2.4 | Clostridium perfringens: PBP 1A [129] |
4.6. Cefpodoxime
4.7. Cefmenoxime
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozłowska, J.; Potaniec, B.; Żarowska, B.; Anioł, M. Synthesis and Biological Activity of Novel O-Alkyl Derivatives of Naringenin and Their Oximes. Molecules 2017, 22, 1485. [Google Scholar] [CrossRef] [Green Version]
- Zhmurenko, L.A.; Litvinova, S.A.; Kutepova, I.S.; Nerobkova, L.N.; Mokrov, G.V.; Rebeko, A.G.; Voronina, T.A.; Gudasheva, T.A. Synthesis of Dibenzofuranone-Oxime Derivatives with Anticonvulsant, Antihypoxic, and Anti-Ischemic Activity. Pharm. Chem. J. 2020, 53, 997–1004. [Google Scholar] [CrossRef]
- Purves, D.; Williams, S.M. (Eds.) Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Robb, E.L.; Baker, M.B. Organophosphate Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Wilson, I.B.; Ginsburg, S. A Powerful Reactivator of Alkylphosphate-Inhibited Acetylcholinesterase. Biochim. Biophys. Acta 1955, 18, 168–170. [Google Scholar] [CrossRef]
- Grob, D.; Johns, R.J. Use of Oximes in the Treatment of Intoxication by Anticholinesterase Compounds in Normal Subjects. Am. J. Med. 1958, 24, 497–511. [Google Scholar] [CrossRef]
- Shahbaz, K. Cephalosporins: Pharmacology and Chemistry. Pharm. Biol. Eval. 2017, 4, 234. [Google Scholar] [CrossRef]
- Lima, L.M.; da Silva, B.N.M.; Barbosa, G.; Barreiro, E.J. β-Lactam Antibiotics: An Overview from a Medicinal Chemistry Perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P. The Cephalosporin C Group. Q. Rev. Chem Soc. 1967, 21, 231–248. [Google Scholar] [CrossRef]
- Bui, T.; Preuss, C.V. Cephalosporins. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Sørensen, M.; Neilson, E.H.J.; Møller, B.L. Oximes: Unrecognized Chameleons in General and Specialized Plant Metabolism. Mol. Plant 2018, 11, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Surowiak, A.K.; Lochyński, S.; Strub, D.J. Unsubstituted Oximes as Potential Therapeutic Agents. Symmetry 2020, 12, 575. [Google Scholar] [CrossRef] [Green Version]
- Tapper, B.A.; Conn, E.E.; Butler, G.W. Conversion of α-Keto-Isovaleric Acid Oxime and Isobutyraldoxime to Linamarin in Flax Seedlings. Arch. Biochem. Biophys. 1967, 119, 593–595. [Google Scholar] [CrossRef]
- Underhill, E.W. Biosynthesis of Mustard Oil Glucosides. Eur. J. Biochem. 1967, 2, 61–63. [Google Scholar] [CrossRef]
- Mano, Y.; Nemoto, K. The Pathway of Auxin Biosynthesis in Plants. J. Exp. Bot. 2012, 63, 2853–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, S. Role of Oximes in Nitrogen Metabolism in Plants. Annu. Rev. Plant Physiol. 1973, 24, 69–88. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef]
- Beckmann, E. Zur Kenntniss der Isonitrosoverbindungen. Berichte Dtsch. Chem. Ges. 1886, 19, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Tinge, J.; Groothaert, M.; op het Veld, H.; Ritz, J.; Fuchs, H.; Kieczka, H.; Moran, W.C. Caprolactam. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 1–31. [Google Scholar] [CrossRef]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and Oximes: Synthesis, Structure, and Their Key Role as NO Donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [Green Version]
- Robertson, G.M. Imines and Their N-Substituted Derivatives: Oximes and Their O-R Substituted Analogues. In Comprehensive Organic Functional Group Transformations; Elsevier: Amsterdam, The Netherlands, 1995; pp. 425–441. [Google Scholar] [CrossRef]
- Ãbele, E.; Lukevics, E. Recent Advances in the Chemistry of Oximes. Org. Prep. Proced. Int. 2000, 32, 235–264. [Google Scholar] [CrossRef]
- Bohle, D.S.; Chua, Z.; Perepichka, I.; Rosadiuk, K. E/ Z Oxime Isomerism in PhC(NOH)CN. Chem. Eur. J. 2013, 19, 4223–4229. [Google Scholar] [CrossRef]
- Sharghi, H.; Sarvari, M.H. Selective Synthesis of E and Z Isomers of Oximes. Synlett 2001, 2001, 99–101. [Google Scholar] [CrossRef]
- Blatt, A.H. The Tautomersim of Oximes. J. Org. Chem. 1938, 3, 91–98. [Google Scholar] [CrossRef]
- Grigg, R.; Gunaratne, H.Q.N. Prototropic Generation of Dipoles. A New Synthesis of Indole-3-Carboxylic Acids. J. Chem. Soc. Chem. Commun. 1984, 10, 661–662. [Google Scholar] [CrossRef]
- Noguchi, M.; Okada, H.; Nishimura, S.; Yamagata, Y.; Takamura, S.; Tanaka, M.; Kakehi, A.; Yamamoto, H. A Simple Oxime–Nitrone Isomerisation and Intramolecular Nitrone-Cycloaddition Reaction of 3-(Alk-2-Enylamino)Propionaldehyde Oximes. J. Chem. Soc. Perkin Trans. 1 1999, 2, 185–192. [Google Scholar] [CrossRef]
- Jung, S.; Choi, K.; Pae, A.N.; Lee, J.K.; Choo, H.; Keum, G.; Cho, Y.S.; Min, S.-J. Facile Diverted Synthesis of Pyrrolidinyl Triazoles Using Organotrifluoroborate: Discovery of Potential MPTP Blockers. Org. Biomol. Chem. 2014, 12, 9674–9682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef]
- Balachandran, N.; To, F.; Berti, P.J. Linear Free Energy Relationship Analysis of Transition State Mimicry by 3-Deoxy-d-Arabino- Heptulosonate-7-Phosphate (DAHP) Oxime, a DAHP Synthase Inhibitor and Phosphate Mimic. Biochemistry 2017, 56, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Rose, K. Facile Synthesis of Homogeneous Artificial Proteins. J. Am. Chem. Soc. 1994, 116, 30–33. [Google Scholar] [CrossRef]
- Stanley, M.; Virdee, S. Genetically Directed Production of Recombinant, Isosteric and Nonhydrolysable Ubiquitin Conjugates. ChemBioChem 2016, 17, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, A.K.; Kuča, K.; Musilek, K.; Gordon, R.K. In Silico Pharmacophore Model for Tabun-Inhibited Acetylcholinesterase Reactivators: A Study of Their Stereoelectronic Properties. Chem. Res. Toxicol. 2010, 23, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Holmstedt, B. Structure-Activity Relationships of the Organophosphorus Anticholinesterase Agents. In Cholinesterases and Anticholinesterase Agents; Koelle, G.B., Eichler, O., Farah, A., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 1963; Volume 15, pp. 428–485. [Google Scholar] [CrossRef]
- Eyer, P. The Role of Oximes in the Management of Organophosphorus Pesticide Poisoning. Toxicol. Rev. 2003, 22, 165–190. [Google Scholar] [CrossRef]
- NATO Advanced Study Institute on Toxicology of Pesticides; Costa, L.G.; Galli, C.L.; Murphy, S.D.; North Atlantic Treaty Organization; Scientific Affairs Division. Toxicology of Pesticides: Experimental, Clinical, and Regulatory Perspectives; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Balali-Mood, M.; Saber, H. Recent Advances in the Treatment of Organophosphorous Poisonings. Iran. J. Med. Sci. 2012, 37, 74–91. [Google Scholar] [PubMed]
- Alozi, M.; Rawas-Qalaji, M. Treating Organophosphates Poisoning: Management Challenges and Potential Solutions. Crit. Rev. Toxicol. 2020, 50, 764–779. [Google Scholar] [CrossRef]
- Jacquet, P.; Daudé, D.; Bzdrenga, J.; Masson, P.; Elias, M.; Chabrière, E. Current and Emerging Strategies for Organophosphate Decontamination: Special Focus on Hyperstable Enzymes. Environ. Sci. Pollut. Res. 2016, 23, 8200–8218. [Google Scholar] [CrossRef]
- Reddy, P.V.L.; Kim, K.-H. A Review of Photochemical Approaches for the Treatment of a Wide Range of Pesticides. J. Hazard. Mater. 2015, 285, 325–335. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Katz, M.J.; Isley, W.C., III; Ghosh, P.; Liao, P.; Bury, W.; Wagner, G.W.; Hall, M.G.; DeCoste, J.B.; Peterson, G.W.; et al. Destruction of Chemical Warfare Agents Using Metal–Organic Frameworks. Nat. Mater. 2015, 14, 512–516. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, J.H.; Kang, B.-K. Decomposition Reaction of Organophosphorus Nerve Agents on Solid Surfaces with Atmospheric Radio Frequency Plasma Generated Gaseous Species. Langmuir 2007, 23, 8074–8078. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Gupta, R.D. Organophosphorus Nerve Agents: Types, Toxicity, and Treatments. J. Toxicol. 2020, 2020, 3007984. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Medintz, I.L.; Walper, S.A. Enzymatic Bioremediation of Organophosphate Compounds—Progress and Remaining Challenges. Front. Bioeng. Biotechnol. 2019, 7, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddleston, M. Are Oximes Still Indicated for Acute Organophosphorus Insecticide Self-Poisoning? J. Med. Toxicol. 2018, 14, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Nepovimova, E.; Kuca, K. Chemical Warfare Agent NOVICHOK—Mini-Review of Available Data. Food Chem. Toxicol. 2018, 121, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Vale, J.A.; Marrs, T.C.; Maynard, R.L. Novichok: A Murderous Nerve Agent Attack in the UK. Clin. Toxicol. 2018, 56, 1093–1097. [Google Scholar] [CrossRef]
- Lyagin, I.; Efremenko, E. Theoretical Evaluation of Suspected Enzymatic Hydrolysis of Novichok Agents. Catal. Commun. 2019, 120, 91–94. [Google Scholar] [CrossRef]
- Jeong, K.; Choi, J. Theoretical Study on the Toxicity of ‘Novichok’ Agent Candidates. R. Soc. Open Sci. 2019, 6, 190414. [Google Scholar] [CrossRef] [Green Version]
- de Castro, A.A.; Assis, L.C.; Soares, F.V.; Kuca, K.; Polisel, D.A.; da Cunha, E.F.F.; Ramalho, T.C. Trends in the Recent Patent Literature on Cholinesterase Reactivators (2016–2019). Biomolecules 2020, 10, 436. [Google Scholar] [CrossRef] [Green Version]
- Worek, F.; Thiermann, H.; Wille, T. Organophosphorus Compounds and Oximes: A Critical Review. Arch. Toxicol. 2020, 94, 2275–2292. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Parmar, M. Pralidoxime. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kharel, H.; Pokhrel, N.B.; Ghimire, R.; Kharel, Z. The Efficacy of Pralidoxime in the Treatment of Organophosphate Poisoning in Humans: A Systematic Review and Meta-Analysis of Randomized Trials. Cureus 2020, 12, e7174. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.G. Organophosphorus Compounds at 80: Some Old and New Issues. Toxicol. Sci. 2018, 162, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, S.; van den Berg, R.M.; Mikler, J.; Klaassen, S.D.; Joosen, M.J.A. Pharmacokinetics of Three Oximes in a Guinea Pig Model and Efficacy of Combined Oxime Therapy. Toxicol. Lett. 2020, 324, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Roberts—Agency for Toxic Substances and Disease Registry. Available online: https://www.atsdr.cdc.gov/csem/cholinesterase/docs/cholinesterase.pdf (accessed on 5 December 2021).
- Kesharwani, M.K.; Ganguly, B.; Das, A.; Bandyopadhyay, T. Differential Binding of Bispyridinium Oxime Drugs with Acetylcholinesterase. Acta Pharmacol. Sin. 2010, 31, 313–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholinesterase Inhibitors: Part 2: What Are Cholinesterase Inhibitors?|Environmental Medicine|ATSDR. Available online: https://www.atsdr.cdc.gov/csem/cholinesterase-inhibitors/inhibitors.html (accessed on 21 December 2021).
- Zhuang, Q.; Young, A.; Callam, C.S.; McElroy, C.A.; Ekici, Ö.D.; Yoder, R.J.; Hadad, C.M. Efforts toward Treatments against Aging of Organophosphorus-Inhibited Acetylcholinesterase: Treatments against Aging of OP-Inhibited Acetylcholinesterase. Ann. N. Y. Acad. Sci. 2016, 1374, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Marrs, T.C.; Rice, P.; Vale, J.A. The Role of Oximes in the Treatment of Nerve Agent Poisoning in Civilian Casualties. Toxicol. Rev. 2006, 25, 297–323. [Google Scholar] [CrossRef]
- De Boer, D.; Nguyen, N.; Mao, J.; Moore, J.; Sorin, E.J. A Comprehensive Review of Cholinesterase Modeling and Simulation. Biomolecules 2021, 11, 580. [Google Scholar] [CrossRef]
- Singh, S.P.; Gupta, D. Discovery of Potential Inhibitor against Human Acetylcholinesterase: A Molecular Docking and Molecular Dynamics Investigation. Comput. Biol. Chem. 2017, 68, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Jończyk, J.; Kukułowicz, J.; Łątka, K.; Malawska, B.; Jung, Y.-S.; Musilek, K.; Bajda, M. Molecular Modeling Studies on the Multistep Reactivation Process of Organophosphate-Inhibited Acetylcholinesterase and Butyrylcholinesterase. Biomolecules 2021, 11, 169. [Google Scholar] [CrossRef]
- Ramalho, T.C.; França, T.C.C.; Rennó, M.N.; Guimarães, A.P.; da Cunha, E.F.F.; Kuča, K. Development of New Acetylcholinesterase Reactivators: Molecular Modeling versus in Vitro Data. Chem. Biol. Interact. 2010, 185, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Jokanovic, M.; Prostran, M. Pyridinium Oximes as Cholinesterase Reactivators. Structure-Activity Relationship and Efficacy in the Treatment of Poisoning with Organophosphorus Compounds. Curr. Med. Chem. 2009, 16, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.K.; Bandyopadhyay, T. Dynamic Mechanism of a Fluorinated Oxime Reactivator Unbinding from AChE Gorge in Polarizable Water. J. Phys. Chem. B 2018, 122, 3876–3888. [Google Scholar] [CrossRef]
- Sepsova, V.; Karasova, J.; Korabecny, J.; Dolezal, R.; Zemek, F.; Bennion, B.; Kuca, K. Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study. Int. J. Mol. Sci. 2013, 14, 16882–16900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Design, Evaluation and Structure-Activity Relationship Studies of the AChE Reactivators against Organophosphorus Pesticides. Med. Res. Rev. 2011, 31, 548–575. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Gerlits, O.; Kong, X.; Cheng, X.; Blumenthal, D.K.; Taylor, P.; Ballatore, C.; Kovalevsky, A.; Radić, Z. Correction: Rational Design, Synthesis, and Evaluation of Uncharged, “Smart” Bis-Oxime Antidotes of Organophosphate-Inhibited Human Acetylcholinesterase. J. Biol. Chem. 2020, 295, 6784. [Google Scholar] [CrossRef]
- Castro, A.T.; Figueroa-Villar, J.D. Molecular Structure, Conformational Analysis and Charge Distribution of Pralidoxime: Ab Initio and DFT Studies. Int. J. Quantum Chem. 2002, 89, 135–146. [Google Scholar] [CrossRef]
- Silva, G.R.; Borges, I.; Figueroa-Villar, J.D. DFT Conformational Studies of the HI-6 Molecule. Int. J. Quantum Chem. 2005, 105, 260–269. [Google Scholar] [CrossRef]
- Chambers, J.E.; Meek, E.C. Novel Centrally Active Oxime Reactivators of Acetylcholinesterase Inhibited by Surrogates of Sarin and VX. Neurobiol. Dis. 2020, 133, 104487. [Google Scholar] [CrossRef]
- Gorecki, L.; Korabecny, J.; Musilek, K.; Nepovimova, E.; Malinak, D.; Kucera, T.; Dolezal, R.; Jun, D.; Soukup, O.; Kuca, K. Progress in Acetylcholinesterase Reactivators and in the Treatment of Organophosphorus Intoxication: A Patent Review (2006–2016). Expert Opin. Ther. Pat. 2017, 27, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Musilek, K.; Jun, D.; Zdarova-Karasova, J.; Nepovimova, E.; Soukup, O.; Hrabinova, M.; Mikler, J.; Franca, T.C.C.; Da Cunha, E.F.F.; et al. A Newly Developed Oxime K203 Is the Most Effective Reactivator of Tabun-Inhibited Acetylcholinesterase. BMC Pharmacol. Toxicol. 2018, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuča, K.; Petroianu, G.A. Oximes as Pretreatment before Acute Exposure to Paraoxon. J. Appl. Toxicol. 2019, 39, 1506–1515. [Google Scholar] [CrossRef]
- Sakurada, K.; Ohta, H. No Promising Antidote 25 Years after the Tokyo Subway Sarin Attack: A Review. Leg. Med. 2020, 47, 101761. [Google Scholar] [CrossRef] [PubMed]
- Norrrahim, M.N.F.; Razak, M.A.I.A.; Shah, N.A.A.; Kasim, H.; Yusoff, W.Y.W.; Abdul Halim, N.; Nor, S.A.M.; Hasnawati Jamal, S.; Khim Ong, K.; Yunus, W.M.Z.W.; et al. Recent Developments on Oximes to Improve the Blood Brain Barrier Penetration for the Treatment of Organophosphorus Poisoning: A Review. RSC Adv. 2020, 10, 4465–4489. [Google Scholar] [CrossRef] [Green Version]
- Jaćević, V.; Nepovimova, E.; Kuča, K. Interspecies and Intergender Differences in Acute Toxicity of K-Oximes Drug Candidates. Chem. Biol. Interact. 2019, 308, 312–316. [Google Scholar] [CrossRef] [PubMed]
- PROTOPAM Chloride (Pralidoxime Chloride) for Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/014134s022lbl.pdf (accessed on 5 December 2021).
- Kassa, J. Review of Oximes in the Antidotal Treatment of Poisoning by Organophosphorus Nerve Agents. J. Toxicol. Clin. Toxicol. 2002, 40, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Worek, F.; Thiermann, H.; Wille, T. Oximes in Organophosphate Poisoning: 60 Years of Hope and Despair. Chem. Biol. Interact. 2016, 259, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Unnisa, L.; Sumakanth, M.; Rao, B.L.; Divi, M.K.; Rao, A. A Simple Process for the Preparation of Pralidoxime Chloride. Indian J. Chem. 2014, 4, 431–435. [Google Scholar]
- Berman, D.S.; Kiat, H.; Maddahi, J. The New 99mTc Myocardial Perfusion Imaging Agents: 99mTc-Sestamibi and 99mTc-Teboroxime. Circulation 1991, 84 (Suppl. 3), I7–I21. [Google Scholar]
- Beanlands, R.S.B.; de Kemp, R.A.; Harmsen, E.; Veinot, J.P.; Hartman, N.G.; Ruddy, T.D. Myocardial Kinetics of Technetium-99m Teboroxime in the Presence of Postischemic Injury, Necrosis and Low Flow Reperfusion. J. Am. Coll. Cardiol. 1996, 28, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.M.; Heel, R.C. Ceftizoxime A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Use. Drugs 1985, 29, 281–329. [Google Scholar] [CrossRef]
- Todd, W.M. Cefpodoxime Proxetil: A Comprehensive Review. Int. J. Antimicrob. Agents 1994, 4, 37–62. [Google Scholar] [CrossRef]
- Chugh, K.; Agrawal, S. Cefpodoxime: Pharmacokinetics and Therapeutic Uses. Indian J. Pediatr. 2003, 70, 227–231. [Google Scholar] [CrossRef]
- Frampton, J.E.; Brogden, R.N.; Langtry, H.D.; Buckley, M.M. Cefpodoxime Proxetil: A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Potential. Drugs 1992, 44, 889–917. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Kondo, M.; Kida, M.; Nakao, M.; Iwahi, T.; Nishi, T.; Noji, Y.; Takeuchi, M.; Nozaki, Y. Cefmenoxime (SCE-1365), a Novel Broad-Spectrum Cephalosporin: In Vitro and in Vivo Antibacterial Activities. Antimicrob. Agents Chemother. 1981, 19, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamm, J.M.; Girolami, R.L.; Shipkowitz, N.L.; Bower, R.R. Antimicrobial Activity of Cefmenoxime (SCE-1365). Antimicrob. Agents Chemother. 1981, 19, 454–460. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, C.H.; Sykes, R.B.; Griffiths, A.; Thornton, J.E. Cefuroxime, a New Cephalosporin Antibiotic: Activity In Vitro. Antimicrob. Agents Chemother. 1976, 9, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J.; Ormrod, D.; Goa, K.L. Cefuroxime Axetil: An Updated Review of Its Use in the Management of Bacterial Infections. Drugs 2001, 61, 1455–1500. [Google Scholar] [CrossRef]
- McSherry, B.A. Technetium-99m-Teboroxime: A New Agent for Myocardial Perfusion Imaging. J. Nucl. Med. Technol. 1991, 19, 5. [Google Scholar]
- Narra, R.K.; Nunn, A.D.; Kuczynski, B.L.; Feld, T.; Wedeking, P.; Eckelman, W.C. A Neutral Technetium-99m Complex for Myocardial Imaging. J. Nucl. Med. 1989, 30, 1830–1837. [Google Scholar]
- Kowalsky, J. Technetium Agents and Thallium for Myocardial Perfusion Imaging. Available online: https://pharmacyce.unm.edu/nuclear_program/freelessonfiles/vol1lesson2.pdf (accessed on 5 December 2021).
- Kane, S.M.; Davis, D.D. Technetium-99m. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ghosh, P.R. FDA Approves Two New Technetium-Labeled Cardiac Agents and a Pharmacologic Alternative to Exercise in Stress-Thallium Studies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1991, 32, 11N–14N. [Google Scholar]
- Okada, D.R.; Johnson, G.; Okada, R.D. Myocardial Clearance of Technetium-99m-Teboroxime in Reperfused Injured Canine Myocardium. EJNMMI Res. 2014, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.S.; Kiat, H.; Van Train, K.F.; Friedman, J.; Garcia, E.V.; Maddahi, J. Comparison of SPECT Using Technetium-99m Agents and Thallium-201 and PET for the Assessment of Myocardial Perfusion and Viability. Am. J. Cardiol. 1990, 66, E72–E79. [Google Scholar] [CrossRef]
- Labonté, C.; Taillefer, R.; Lambert, R.; Basile, F.; TonThat, T.; Jarry, M.; Léveillé, J. Comparison between Technetium-99m-Teboroxime and Thallium-201 Dipyridamole Planar Myocardial Perfusion Imaging in Detection of Coronary Artery Disease. Am. J. Cardiol. 1992, 69, 90–96. [Google Scholar] [CrossRef]
- Heatley, N.G. Alexander Fleming. The Man and the Myth. Med. Hist. 1984, 28, 453–455. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A. Classics in Infectious Diseases: On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. Influenzae by Alexander Fleming, Reprinted from the British Journal of Experimental Pathology 10:226-236, 1929. Rev. Infect. Dis. 1980, 2, 129–139. [Google Scholar] [PubMed]
- Vardanyan, R.S.; Hruby, V.J. Antibiotics. In Synthesis of Essential Drugs; Elsevier: Amsterdam, The Netherlands, 2006; pp. 425–498. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Dellamonica, P. Cefuroxime Axetil. Int. J. Antimicrob. Agents 1994, 4, 23–36. [Google Scholar] [CrossRef]
- Barza, M. Pharmacokinetics of Newer Cephalosporins After Subconjunctival and Intravitreal Injection in Rabbits. Arch. Ophthalmol. 1993, 111, 121. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, Y.; He, Z.; Liu, H.; Xue, K. Effects of Cefuroxime Axetil Combined with Xingpi Yanger Granules on the Serum Gastrin, Motilin, and Somatostatin Levels in Children with Upper Respiratory Tract Infection Accompanied by Diarrhea: Results of a Randomized Trial. Transl. Pediatr. 2021, 10, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Cefuroxime Advanced Patient Information. Available online: https://www.drugs.com/cons/cefuroxime.html (accessed on 12 October 2021).
- Fu, K.P.; Neu, H.C. Antibacterial Activity of Ceftizoxime, a Beta-Lactamase-Stable Cephalosporin. Antimicrob. Agents Chemother. 1980, 17, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cefizox Injection—FDA Prescribing Information, Side Effects and Uses. Available online: https://www.drugs.com/pro/cefizox-injection.html (accessed on 5 December 2021).
- Platt, R. Adverse Effects of Third-Generation Cephalosporins. J. Antimicrob. Chemother. 1982, 10 (Suppl. C), 135–140. [Google Scholar] [CrossRef]
- Stearne, L.E.T.; van Boxtel, D.; Lemmens, N.; Goessens, W.H.F.; Mouton, J.W.; Gyssens, I.C. Comparative Study of the Effects of Ceftizoxime, Piperacillin, and Piperacillin-Tazobactam Concentrations on Antibacterial Activity and Selection of Antibiotic-Resistant Mutants of Enterobacter Cloacae and Bacteroides Fragilis In Vitro and In Vivo in Mixed-Infection Abscesses. Antimicrob. Agents Chemother. 2004, 48, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zheng, X.; Zhong, W.; Chen, J.; Jiang, J.; Hu, P. Validation and Application of an LC–MS-MS Method for the Determination of Ceftizoxime in Human Serum and Urine. J. Chromatogr. Sci. 2016, 54, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Neu, H.C. Ceftizoxime: A Beta-Lactamase-Stable, Broad-Spectrum Cephalosporin. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1984, 4, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.E.; Blair, A.D.; Burgess, E.D.; Parks, D. Pharmacokinetics of Ceftizoxime. J. Antimicrob. Chemother. 1982, 10 (Suppl. C), 91–97. [Google Scholar] [CrossRef]
- Human Metabolome Database: Showing Metabocard for Ceftizoxime (HMDB0015427). Available online: https://hmdb.ca/metabolites/HMDB0015427 (accessed on 5 December 2021).
- Yanagawa, A.; Shimada, J.; Mori, N.; Sugihara, T.; Sakai, A.; Yamaji, S.; Yano, K.; Kitamura, T.; Kano, T. Effects of Gastrointestinal Stimulant and Suppressant Pretreatment on the Pharmacokinetics of AS-924, a Novel Ester-Type Cephem Antibiotic. Int. J. Antimicrob. Agents 2001, 18, 483–487. [Google Scholar] [CrossRef]
- Łęski, T.A.; Tomasz, A. Role of Penicillin-Binding Protein 2 (PBP2) in the Antibiotic Susceptibility and Cell Wall Cross-Linking of Staphylococcus Aureus: Evidence for the Cooperative Functioning of PBP2, PBP4, and PBP2A. J. Bacteriol. 2005, 187, 1815–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borin, M.T. A Review of the Pharmacokinetics of Cefpodoxime Proxetil. Drugs 1991, 42 (Suppl. 3), 13–21. [Google Scholar] [CrossRef]
- Liu, P.; Rand, K.H.; Obermann, B.; Derendorf, H. Pharmacokinetic-Pharmacodynamic Modelling of Antibacterial Activity of Cefpodoxime and Cefixime in in Vitro Kinetic Models. Int. J. Antimicrob. Agents 2005, 25, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Katara, R.; Ramteke, S. Enhancement of Bioavailability of Cefpodoxime Proxetil Using Different Polymeric Microparticles. AAPS PharmSciTech 2010, 11, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Boaretti, M.; Lleó, M.M.; Canepari, P. In Vitro Activity, Beta-Lactamase Stability and PBP Affinity of RU 51,746-2, the Active Metabolite of the New Orally Absorbed Cephalosporin Ester, RU 51807. J. Chemother. Florence Italy 1991, 3 (Suppl. 1), 57–61. [Google Scholar]
- Pangon, B.; Joly, V.; Vallois, J.M.; Abel, L.; Buré, A.; Brion, N.; Contrepois, A.; Carbon, C. Comparative Efficacy of Cefotiam, Cefmenoxime, and Ceftriaxone in Experimental Endocarditis and Correlation with Pharmacokinetics and in Vitro Efficacy. Antimicrob. Agents Chemother. 1987, 31, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Granneman, G.R.; Sennello, L.T.; Steinberg, F.J.; Sonders, R.C. Intramuscular and Intravenous Pharmacokinetics of Cefmenoxime, a New Broad-Spectrum Cephalosporin, in Healthy Subjects. Antimicrob. Agents Chemother. 1982, 21, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Human Metabolome Database: Showing Metabocard for Cefmenoxime (HMDB0014412). Available online: https://hmdb.ca/metabolites/HMDB0014412 (accessed on 5 December 2021).
- Perry, C.M.; Brogden, R.N. Cefuroxime Axetil: A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1996, 52, 125–158. [Google Scholar] [CrossRef] [PubMed]
- Sommers, D.K.; Van Wyk, M.; Williams, P.E.; Harding, S.M. Pharmacokinetics and Tolerance of Cefuroxime Axetil in Volunteers during Repeated Dosing. Antimicrob. Agents Chemother. 1984, 25, 344–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Ohtani, K.; Hirakawa, H.; Ohshima, K.; Yamashita, A.; Shiba, T.; Ogasawara, N.; Hattori, M.; Kuhara, S.; Hayashi, H. Complete Genome Sequence of Clostridium Perfringens, an Anaerobic Flesh-Eater. Proc. Natl. Acad. Sci. USA 2002, 99, 996–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahwa, R.; Rana, A.S.; Dhiman, S.; Negi, P.; Singh, I. Cefpodoxime Proxetil: An Update on Analytical, Clinical and Pharmacological Aspects. J. Curr. Chem. Pharm. 2015, 5, 56–66. [Google Scholar]
- Chocas, E.C.; Paap, C.M.; Godley, P.J. Cefpodoxime Proxetil: A New, Broad-Spectrum, Oral Cephalosporin. Ann. Pharmacother. 1993, 27, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Cefpodoxime Proxetil (Vantin®). Available online: https://www.reliasmedia.com/articles/139852-cefpodoxime-proxetil-vantin (accessed on 12 October 2021).
- El-Shabrawi, M.H.; Tolba, O.A.; El-Adly, T.Z. Efficacy and Safety of Cefpodoxime in the Treatment of Acute Otitis Media in Children. Egypt. Pediatr. Assoc. Gaz. 2016, 64, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Cefpodoxime Uses, Side Effects & Warnings. Available online: https://www.drugs.com/mtm/cefpodoxime.html (accessed on 12 October 2021).
- Rodríguez, J. An Improved Method for Preparation of Cefpodoxime Proxetil. Il Farm. 2003, 58, 363–369. [Google Scholar] [CrossRef]
- Fuchs, P.C.; Jones, R.N.; Thornsberry, C.; Barry, A.L.; Gerlach, E.H.; Sommers, H.M. Cefmenoxime (SCE-1365), a New Cephalosporin: In Vitro Activity, Comparison with Other Antimicrobial Agents, Beta-Lactamase Stability, and Disk Diffusion Testing with Tentative Interpretive Criteria. Antimicrob. Agents Chemother. 1981, 20, 747–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambertoglio, J.G.; Alexander, D.P.; Barriere, S.L. Cefmenoxime Pharmacokinetics in Healthy Volunteers and Subjects with Renal Insufficiency and on Hemodialysis. Antimicrob. Agents Chemother. 1984, 26, 845–849. [Google Scholar] [CrossRef] [Green Version]
- Sica, D.A.; Polk, R.E.; Kerkering, T.M.; Patterson, P.; Baggett, J. Cefmenoxime Kinetics during Continuous Ambulatory Peritoneal Dialysis. Eur. J. Clin. Pharmacol. 1986, 30, 713–717. [Google Scholar] [CrossRef]
- Cefmax Generic. Price of Cefmax. Uses, Dosage, Side Effects. Available online: https://www.ndrugs.com/?s=cefmax (accessed on 12 October 2021).
- Zhang, X. Cefmenoxime Compound and Synthetic Method Thereof. PCT Int. Appl. CN1709880, 20 June 2007. [Google Scholar]














| Oxime | EC50 (mg/L) | Tmax (min) | Cmax (µM) | T1/2 min | Status |
|---|---|---|---|---|---|
| 2-PAM [62] | 4.67 | 10 | 37–132 | 37.6–55 | Approved |
| Obidoxime [73,74,75] | 57.0 | 4.5 | 78 | 28.4 | Approved |
| HI-6 [37,73,76] | 304 | 5.8 | 138 | 25.7 | Approved |
| RS194B [77] | ND | 5 | 112 | 52 | Candidate |
| K027 [76,78] | 229 | 5 | 586 | 60 | Candidate |
| K203 [79] | ND | 22 | 242 | 101 | Candidate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhuguru, J.; Zviagin, E.; Skouta, R. FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals 2022, 15, 66. https://doi.org/10.3390/ph15010066
Dhuguru J, Zviagin E, Skouta R. FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals. 2022; 15(1):66. https://doi.org/10.3390/ph15010066
Chicago/Turabian StyleDhuguru, Jyothi, Eugene Zviagin, and Rachid Skouta. 2022. "FDA-Approved Oximes and Their Significance in Medicinal Chemistry" Pharmaceuticals 15, no. 1: 66. https://doi.org/10.3390/ph15010066
APA StyleDhuguru, J., Zviagin, E., & Skouta, R. (2022). FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals, 15(1), 66. https://doi.org/10.3390/ph15010066