
Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex


Abstract
:
1. Introduction
2. Results and Discussion
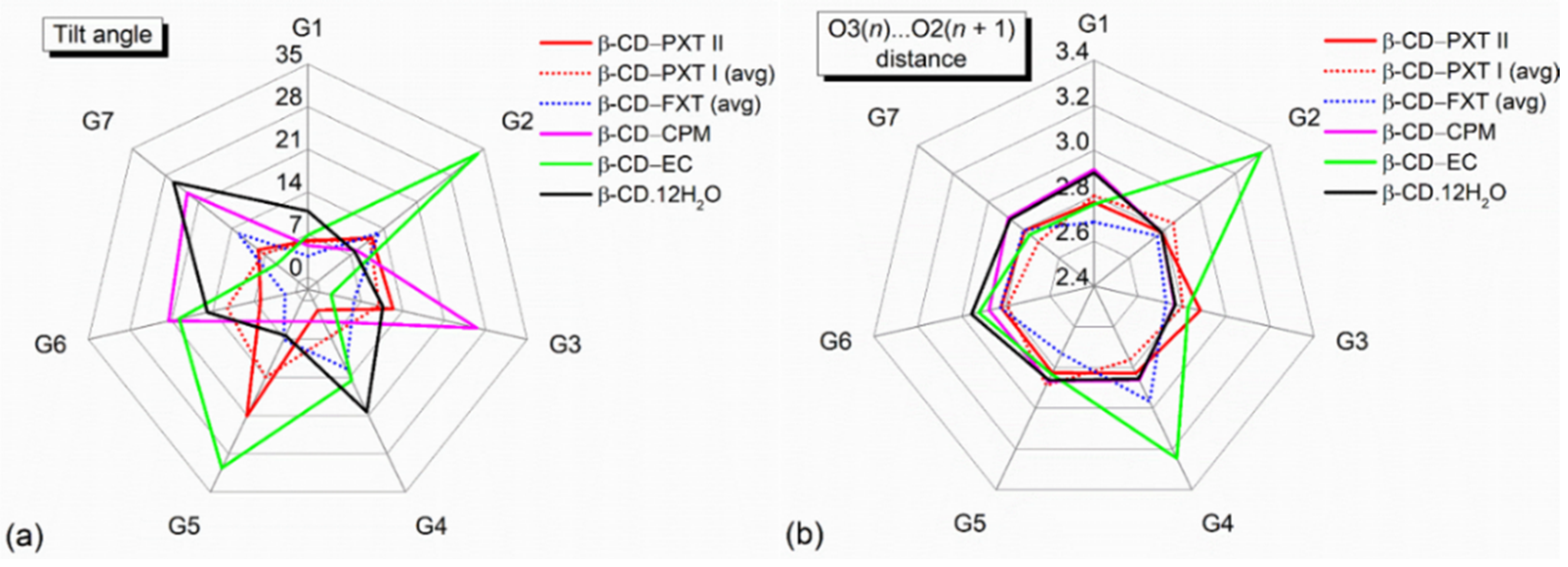
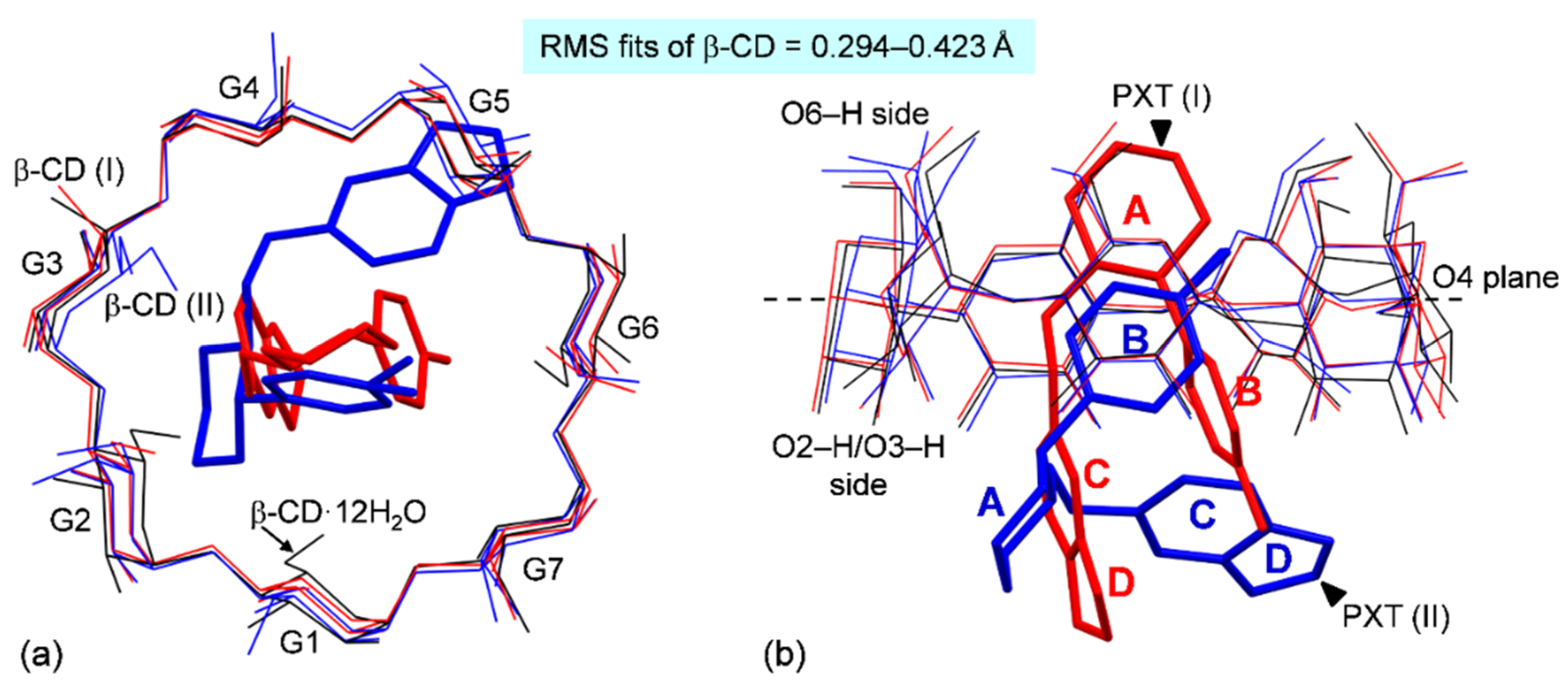
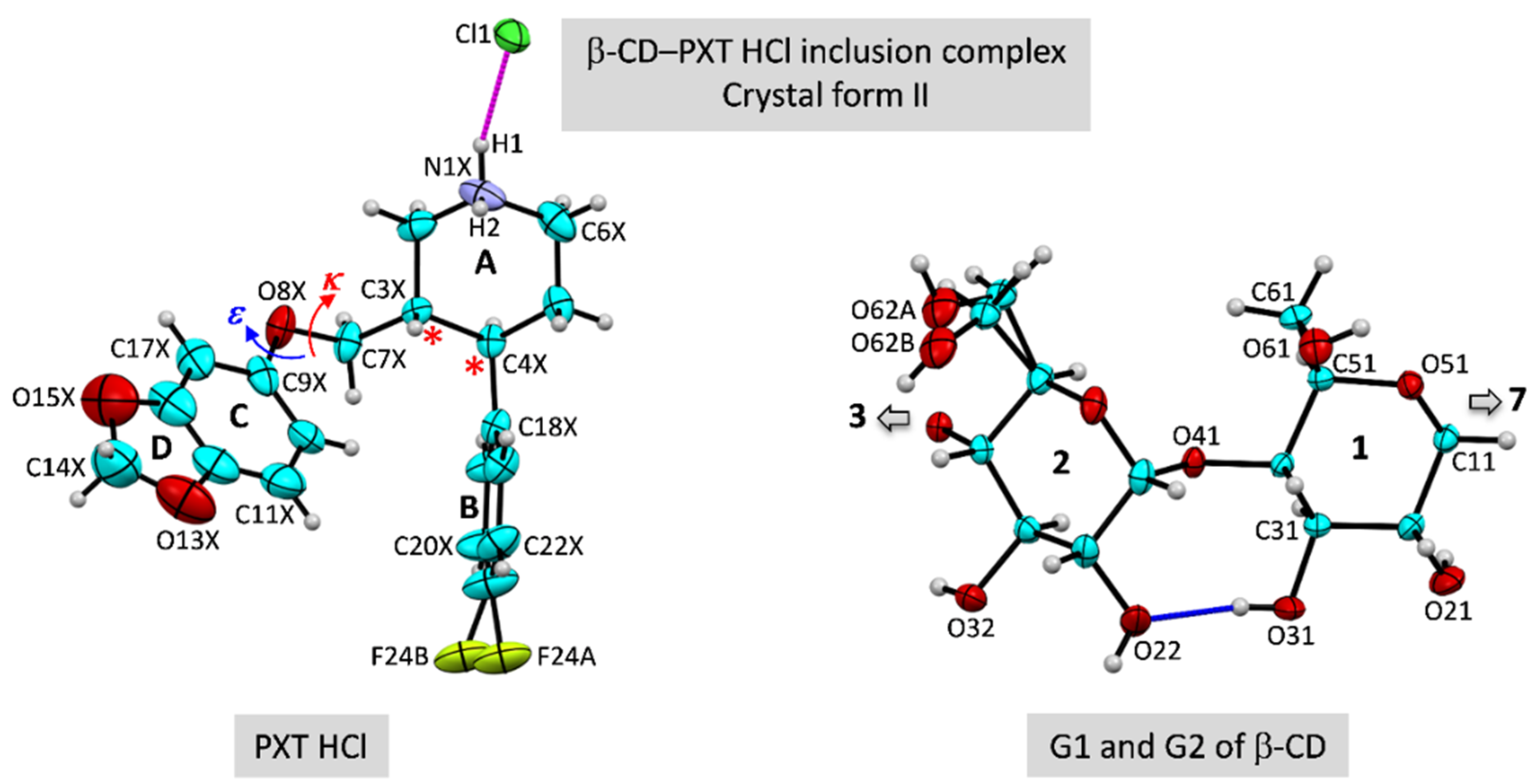
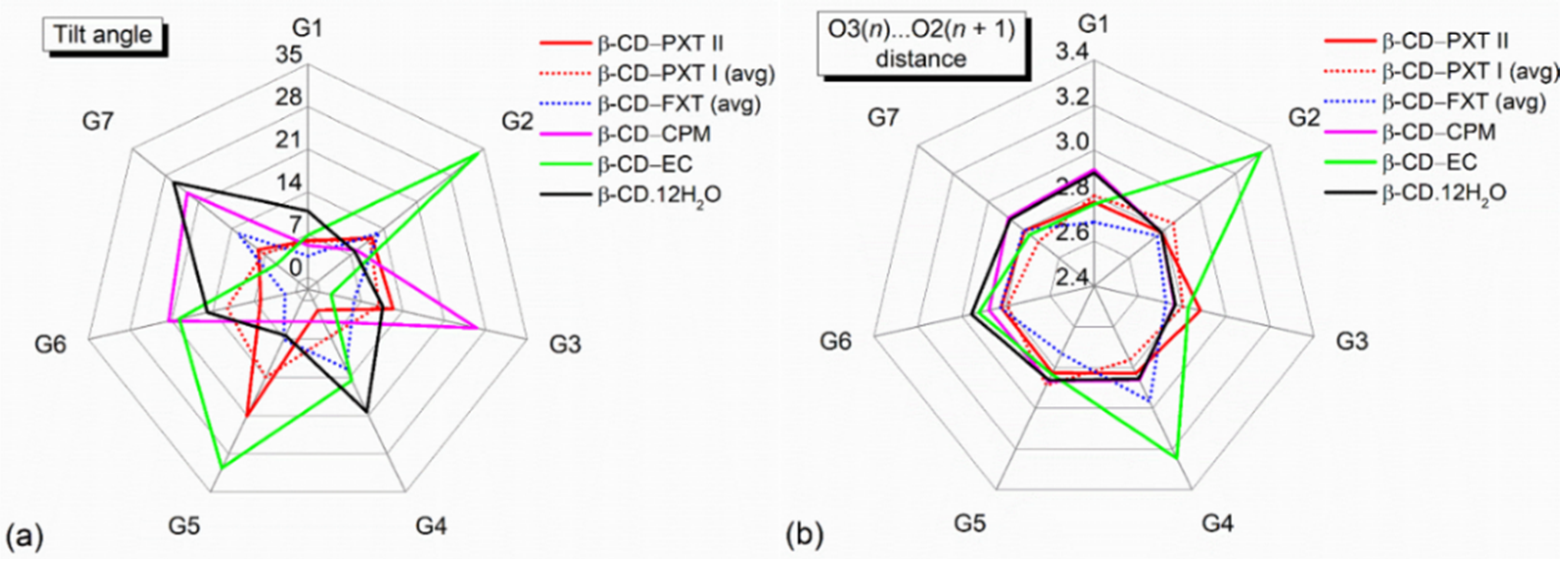
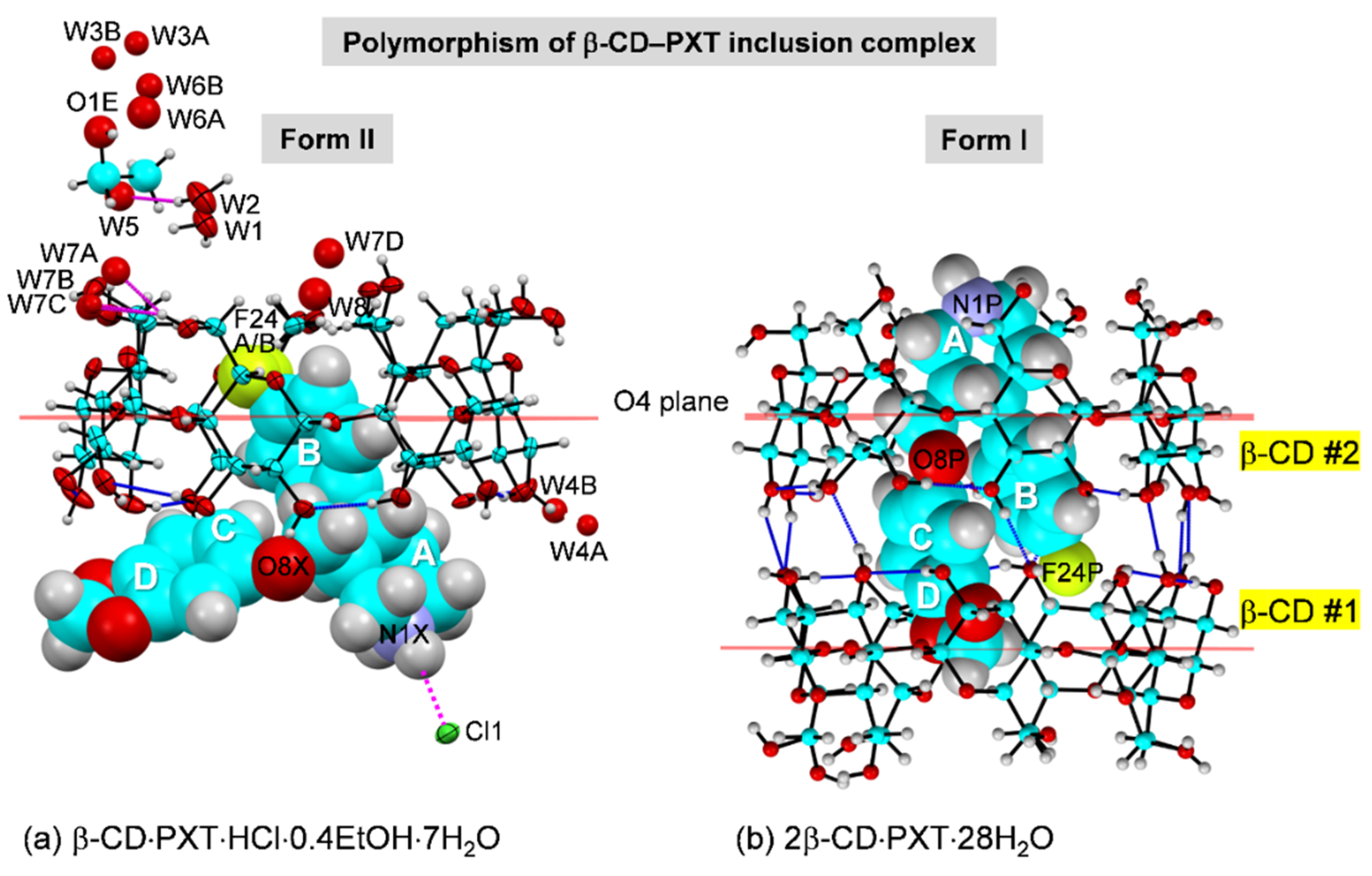
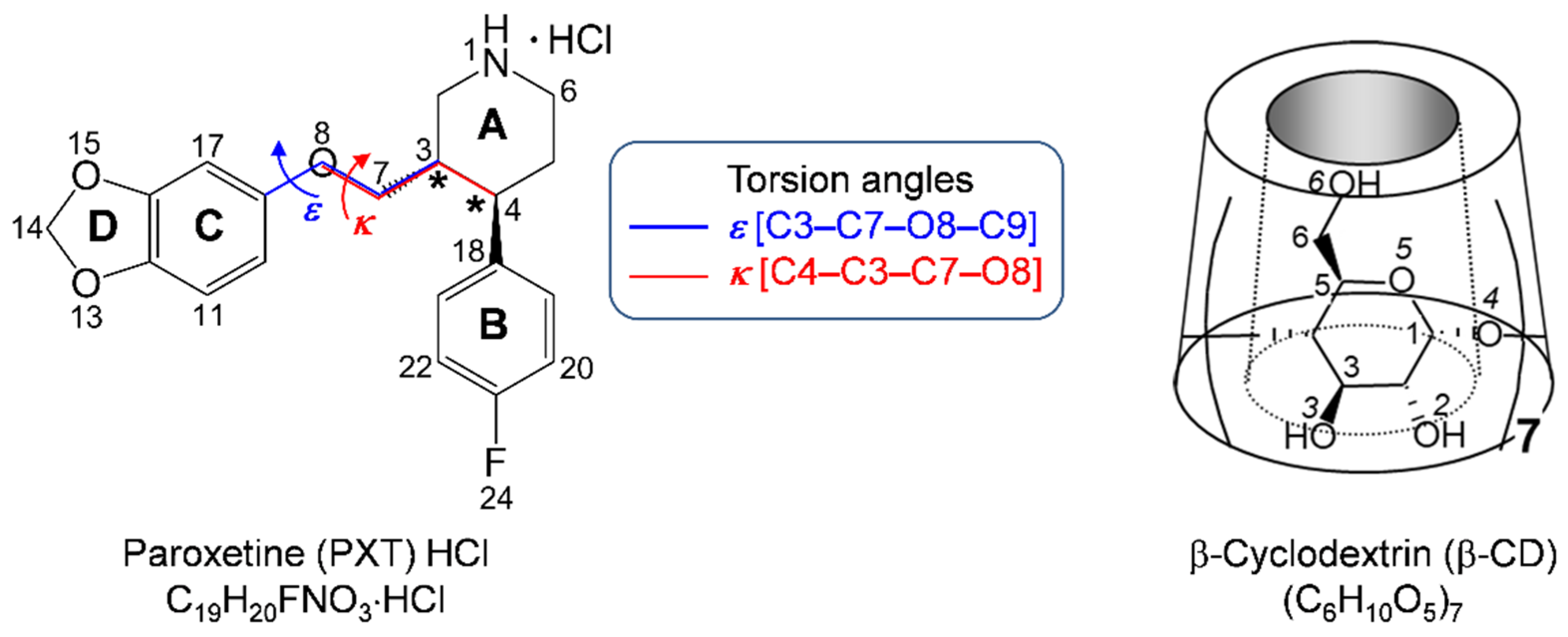
2.1. Marginal β-CD Structural Changes upon Inclusion of PXT
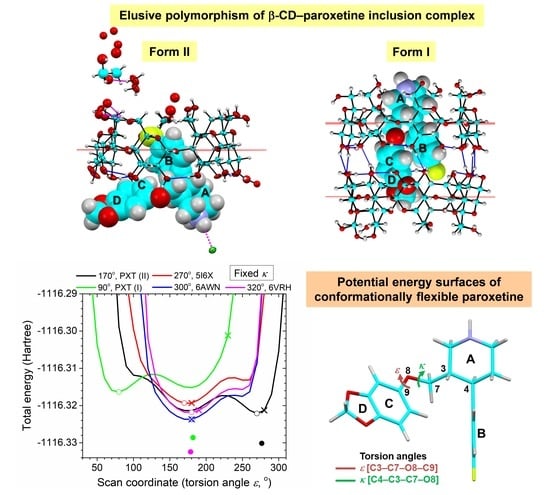
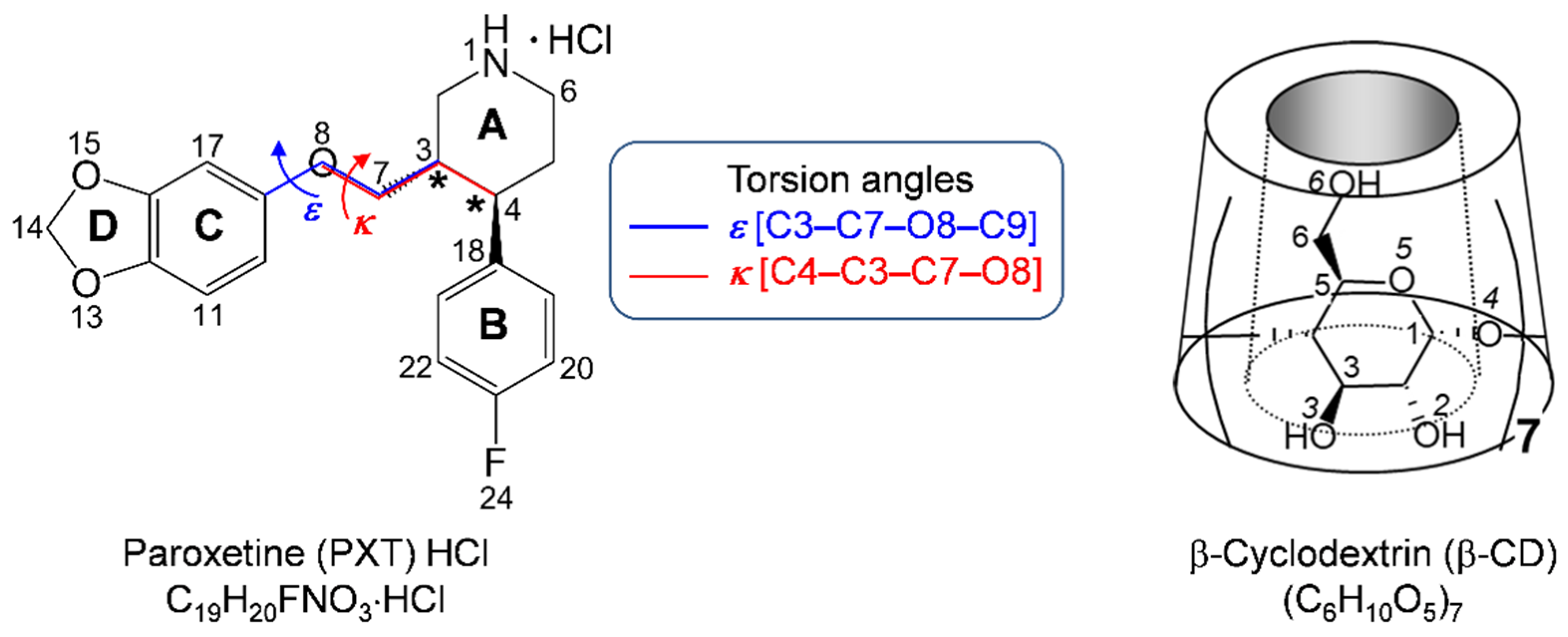
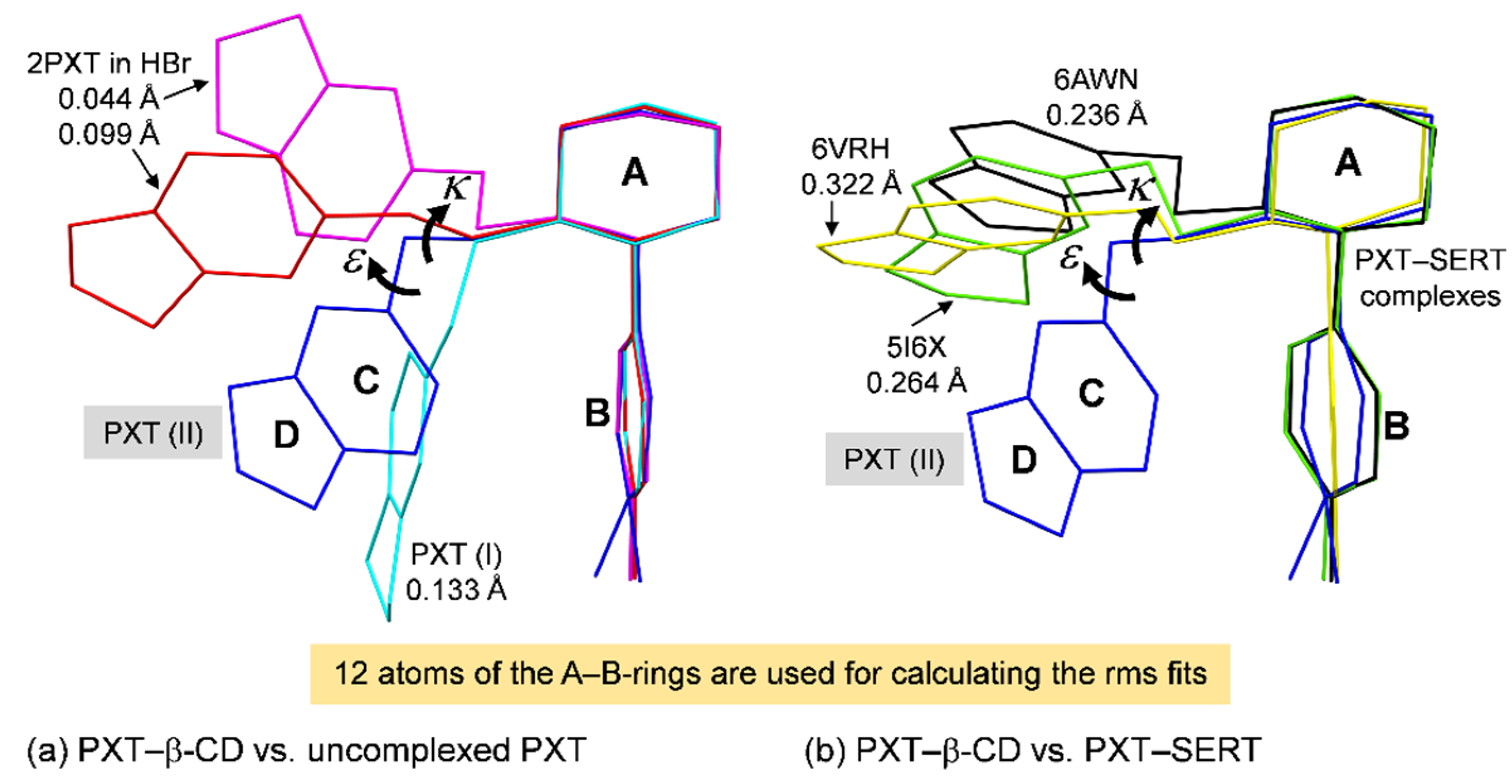
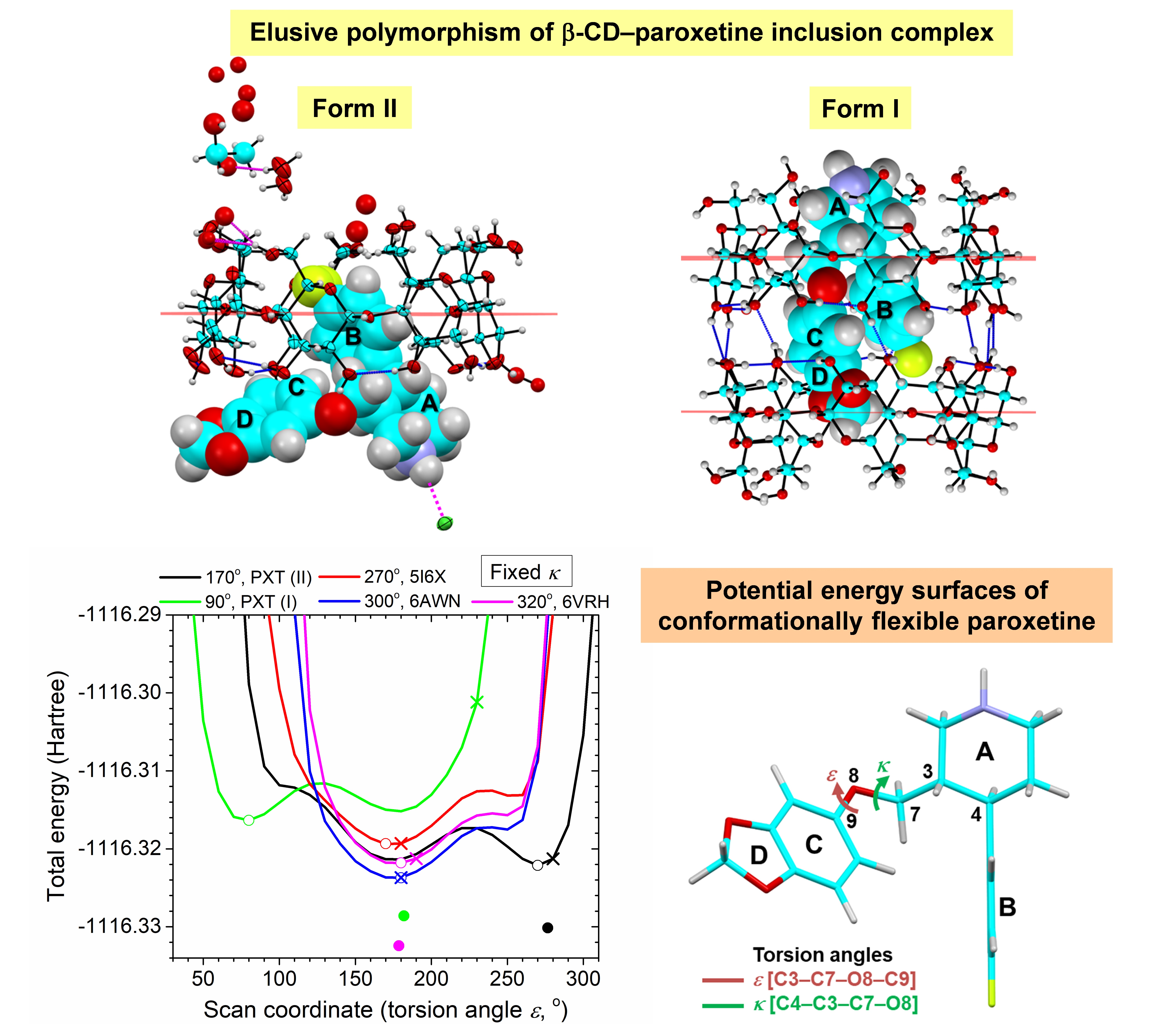
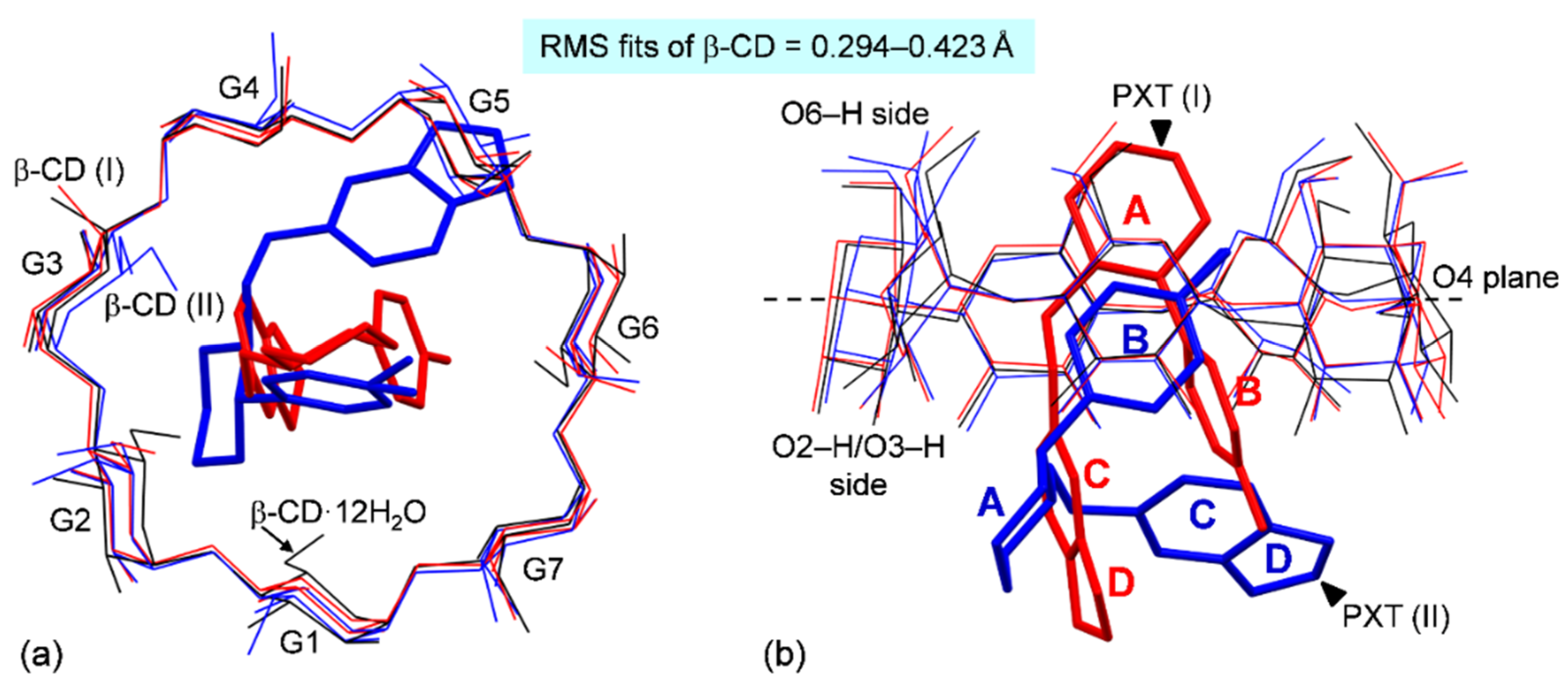
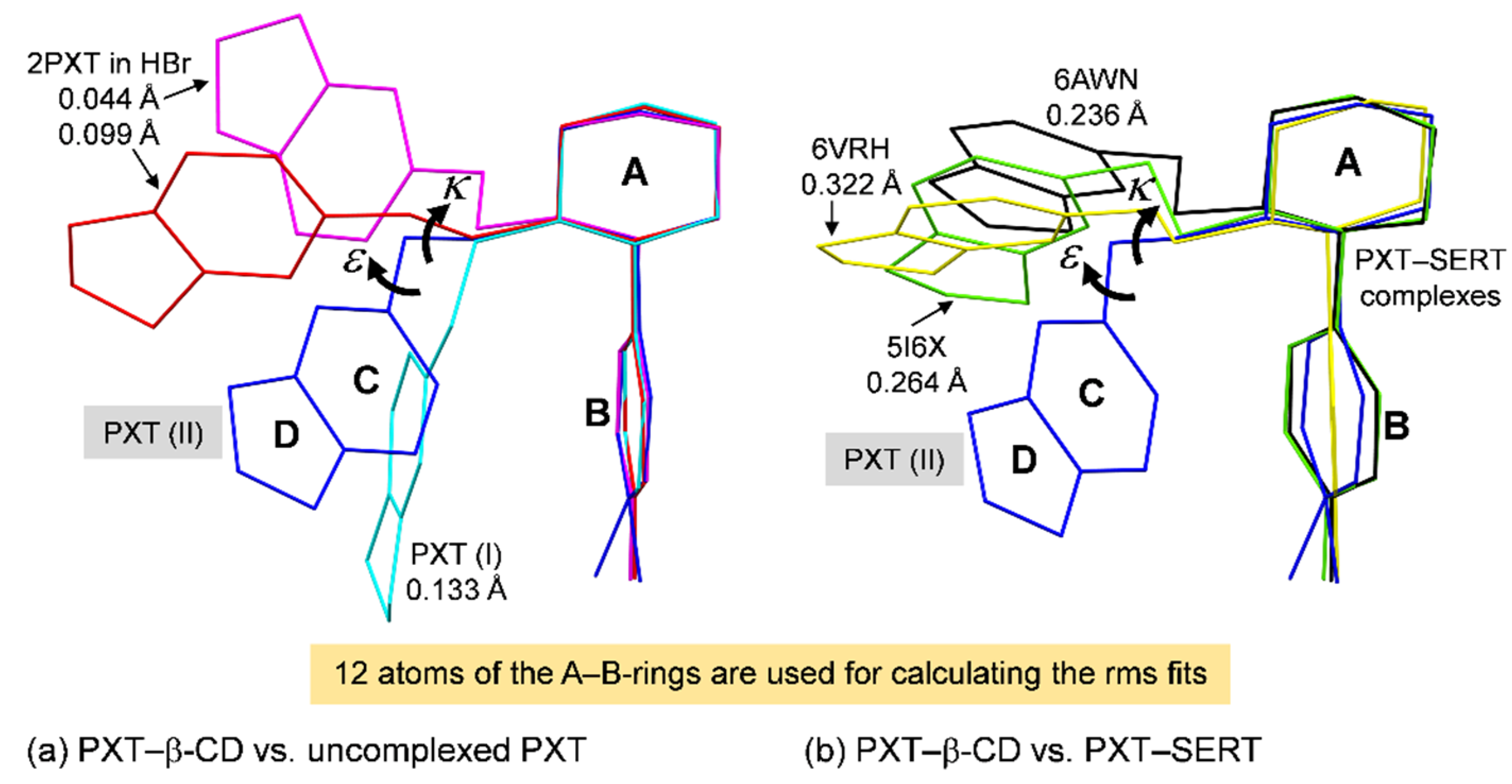
2.2. Variation of Inclusion Scenarios of Conformationally Flexible PXT
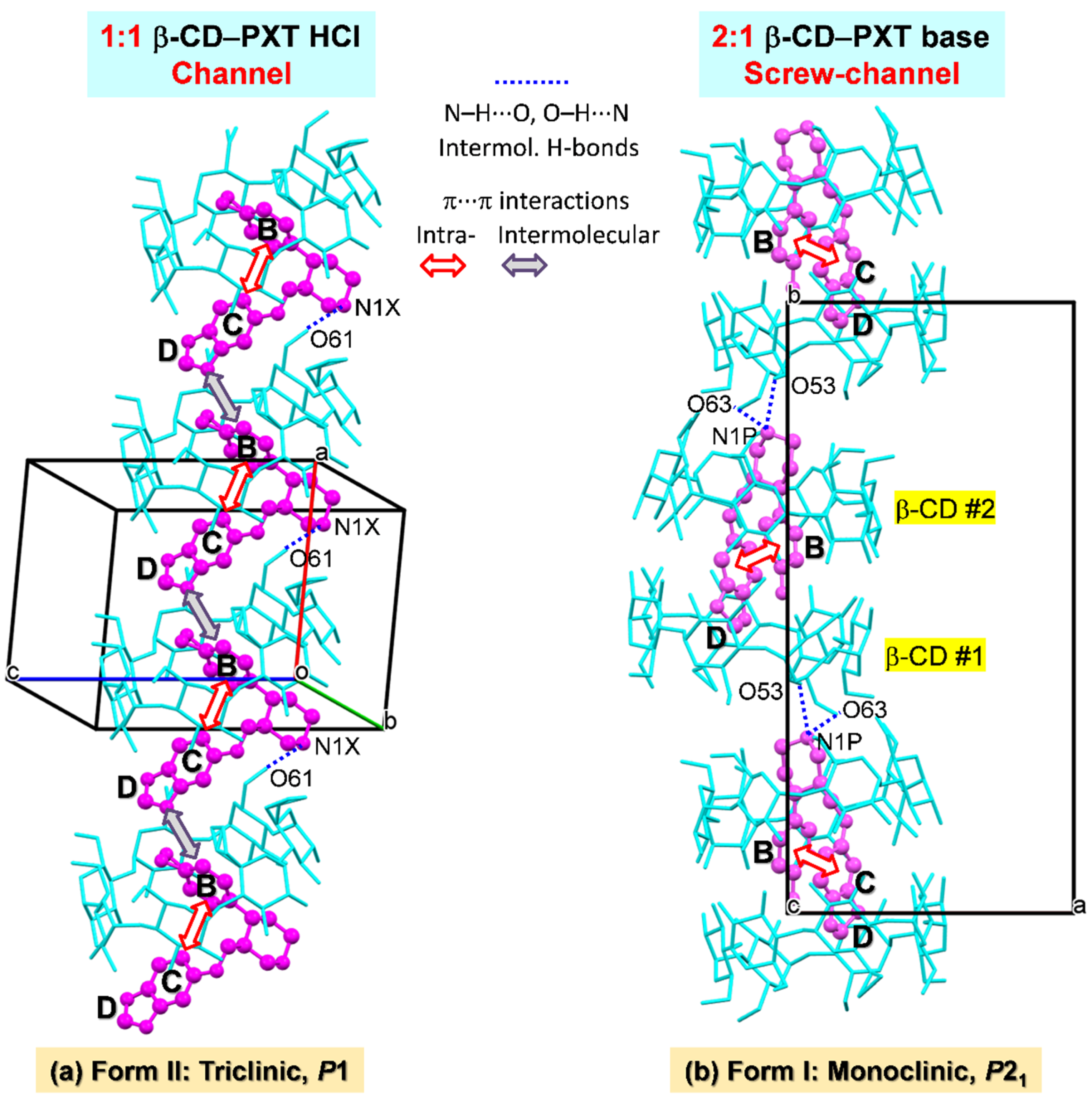
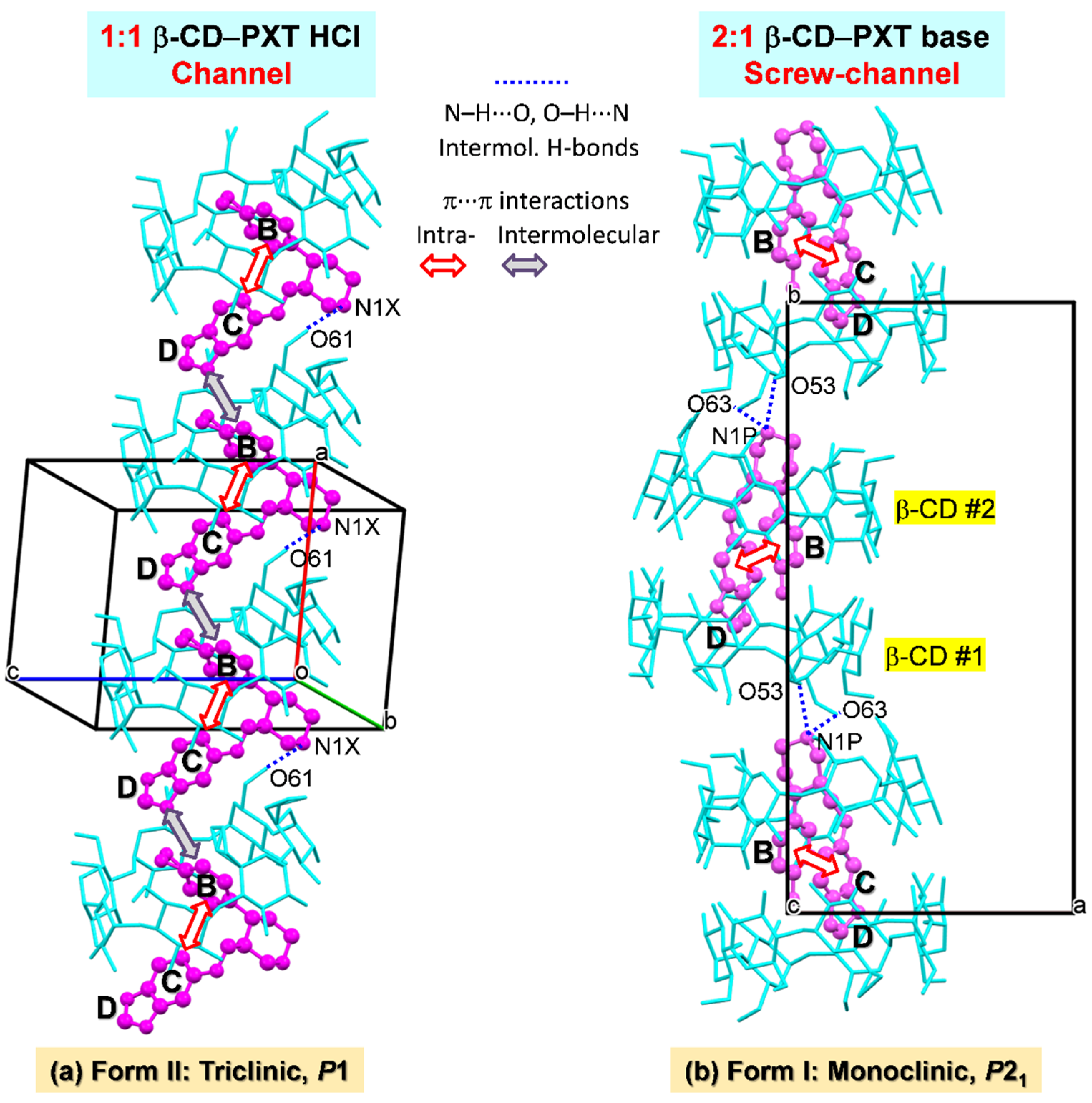
2.3. Similar Channel-Type Packing through Different Host–Guest Ratios and Inclusion Modes
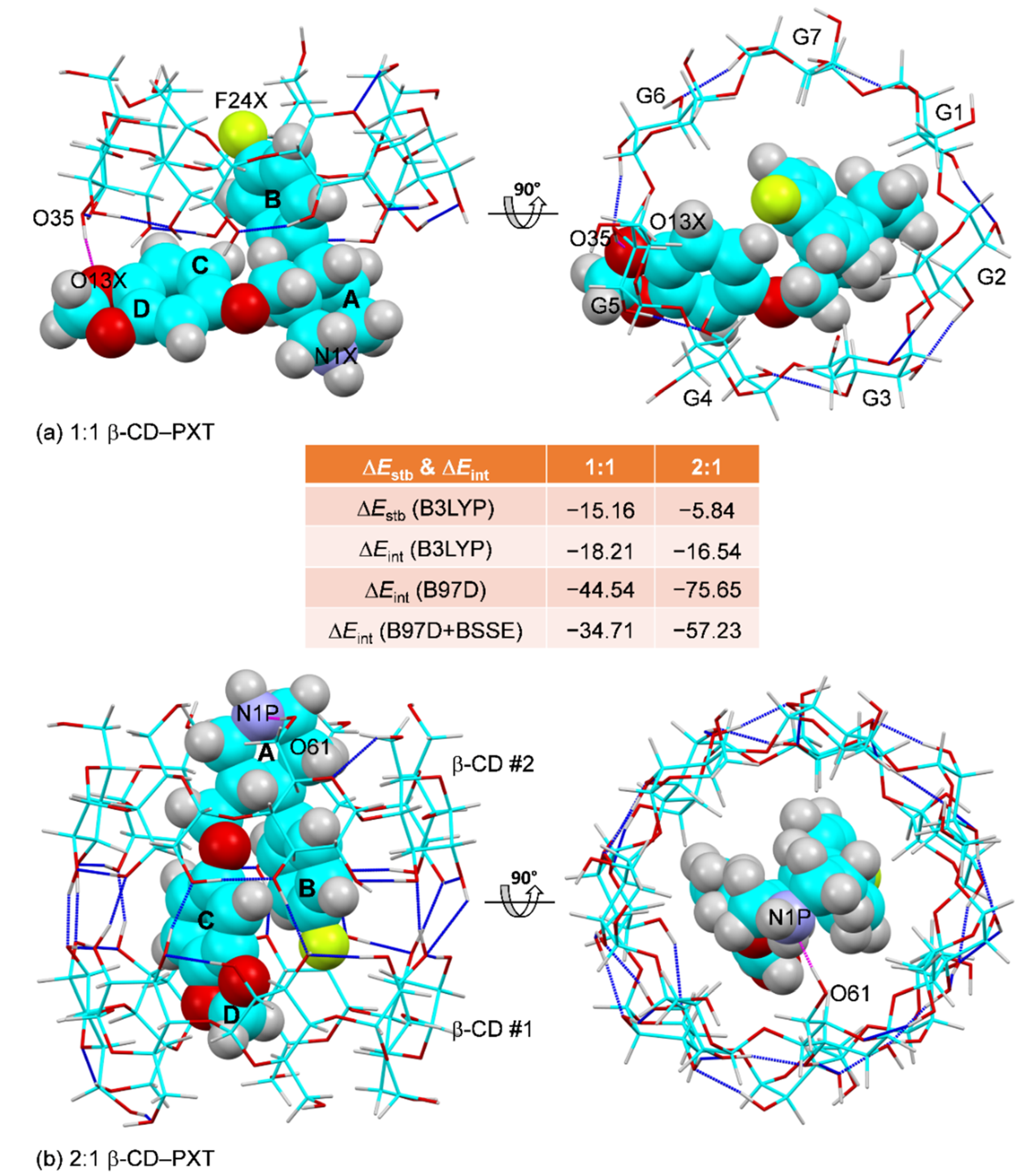
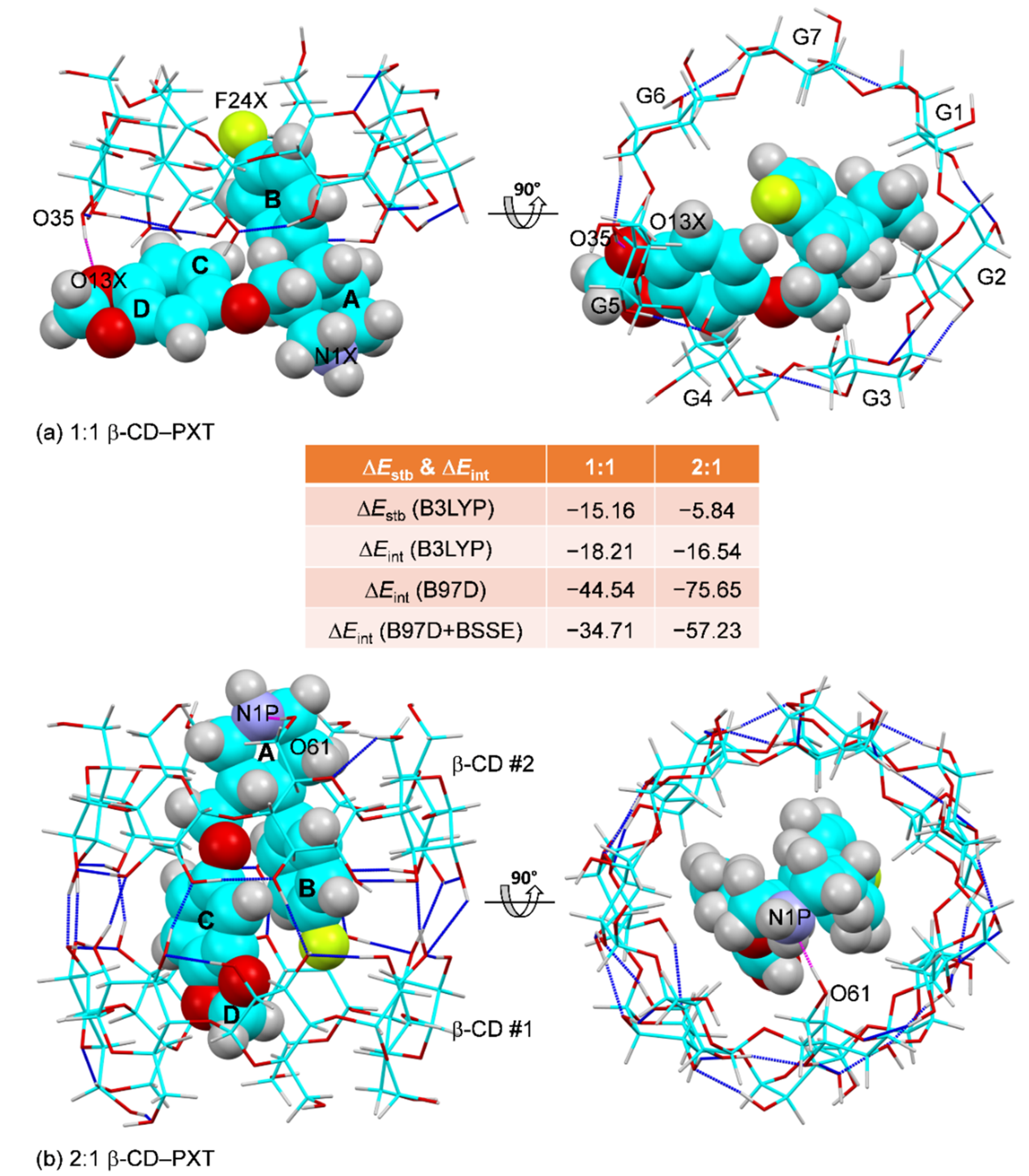
2.4. Thermodynamic Stabilities of the 1:1 and 2:1 β-CD–PXT Inclusion Complexes
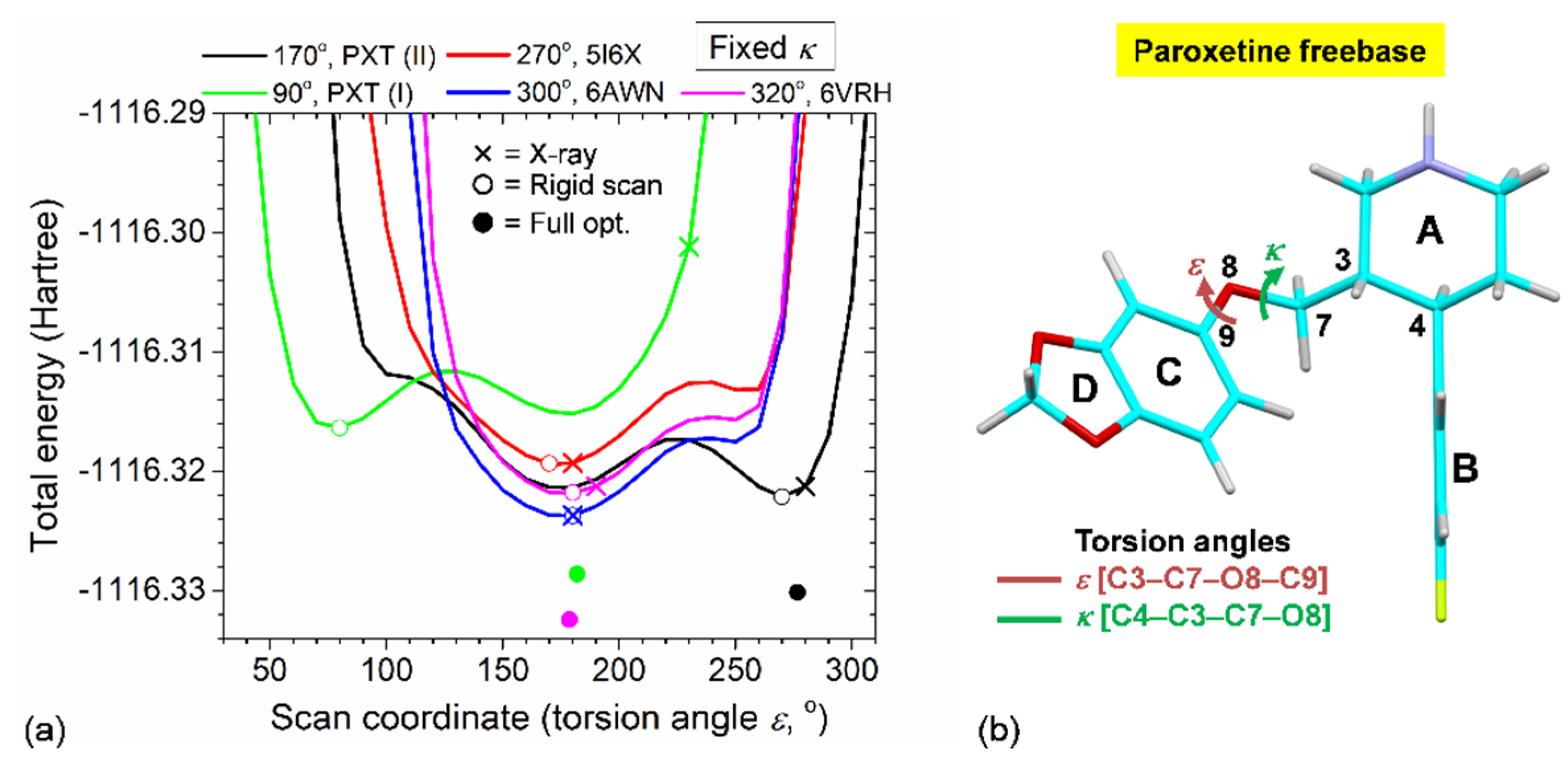
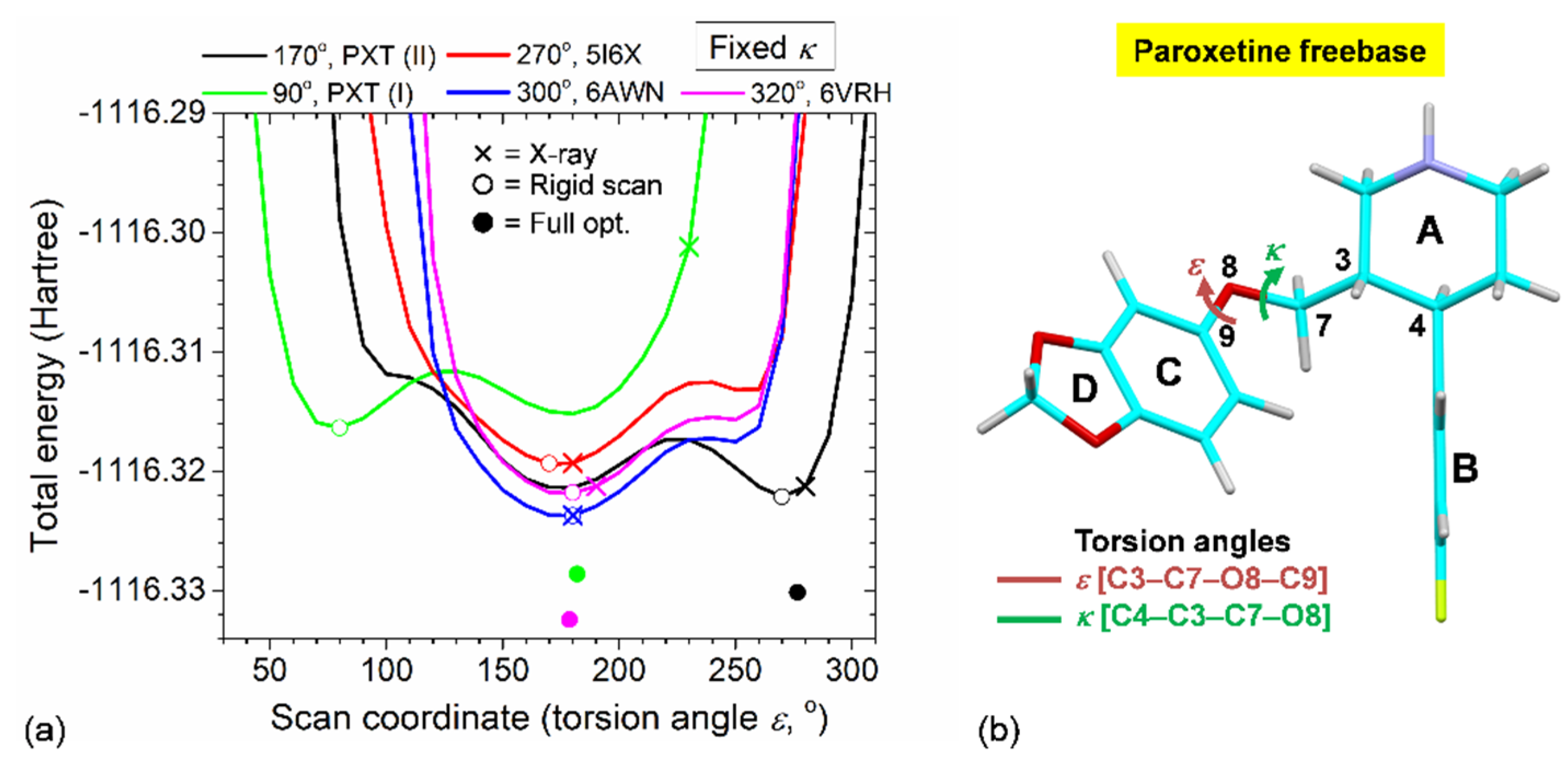
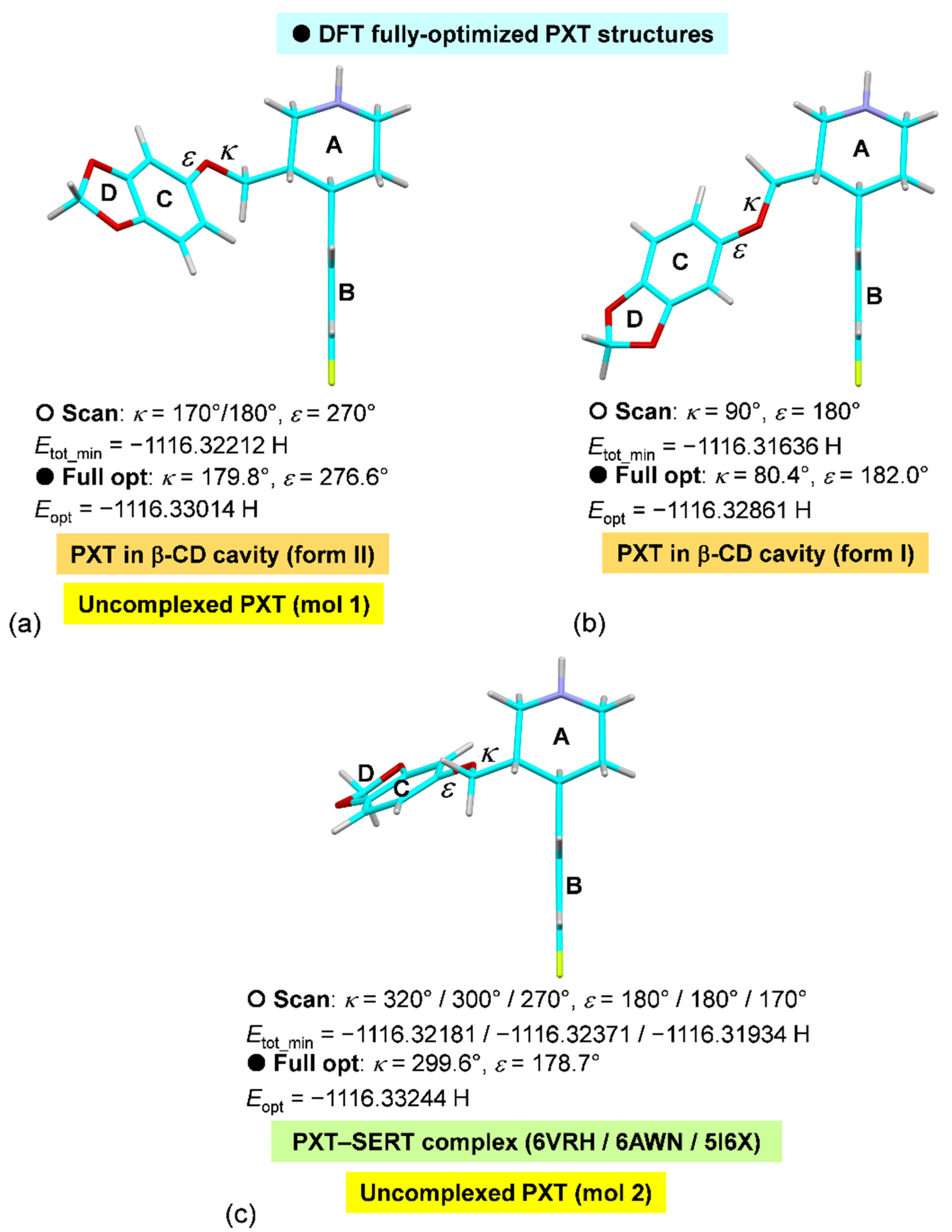
2.5. Rationale of PXT Conformational Flexibility through Potential Energy Surfaces (PESs)
3. Materials and Methods
3.1. Materials
3.2. X-ray Crystallography
3.2.1. Single-Crystal Preparation
3.2.2. X-ray Diffraction Experiment
3.2.3. Structure Solution and Refinement
3.3. DFT Calculations
3.3.1. Full-Geometry Optimization of 1:1 and 2:1 β-CD-PXT Complexes
3.3.2. Dispersion and BSSE Corrections of Interaction Energies
3.3.3. Construction of the Potential Energy Surfaces (PESs) of PXT Conformers
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Depression. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 29 November 2021).
- Wang, Y.; Kala, M.P.; Jafar, T.H. Factors associated with psychological distress during the coronavirus disease 2019 (COVID-19) pandemic on the predominantly general population: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0244630. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Director-General’s Opening Remarks at the Media Briefing on COVID-19—3 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---3-march-2020 (accessed on 29 November 2021).
- Worldometers. COVID Live Update. 7 November 2021. Available online: https://www.worldometers.info/coronavirus/ (accessed on 29 November 2021).
- Mohebbi, N.; Talebi, A.; Moghadamnia, M.; Taloki, Z.N.; Shakiba, A. Drug interactions of psychiatric and COVID-19 medications. Basic Clin. Neurosci. 2020, 11, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Lenze, E.J.; Mattar, C.; Zorumski, C.F.; Stevens, A.; Schweiger, J.; Nicol, G.E.; Reiersen, A.M. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: A randomized clinical trial. JAMA 2020, 324, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Hoertel, N.; Sánchez-Rico, M.; Vernet, R.; Beeker, N.; Jannot, A.S.; Neuraz, A.; Limosin, F. Association between antidepressant use and reduced risk of intubation or death in hospitalized patients with COVID-19: Results from an observational study. Mol. Psychiatry 2021, 26, 5199–5212. [Google Scholar] [CrossRef] [PubMed]
- Meikle, C.K.S.; Creeden, J.F.; McCullumsmith, C.; Worth, R.G. SSRIs: Applications in inflammatory lung disease and implications for COVID-19. Neuropsychopharmacol. Rep. 2021, 41, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.; dos Santos Moreira-Silva, E.A.; Silva, D.C.M.; Thabane, L.; Milagres, A.C.; Ferreira, T.S.; Mills, E.J. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: The TOGETHER randomised, platform clinical trial. Lancet Glob. Health 2021, 10, E42–E51. [Google Scholar] [CrossRef]
- Schloer, S.; Brunotte, L.; Mecate-Zambrano, A.; Zheng, S.; Tang, J.; Ludwig, S.; Rescher, U. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-CoV-2 infection in vitro. Br. J. Pharmacol. 2021, 178, 2339–2350. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Depression Medicines. 2019. Available online: https://www.fda.gov/consumers/free-publications-women/depression-medicines (accessed on 29 November 2021).
- Uekama, K.; Irie, T.; Otagiri, M.; Hoshino, T.; Yamada, Y.; Ohtani, Y. Protective effects of cyclodextrins on the haemolysis induced with imipramine in vitro. Membrane 1983, 8, 315–321. [Google Scholar] [CrossRef]
- 13. Hoshino, T.; Hirayama, F.; Uekama, K.; Yamasaki, M. Reduction of protriptyline-photosensitized hemolysis by β-cyclodextrin complexations. Int. J. Pharm. 1989, 50, 45–52. [Google Scholar] [CrossRef]
- Passos, J.J.; de Sousa, F.B.; Lula, I.S.; Barreto, E.A.; Lopes, J.F.; de Almeida, W.B.; Sinisterra, R.D. Multi-equilibrium system based on sertraline and β-cyclodextrin supramolecular complex in aqueous solution. Int. J. Pharm. 2011, 421, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.J.; de Sousa, F.B.; Mundim, I.M.; Bonfim, R.R.; Melo, R.; Viana, A.F.; Sinisterra, R.D. In vivo evaluation of the highly soluble oral β-cyclodextrin–Sertraline supramolecular complexes. Int. J. Pharm. 2012, 436, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Buko, V.; Palecz, B.; Belica-Pacha, S.; Zavodnik, I. The Supramolecular Complex of Sertraline with Cyclodextrins: Physicochemical and Pharmacological Properties. In Nano-and Microscale Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2017; pp. 343–356. [Google Scholar] [CrossRef]
- Abouhussein, D.M.; el Nabarawi, M.A.; Shalaby, S.H.; El-Bary, A.A. Sertraline-Cyclodextrin Complex Orodispersible Sublingual Tablet: Optimization, Stability, and Pharmacokinetics. J. Pharm. Innov. 2021, 16, 53–66. [Google Scholar] [CrossRef]
- Morin-Crini, N.; Fourmentin, S.; Fenyvesi, É.; Lichtfouse, E.; Torri, G.; Fourmentin, M.; Crini, G. 130 years of cyclodextrin discovery for health, food, agriculture, and the industry: A review. Environ. Chem. Lett. 2021, 19, 2581–2617. [Google Scholar] [CrossRef]
- Adeoye, O.; Cabral-Marques, H. Cyclodextrin nanosystems in oral drug delivery: A mini review. Int. J. Pharm. 2017, 531, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Matencio, A.; Navarro-Orcajada, S.; García-Carmona, F.; López-Nicolás, J.M. Applications of cyclodextrins in food science. A review. Trends Food Sci. Technol. 2020, 104, 132–143. [Google Scholar] [CrossRef]
- Bakshi, P.R.; Londhe, V.Y. Widespread applications of host-guest interactive cyclodextrin functionalized polymer nanocomposites: Its meta-analysis and review. Carbohydr. Polym. 2020, 242, 116430. [Google Scholar] [CrossRef]
- Fourmentin, S.; Crini, G.; Lichtfouse, E. (Eds.) Cyclodextrin Applications in Medicine, Food, Environment and Liquid Crystals; Springer: Cham, Switzerland, 2018; Volume 17. [Google Scholar]
- Diniz, T.C.; Pinto, T.C.C.; Menezes, P.D.P.; Silva, J.C.; Teles, R.B.D.A.; Ximenes, R.C.C.; Almeida, J.R.G.D.S. Cyclodextrins improving the physicochemical and pharmacological properties of antidepressant drugs: A patent review. Expert Opin. Ther. Pat. 2018, 28, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, P.H. Polymorphism of active pharmaceutical ingredients. Chem. Eng. Technol. 2006, 29, 233–237. [Google Scholar] [CrossRef]
- Aree, T.; Chaichit, N.; Engkakul, C. Polymorphism in β-cyclodextrin–benzoic acid inclusion complex: A kinetically controlled crystal growth according to the Ostwald’s rule. Carbohydr. Res. 2008, 343, 2451–2458. [Google Scholar] [CrossRef]
- Fernandes, J.A.; Ramos, A.I.; Ribeiro-Claro, P.; Paz, F.A.A.; Braga, S.S. Studies on polymorph conversion in a new cyclodextrin inclusion compound. CrystEngComm 2015, 17, 937–946. [Google Scholar] [CrossRef]
- Censi, R.; di Martino, P. Polymorph impact on the bioavailability and stability of poorly soluble drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilfiker, R.; Von Raumer, M. (Eds.) Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals 2e. International Union of Crystallography; Oxford University Press: Oxford, UK, 2020; Volume 30. [Google Scholar]
- Aree, T. β-Cyclodextrin encapsulation of nortriptyline HCl and amitriptyline HCl: Molecular insights from single-crystal X-ray diffraction and DFT calculation. Int. J. Pharm. 2020, 575, 118899. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. β-Cyclodextrin inclusion complexation with tricyclic antidepressants desipramine and imipramine: A structural chemistry perspective. J. Pharm. Sci. 2020, 109, 3086–3094. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Supramolecular Complexes of β-Cyclodextrin with Clomipramine and Doxepin: Effect of the Ring Substituent and Component of Drugs on Their Inclusion Topologies and Structural Flexibility. Pharmaceuticals 2020, 13, 278. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Distinctive Supramolecular Features of β-Cyclodextrin Inclusion Complexes with Antidepressants Protriptyline and Maprotiline: A Comprehensive Structural Investigation. Pharmaceuticals 2021, 14, 812. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Advancing insights on β-cyclodextrin inclusion complexes with SSRIs through lens of X-ray diffraction and DFT calculation. Int. J. Pharm. 2021, 609, 121113. [Google Scholar] [CrossRef]
- Caira, M.R.; de Vries, E.; Nassimbeni, L.R.; Jacewicz, V.W. Inclusion of the antidepressant paroxetine in β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2003, 46, 37–42. [Google Scholar] [CrossRef]
- Tavoulari, S.; Forrest, L.R.; Rudnick, G. Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J. Neurosci. 2009, 29, 9635–9643. [Google Scholar] [CrossRef]
- Bernini, A.; Spiga, O.; Ciutti, A.; Scarselli, M.; Bottoni, G.; Mascagni, P.; Niccolai, N. NMR studies of the inclusion complex between β-cyclodextrin and paroxetine. Eur. J. Pharm. Sci. 2004, 22, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Gouaux, E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. [Google Scholar] [CrossRef]
- French, A.D.; Johnson, G.P. Linkage and pyranosyl ring twisting in cyclodextrins. Carbohydr. Res. 2007, 342, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Lindner, K.; Saenger, W. Crystal and molecular structure of cyclohepta-amylose dodecahydrate. Carbohydr. Res. 1982, 99, 103–115. [Google Scholar] [CrossRef]
- Cremer, D.T.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Aree, T.; Jongrungruangchok, S. Crystallographic evidence for β-cyclodextrin inclusion complexation facilitating the improvement of antioxidant activity of tea (+)-catechin and (−)-epicatechin. Carbohydr. Polym. 2016, 140, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Caira, M.R.; Griffith, V.J.; Nassimbeni, L.R.; van Oudtshoorn, B. Synthesis and X-ray crystal structure of β-cyclodextrin diclofenac sodium undecahydrate, a β-CD complex with a unique crystal packing arrangement. J. Chem. Soc. Chem. Commun. 1994, 9, 1061–1062. [Google Scholar] [CrossRef]
- Harata, K. The structure of the cyclodextrin complex. V. Crystal structures of α-cyclodextrin complexes with p-nitrophenol and p-hydroxybenzoic acid. Bull. Chem. Soc. Jpn. 1977, 50, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Law, C.J.; Reith, M.E.; Wang, D.N. Antidepressant specificity of serotonin transporter suggested by three LeuT–SSRI structures. Nat. Struct. Mol. Biol. 2009, 16, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Navratna, V.; Antermite, D.; Yang, D.; Bull, J.A.; Gouaux, E. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. eLife 2020, 9, e56427. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, Y.L.; Gao, L.N.; Jiang, H.L. Effects of β-cyclodextrin on the intestinal absorption of berberine hydrochloride, a P-glycoprotein substrate. Int. J. Biol. Macromol. 2013, 59, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E.; Orlandi, G. Large amplitude out-of-plane vibrations of 1,3-benzodioxole in the S0 and S1 states: An analysis of fluorescence and excitation spectra by ab initio calculations. J. Phys. Chem. 2005, 109, 6471–6482. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Young, J. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef]
- Carvalho, P.S., Jr.; de Melo, C.; Ayala, A.P.; da Silva, C.C.; Ellena, J. Reversible solid-state hydration/dehydration of paroxetine HBr hemihydrate: Structural and thermochemical studies. Cryst. Growth Des. 2016, 16, 1543–1549. [Google Scholar] [CrossRef]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Ganazzoli, F.; Malpezzi, L.; Mele, A.; Panzeri, W.; Raffaini, G. Inclusion complexes of β-cyclodextrin with tricyclic drugs: An X-ray diffraction, NMR and molecular dynamics study. Beilstein J. Org. Chem. 2017, 13, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruker. APEX2, SADABS and SHELXTL; Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Bruker. SAINT and XPREP; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K. Features and development of Coot. Acta Cryst. 2010, D66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D.E. Protein shape and biological control. Sci. Am. 1973, 229, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Aree, T.; Jongrungruangchok, S. Enhancement of antioxidant activity of green tea epicatechins in β-cyclodextrin cavity: Single-crystal X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2016, 151, 1139–1151. [Google Scholar] [CrossRef]
- Aree, T.; Jongrungruangchok, S. Structure—Antioxidant activity relationship of β-cyclodextrin inclusion complexes with olive tyrosol, hydroxytyrosol and oleuropein: Deep insights from X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2018, 199, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Understanding structures and thermodynamics of β-cyclodextrin encapsulation of chlorogenic, caffeic and quinic acids: Implications for enriching antioxidant capacity and masking bitterness in coffee. Food Chem. 2019, 293, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Cryst. 2010, B66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.E.A.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Nakatsuji, H. Gaussian 09, revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Boys, S.F.; Bernardi, F.J.M.P. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphism of β-CD–PXT complex | PXT (II) | PXT (I) a |
|---|---|---|
| (1) Geometrical parameters | ||
| D-ring puckering b | ||
| Q (Å) | 0.103(15) | 0.219 |
| φ (°) | 164(8) | 325.7 |
| Conformation | Twist | Envelope |
| B vs. C interplanar angle (°) | 65.7(2) | 9.0 |
| Centroid–centroid distance (Å) | ||
| A–B | 4.282 | 4.347 |
| A–C | 5.580 | 6.100 |
| B–C | 5.457 | 3.637 |
| Selected torsion angles (°) c | ||
| C5–C4–C18–C19 | −129.7(4) | −120.5 |
| C2–C3–C7–O8 | −68.3(6) | −150.4 |
| C7–O8–C9–C10 | 3.1(6) | −9.9 |
| C4–C3–C7–O8, κ | 167.7(4) | 87.2 |
| C3–C7–O8–C9, ε | −83.4(6) | −131.7 |
| (2) Inclusion structure | ||
| Ring embedded in CD cavity d | B | A |
| Interplanar angle (°) | ||
| A-ring vs. β-CD O4 plane | 50.2(2) g | 72.2 g |
| B-ring vs. β-CD O4 plane | 76.3(1) | 69.3 |
| C-ring vs. β-CD O4 plane | 39.0(2) | 78.3 |
| Distance from drug to β-CD (Å) | ||
| A/B-ring centroid to O4 centroid (diagonal) e | −1.107 | 2.029 |
| A/B-ring centroid to O4 plane (vertical) | −0.871 | 1.640 |
| Chiral center C4X/C4P to O4 centroid (diagonal) f | −3.733 | −0.931 |
| Interaction | D–H | H···A | D···A | ∠(DHA) | Interaction | D–H | H···A | D···A | ∠(DHA) |
|---|---|---|---|---|---|---|---|---|---|
| X-ray | |||||||||
| 1:1 β-CD–PXT HCl | 2:1 β-CD–PXT base | ||||||||
| (form II) | (form I) d | ||||||||
| N1X–H1···O61 i b | 0.89 | 1.95 | 2.792(6) | 158.1 | O63_1–H···N1P iii b | 0.99 | 1.863 | 2.846 | 169.3 |
| N1X–H1···Cl1 | 0.89 | 2.38 | 3.131(5) | 141.6 | N1P–H···O53_1 iv | 1.02 | 2.534 | 3.172 | 120.5 |
| O66–H···Cl1 ii | 0.82 | 2.29 | 3.084(5) | 163.4 | C36_1–H···Cg3(B) e | 1.00 | 3.628 | 4.519 | 149.6 |
| O1W–H2···Cl1 ii | 0.96 | 2.28 | 3.154(5) | 151.0 | C37_1–H···Cg2(C) | 1.00 | 3.685 | 4.641 | 160.6 |
| O25–H···Cg2(C) c | 0.82 | 3.410 | 4.228 | 176.0 | C35_2–H···Cg3(B) | 1.00 | 3.717 | 4.620 | 151.5 |
| C31–H···Cg3(B) | 0.98 | 3.623 | 4.548 | 158.2 | C36_2–H···Cg3(B) | 1.00 | 3.262 | 4.248 | 168.4 |
| DFT f | |||||||||
| 1:1 β-CD–PXT base | 2:1 β-CD–PXT base | ||||||||
| O35–H···O13X | 0.98 | 1.97 | 2.92 | 162.3 | O61_2–H···N1P | 0.99 | 1.91 | 2.90 | 172.8 |
| O25–H···Cg2(C) c | 0.98 | 3.74 | 4.34 | 121.8 | C36_1–H···Cg2(C) c | 1.10 | 3.57 | 4.65 | 168.3 |
| C31–H···Cg3(B) | 1.10 | 3.66 | 4.74 | 165.3 | C36_2–H···Cg3(B) | 1.10 | 3.50 | 4.55 | 161.1 |
| X-ray | Free HBr Form | In β-CD Cavity | SERT–PXT Complex | ||||
|---|---|---|---|---|---|---|---|
| Mol 1 f | Mol 2 f | PXT (I) g | PXT (II) | 5I6X h | 6AWN i | 6VRH j | |
| Molecular shape | L | L | U | V | L | L | L |
| Ring-D puckering a | |||||||
| Q (Å) | 0.198 | 0.246 | 0.219 | 0.103(15) | 0.193 | - | 0.097 |
| φ (°) | 144.5 | 141.6 | 325.7 | 164(8) | 144.4 | - | 215.9 |
| Conformation | Env. | Env. | Env. | Twist | Env. | Planar | Env. |
| B vs. C interplanar angle (°) | 75.3 | 42.3 | 9.0 | 65.7(2) | 58.3 | 56.0 | 87.6 |
| Centroid–centroid dist. (Å) | |||||||
| A–B | 4.313 | 4.279 | 4.347 | 4.282 | 4.318 | 4.312 | 4.284 |
| A–C | 6.342 | 6.042 | 6.100 | 5.580 | 6.175 | 6.139 | 6.070 |
| B–C | 7.197 | 6.341 | 3.637 | 5.457 | 6.018 | 6.161 | 6.067 |
| Selected torsion angles (°) | |||||||
| C5–C4–C18–C19 | −119.6 | −130.5 | −120.5 | −129.7(4) | −154.3 | −150.7 | −94.1 |
| C2–C3–C7–O8 | −58.2 | 64.8 | −150.4 | −68.3(6) | 30.4 | 64.8 | 85.7 |
| C7–O8–C9–C10 | −147.4 | −40.9 | −9.9 | 3.1(6) | 102.8 | 64.5 | −139.8 |
| ✕ C4–C3–C7–O8, κ b | 179.7 | −60.0 | 87.2 | 167.7(4) | −91.1 | −55.7 | −36.9 |
| ✕ C3–C7–O8–C9, ε b | 173.4 | −176.3 | −131.7 | −83.4(6) | −177.1 | −176.2 | −172.0 |
| RMS fit (Å) c | 0.099 | 0.044 | 0.133 | 0 | 0.264 | 0.236 | 0.322 |
| DFT | |||||||
| (i) PES rigid scan | |||||||
| ○ κ (fixed) b | 180 | 300 | 90 | 170 | 270 | 300 | 320 |
| ○ εmin (scanned 30–310°) d | 270 | 180 | 80 | 270 | 170 | 180 | 180 |
| Etot_min (Hartree) e | −0.31681 | −0.32371 | −0.31636 | −0.32212 | −0.31934 | −0.32371 | −0.32181 |
| (ii) Full optimization | |||||||
| ● κ b | 179.8 | 299.6 | 80.4 | 179.8 | 299.6 | 299.6 | 299.6 |
| ● ε b | 276.6 | 178.7 | 182.0 | 276.6 | 178.7 | 178.7 | 178.7 |
| Eopt (Hartree) e | −0.33014 | −0.33244 | −0.32861 | −0.33014 | −0.33244 | −0.33244 | −0.33244 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aree, T. Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex. Pharmaceuticals 2022, 15, 98. https://doi.org/10.3390/ph15010098
Aree T. Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex. Pharmaceuticals. 2022; 15(1):98. https://doi.org/10.3390/ph15010098
Chicago/Turabian StyleAree, Thammarat. 2022. "Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex" Pharmaceuticals 15, no. 1: 98. https://doi.org/10.3390/ph15010098
APA StyleAree, T. (2022). Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex. Pharmaceuticals, 15(1), 98. https://doi.org/10.3390/ph15010098