
Development of 3D-Printed, Liquisolid and Directly Compressed Glimepiride Tablets, Loaded with Black Seed Oil Self-Nanoemulsifying Drug Delivery System: In Vitro and In Vivo Characterization

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Development of Glimepiride Liquisolid and Compressed Tablets
2.2. Development of Three-Dimensional Printed Tablets
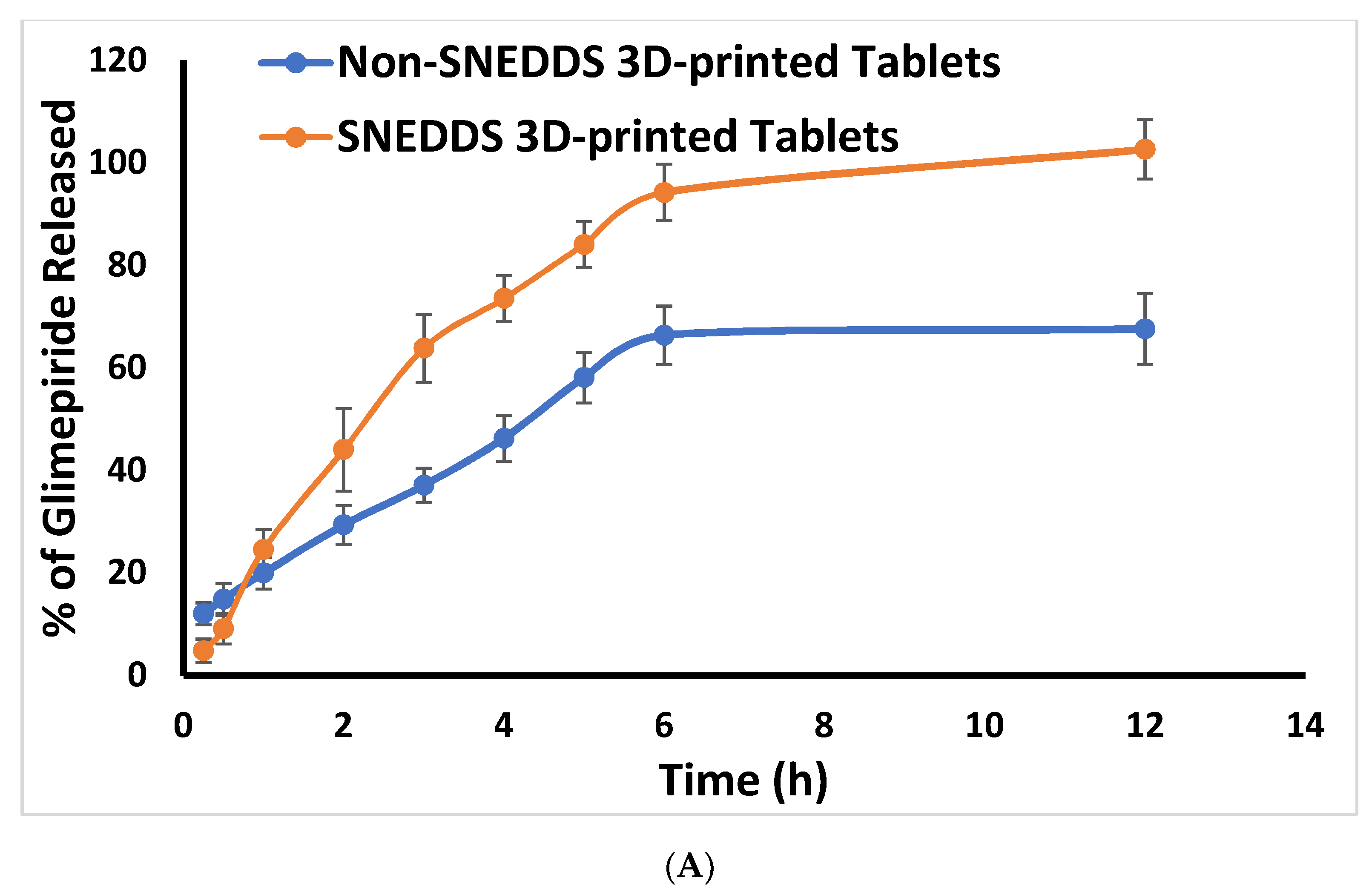
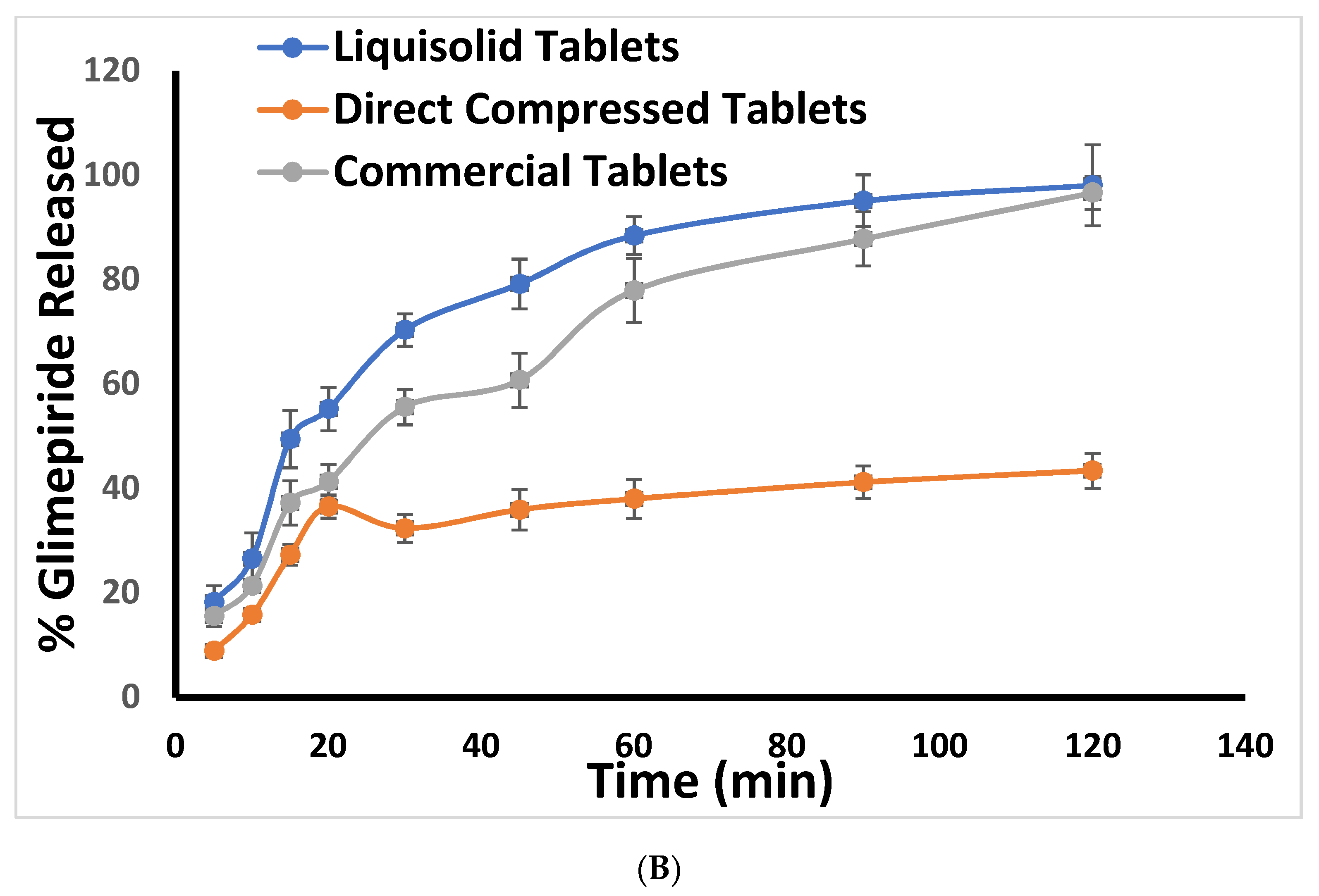
2.3. In Vitro Release Study
2.4. Scanning Electron Microscope (SEM)
2.5. In Vivo Study
3. Materials and Methods
3.1. Materials
3.2. Preparation and Characterization of Glimepiride Loaded SNEDDS
3.3. Preparation and Characterization of Glimepiride Liquisolid Tablets
3.4. Development of Three-Dimensional Printed Tablets
3.5. In Vitro Release Study
3.6. Scanning Electron Microscope (SEM)
3.7. In Vivo Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lura, A.; Tardy, G.; Kleinebudde, P.; Breitkreutz, J. Tableting of mini-tablets in comparison with conventionally sized tablets: A comparison of tableting properties and tablet dimensions. Int. J. Pharm. X 2020, 2, 100061. Available online: https://pubmed.ncbi.nlm.nih.gov/33294842/ (accessed on 29 November 2021). [CrossRef]
- Chandrasekhar, R.; Hassan, Z.; AlHusban, F.; Smith, A.M.; Mohammed, A.R. The role of formulation excipients in the development of lyophilised fast-disintegrating tablets. Eur. J. Pharm. Biopharm. 2009, 72, 119–129. Available online: http://linkinghub.elsevier.com/retrieve/pii/S0939641108004517 (accessed on 29 November 2021). [CrossRef]
- Kharshoum, R.M.; Sanad, R.A.; Abdelhaleem Ali, A.M. Comparative pharmacokinetic study of two lyophilized orally disintegrating tablets formulations of vinpocetine in human volunteers. Int. J. Drug Deliv. 2013, 5, 167. Available online: http://www.arjournals.org/index.php/ijdd/index (accessed on 29 November 2021).
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of tablets containing multiple drugs with defined release profiles. Int. J. Pharm. 2015, 494, 643–650. [Google Scholar] [CrossRef]
- Aguilar-de-Leyva, Á.; Linares, V.; Casas, M.; Caraballo, I. 3D Printed Drug Delivery Systems Based on Natural Products. Pharmaceutics 2020, 12, 620. [Google Scholar] [CrossRef]
- Jamróz, W.; Szafraniec, J.; Kurek, M.; Jachowicz, R. 3D Printing in Pharmaceutical and Medical Applications—Recent Achievements and Challenges. Pharm. Res. 2018, 35, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aimar, A.; Palermo, A.; Innocenti, B. The Role of 3D Printing in Medical Applications: A State of the Art. J. Healthc. Eng. 2019, 2019, 5340616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katstra, W.E.; Palazzolo, R.D.; Rowe, C.W.; Giritlioglu, B.; Teung, P.; Cima, M.J. Oral dosage forms fabricated by Three Dimensional Printing(TM). J. Control. Release 2000, 66, 1–9. [Google Scholar] [CrossRef]
- Badian, M.; Korn, A.; Lehr, K.H.; Malerczyk, V.; Waldhäusl, W. Absolute bioavailability of glimepiride (Amaryl) after oral administration. Drug Metabol. Drug Interact. 2016, 11, 331–340. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12369756 (accessed on 29 November 2021). [CrossRef]
- Ahmed, T.A.; Suhail, M.A.A.; Hosny, K.M.; Abd-Allah, F.I. Clinical pharmacokinetic study for the effect of glimepiride matrix tablets developed by quality by design concept. Drug Dev. Ind. Pharm. 2018, 44, 66–81. Available online: https://www.tandfonline.com/doi/full/10.1080/03639045.2017.1371740 (accessed on 29 November 2021). [CrossRef]
- Ning, X.; Sun, J.; Han, X.; Wu, Y.; Yan, Z.; Han, J.; He, Z. Strategies to improve dissolution and oral absorption of glimepiride tablets: Solid dispersion versus micronization techniques. Drug Dev. Ind. Pharm. 2011, 37, 727–736. Available online: https://pubmed.ncbi.nlm.nih.gov/21204747/ (accessed on 29 November 2021). [CrossRef]
- Qushawy, M.; Nasr, A.; Swidan, S.; Mortagi, Y. Development and characterization of glimepiride novel solid nanodispersion for improving its oral bioavailability. Sci. Pharm. 2020, 88, 52. [Google Scholar] [CrossRef]
- Pal, A.; Roy, S.; Kumar, A.; Mahmood, S.; Khodapanah, N.; Thomas, S.; Agatemor, C.; Ghosal, K. Physicochemical Characterization, Molecular Docking, and In Vitro Dissolution of Glimepiride–Captisol Inclusion Complexes. ACS Omega 2020, 5, 19968–19977. Available online: https://pubs.acs.org/doi/abs/10.1021/acsomega.0c01228 (accessed on 29 November 2021). [CrossRef]
- Li, H.; Pan, T.; Cui, Y.; Li, X.; Gao, J.; Yang, W.; Shen, S. Improved oral bioavailability of poorly water-soluble glimepiride by utilizing microemulsion technique. Int. J. Nanomed. 2016, 11, 3777–3788. Available online: https://pubmed.ncbi.nlm.nih.gov/27540291/ (accessed on 29 November 2021).
- Al-Madhagi, W.; Abdulbari Albarakani, A.; Khaled Alhag, A.; Ahmed Saeed, Z.; Mansour Noman, N.; Mohamed, K. Formulation and Evaluation of New Glimepiride Sublingual Tablets. J. Pharm. 2017, 2017, 690473. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, A.; Haji Idrus, R.; Mokhtar, M.H. Effects of Nigella Sativa on Type-2 Diabetes Mellitus: A Systematic Review. Int. J. Environ. Res. Public Health 2019, 16, 4911. Available online: https://pubmed.ncbi.nlm.nih.gov/31817324/ (accessed on 29 November 2021). [CrossRef] [Green Version]
- Shoaei-Hagh, P.; Kamelan Kafi, F.; Najafi, S.; Zamanzadeh, M.; Heidari Bakavoli, A.; Ramezani, J.; Soltanian, S.; Asili, J.; Hosseinzadeh, H.; Eslami, S.; et al. A randomized, double-blind, placebo-controlled, clinical trial to evaluate the benefits of Nigella sativa seeds oil in reducing cardiovascular risks in hypertensive patients. Phyther. Res. 2021, 35, 4388–4400. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Husain, A.; Mujeeb, M.; Khan, S.A.; Najmi, A.K.; Siddique, N.A.; Damanhouri, Z.A.; Anwar, F. A review on therapeutic potential of Nigella sativa: A miracle herb. Asian Pac. J. Trop Biomed. 2013, 3, 337–352. Available online: https://pubmed.ncbi.nlm.nih.gov/23646296/ (accessed on 29 November 2021). [CrossRef] [Green Version]
- Journal, A.I.; Usmani, A.; Mishra, A.; Jafri, A. Development and evaluation of doxorubicin self nanoemulsifying drug delivery system with Nigella Sativa oil against human hepatocellular carcinoma Development and evaluation of doxorubicin self nanoemulsifying drug delivery. Artif. Cells Nanomed. Biotechnol. 2019, 47, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Halder, S.; Islam, A.; Muhit, M.A.; Shill, M.C.; Haider, S.S. Self-emulsifying drug delivery system of black seed oil with improved hypotriglyceridemic effect and enhanced hepatoprotective function. J. Funct. Foods 2021, 78, 104391. [Google Scholar] [CrossRef]
- Mohsin, K.; Long, M.A.; Pouton, C.W. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: Precipitation of drug after dispersion of formulations in aqueous solution. J. Pharm. Sci. 2009, 98, 3582–3595. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. Available online: https://pubmed.ncbi.nlm.nih.gov/16815001/ (accessed on 29 November 2021). [CrossRef]
- Shahba, A.A.-W.; Mohsin, K.; Alanazi, F.K. Novel Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Oral Delivery of Cinnarizine: Design, Optimization, and In-Vitro Assessment. AAPS PharmSciTech 2012, 13, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baloch, J.; Sohail, M.F.; Sarwar, H.S.; Kiani, M.H.; Khan, G.M.; Jahan, S.; Rafay, M.; Chaudhry, M.T.; Yasinzai, M.; Shahnaz, G. Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Improved Oral Bioavailability of Chlorpromazine: In Vitro and In Vivo Evaluation. Medicina 2019, 55, 210. Available online: https://pubmed.ncbi.nlm.nih.gov/31137751/ (accessed on 29 November 2021). [CrossRef] [Green Version]
- Abdallah, H.M.; El-Bassossy, H.M.; El-Halawany, A.M.; Ahmed, T.A.; Mohamed, G.A.; Malebari, A.M.; Hassan, N.A. Self-Nanoemulsifying Drug Delivery System Loaded with Psiadia punctulata Major Metabolites for Hypertensive Emergencies: Effect on Hemodynamics and Cardiac Conductance. Front. Pharmacol. 2021, 12, 681070. [Google Scholar] [CrossRef] [PubMed]
- The United States Pharmacopeia TNF. USP 28/NF 23; US Pharmacopoeial Convention Inc.: Rockville, MD, USA, 2005.
- Zidan, A.; Alayoubi, A.; Coburn, J.; Asfari, S.; Ghammraoui, B.; Cruz, C.N.; Ashraf, M. Extrudability analysis of drug loaded pastes for 3D printing of modified release tablets. Int. J. Pharm. 2019, 554, 292–301. [Google Scholar] [CrossRef]
- Rahman, L.; Rowe, P.; Cheyne, A.; Wilson, D.I. Ram extrusion of potato starch dough through multi-holed dies. Food Bioprod. Process Trans. Inst. Chem. Eng. Part C 2002, 80, 12–19. [Google Scholar] [CrossRef]
- Zhang, M.; Rough, S.L.; Ward, R.; Seiler, C.; Wilson, D.I. A comparison of ram extrusion by single-holed and multi-holed dies for extrusion-spheronisation of microcrystalline-based pastes. Int. J. Pharm 2011, 416, 210–222. Available online: https://pubmed.ncbi.nlm.nih.gov/21742021/ (accessed on 29 November 2021). [CrossRef]
- Oh, C.M.; Heng, P.W.S.; Chan, L.W. Influence of Hydroxypropyl Methylcellulose on Metronidazole Crystallinity in Spray-Congealed Polyethylene Glycol Microparticles and Its Impact with Various Additives on Metronidazole Release. AAPS PharmSciTech 2015, 16, 1357–1367. Available online: https://link.springer.com/article/10.1208/s12249-015-0320-2 (accessed on 29 November 2021). [CrossRef] [Green Version]
- Kurakula, M.; Rao, G.K. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): As excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. Available online: https://pubmed.ncbi.nlm.nih.gov/32905026/ (accessed on 29 November 2021). [CrossRef]
- Hardy, I.J.; Windberg-Baarup, A.; Neri, C.; Byway, P.V.; Booth, S.W.; Fitzpatrick, S. Modulation of drug release kinetics from hydroxypropyl methyl cellulose matrix tablets using polyvinyl pyrrolidone. Int. J. Pharm. 2007, 337, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, R.H.; Kassem, M.A. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2008, 69, 993–1003. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0939641108000556 (accessed on 29 November 2021). [CrossRef] [PubMed]
- Lu, M.; Xing, H.; Jiang, J.; Chen, X.; Yang, T.; Wang, D.; Ding, P. Liquisolid technique and its applications in pharmaceutics. Asian J. Pharm. Sci. 2017, 12, 115–123. Available online: https://www.sciencedirect.com/science/article/pii/S1818087616301726 (accessed on 29 November 2021). [CrossRef] [Green Version]
- Ahmed, T.A.; Felimban, R.I.; Tayeb, H.H.; Rizg, W.Y.; Alnadwi, F.H.; Alotaibi, H.A.; Alhakamy, N.A.; Abd-Allah, F.I.; Mohamed, G.A.; Zidan, A.S.; et al. Development of Multi-Compartment 3D-Printed Tablets Loaded with Self-Nanoemulsified Formulations of Various Drugs: A New Strategy for Personalized Medicine. Pharmaceutics 2021, 13, 1733. [Google Scholar] [CrossRef]
- Roy, A.; Roy, K.; Roy, S.; Deb, J.; Ghosh, A.; Ali, K.A. Response surface optimization of sustained release metformin-hydrochloride matrix tablets: Influence of some hydrophillic polymers on the release. ISRN Pharm. 2012, 2012, 364261. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22988527 (accessed on 29 November 2021). [CrossRef] [Green Version]
- Ahmed, T.A. Study the pharmacokinetics, pharmacodynamics and hepatoprotective activity of rosuvastatin from drug loaded lyophilized orodispersible tablets containing transfersomes nanoparticles. J. Drug Deliv. Sci. Technol. 2021, 63, 102489. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Elimam, H.; Alrifai, A.O.; Nadhrah, H.M.; Masoudi, L.Y.; Sairafi, W.O.; Khalid, M. Rosuvastatin lyophilized tablets loaded with flexible chitosomes for improved drug bioavailability, anti-hyperlipidemic and anti-oxidant activity. Int. J. Pharm. 2020, 588, 119791. [Google Scholar] [CrossRef]
- Sheehan, C. General Chapters: <1174> POWDER FLOW. In USP29-NF24; 2013; p. 3017. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_c1174.html (accessed on 29 November 2021).
- USP 41-NF 36. The united States Pharmacopeia National Formulary. In Twinbrook Parkway; The United States Pharmacopeial Convention: Rockville, MD, USA, 2018.
- Zhao, F.; Malayev, V.; Rao, V.; Hussain, M. Effect of sodium lauryl sulfate in dissolution media on dissolution of hard gelatin capsule shells. Pharm. Res. 2004, 21, 144–148. Available online: https://pubmed.ncbi.nlm.nih.gov/14984269/ (accessed on 29 November 2021). [CrossRef]
- Ahmed, O.A.A.; Ahmed, T.A.; Abdel-naim, A.B.; Khedr, A.; Banjar, Z.M.; Afouna, M.I. Enhancement of In Vitro Skin Transport and In Vivo Hypoglycemic Efficacy of Glimepiride Transdermal Patches. Trop J. Pharm. Res. 2014, 13, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- El-Say, K.M.; Ahmed, T.A.; Ahmed, O.A.A.; Elimam, H. Enhancing the Hypolipidemic Effect of Simvastatin in Poloxamer-Induced Hyperlipidemic Rats via Liquisolid Approach: Pharmacokinetic and Pharmacodynamic Evaluation. AAPS Pharm. Sci. Tech. 2020, 21, 223. [Google Scholar] [CrossRef]
- Medica Mondiale, A. The Helsinki Declaration. Assist. Inferm. Ric. 2010, 29, 41–44. Available online: https://pubmed.ncbi.nlm.nih.gov/20514811/ (accessed on 29 November 2021).
- Stanciu, C. Ethics, research, and Helsinki declaration. Rev. Medico-Chir. Soc. Med. Nat. Din Iaş̧i. 2001, 105, 626–627. [Google Scholar]
- Mahajan, T.C.; Deshmukh, S.B.; Badgujar, V.L.; Chhajed, P.N.; Patil, J.A. RP-HPLC Method Development and Validation for estimation of Metformin, Voglibose, Glimepiride in Bulk and Combined Tablet Dosage Forms. Int. J. Chem. Pharm. Sci. 2016, 4, 697–702. Available online: www.pharmaresearchlibrary.com/ijcps (accessed on 29 November 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Three-Dimensional Printed Tablets | Liquisolid Tablets | Directly Compressed Tablets | ||
|---|---|---|---|---|---|
| Non-SNEDDS | SNEDDS | ||||
| Composition | Drug carrier/vehicle | Distilled water | SNEDDS | SNEDDS | - |
| Carrier (mg) | - | 100 | 100 | - | |
| Neusilin (mg) | 149.53 | 149.53 | 149.53 | 149.53 | |
| Avicel (mg) | 0.47 | 0.47 | 0.47 | 0.47 | |
| FujiSil (mg) | 10 | 10 | 10 | 10 | |
| PVPK90 | 210 | 210 | - | - | |
| Ac-Di-Sol (mg) | 21.6 | 37.6 | 20.8 | 12.8 | |
| Characteristics | Actual Weight (mg) | 432.53 ± 21.76 | 551.68 ± 31.02 | 291.14 ± 2.54 | 181.683 ± 1.076 |
| Thickness (mm) | 2.647 ± 0.160 | 3.239 ± 0.178 | 4.067 ± 0.046 | 3.996 ± 0.026 | |
| Diameter (mm) | 13.289 ± 0.485 | 14.855 ± 0.319 | 9 ± 0 | 9 ± 0 | |
| Friability (%) | 0.159 | 0.117 | 0.019 | 0.384 | |
| Drug content (mg) | 1.98 ± 0.201 | 1.92 ± 0.219 | 2.080 ± 0.058 | 1.89 ± 0.46 | |
| Disintegration time (min) | >60 | >60 | 3.59 ± 0.68 | 8.40 ± 0.05 | |
| Pharmacokinetic Parameters | Three-Dimensional Printed Tablets | Liquisolid Tablets | Directly Compressed Tablets |
|---|---|---|---|
| Tmax [h] | 2 ± 0 | 2 ± 0 | 4 ± 0 |
| Cmax [ng/mL] | 2223.671 ± 96.877 | 2310.333 ± 20.793 | 1013.333 ± 7.214 |
| Kab [h−1] | 0.894 ± 0.004 | 0.9021 ± 0.0008 | 1.547055 ± 0.029302 |
| t½ab [h] | 0.776 ± 0.003 | 0.7680 ± 0.0007 | 0.448056 ± 0.008503 |
| Kel [h−1] | 0.098 ± 0.004 | 0.1071 ± 0.0015 | 0.086805 ± 0.007111 |
| t½el [h] | 7.083 ± 0.272 | 6.456 ± 0.089 | 8.0195 ± 0.6672 |
| Vd [(mg)/(ng/mL)] | 0.0023 ± 0.0008 | 0.0023 ± 0.0001 | 0.00614 ± 0.00031 |
| TCR [ml/min] | 0.0037 ± 0.0004 | 0.0041 ± 0.0001 | 0.0089 ± 0.0003 |
| AUC(0–24) [ng/mL × h] | 17,061.58 ± 469.14 | 15,965.751 ± 260.806 | 7330.917 ± 170.392 |
| AUC(24–end) [ng/mL × h] | 1573.996 ± 1360.753 | 2105.403 ± 29.163 | 2615.314 ± 217.465 |
| AUC(0–end) [ng/mL × h] | 18,635.582 ± 1157.246 | 18,071.153 ± 246.925 | 9946.230 ± 362.789 |
| AUMC(0–24) [ng/mL × h2] | 131,177.601 ± 3105.725 | 115,564.667 ± 2328.201 | 56,315.921 ± 2452.962 |
| AUMC(24–end) [ng/mL × h2] | 38,034.13 ± 32,210.89 | 50,529.667 ± 699.902 | 62,767.530 ± 5219.163 |
| AUMC(0–end) [ng/mL × h2] | 169,211.711 ± 31,109.462 | 166,094.334 ± 2230.636 | 119,083.413 ± 7549.560 |
| MRT [h] | 9.035 ± 1.175 | 9.191 ± 0.056 | 11.965 ± 0.339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, T.A.; Alotaibi, H.A.; Alharbi, W.S.; Safo, M.K.; El-Say, K.M. Development of 3D-Printed, Liquisolid and Directly Compressed Glimepiride Tablets, Loaded with Black Seed Oil Self-Nanoemulsifying Drug Delivery System: In Vitro and In Vivo Characterization. Pharmaceuticals 2022, 15, 68. https://doi.org/10.3390/ph15010068
Ahmed TA, Alotaibi HA, Alharbi WS, Safo MK, El-Say KM. Development of 3D-Printed, Liquisolid and Directly Compressed Glimepiride Tablets, Loaded with Black Seed Oil Self-Nanoemulsifying Drug Delivery System: In Vitro and In Vivo Characterization. Pharmaceuticals. 2022; 15(1):68. https://doi.org/10.3390/ph15010068
Chicago/Turabian StyleAhmed, Tarek A., Hanadi A. Alotaibi, Waleed S. Alharbi, Martin K. Safo, and Khalid M. El-Say. 2022. "Development of 3D-Printed, Liquisolid and Directly Compressed Glimepiride Tablets, Loaded with Black Seed Oil Self-Nanoemulsifying Drug Delivery System: In Vitro and In Vivo Characterization" Pharmaceuticals 15, no. 1: 68. https://doi.org/10.3390/ph15010068
APA StyleAhmed, T. A., Alotaibi, H. A., Alharbi, W. S., Safo, M. K., & El-Say, K. M. (2022). Development of 3D-Printed, Liquisolid and Directly Compressed Glimepiride Tablets, Loaded with Black Seed Oil Self-Nanoemulsifying Drug Delivery System: In Vitro and In Vivo Characterization. Pharmaceuticals, 15(1), 68. https://doi.org/10.3390/ph15010068