
Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
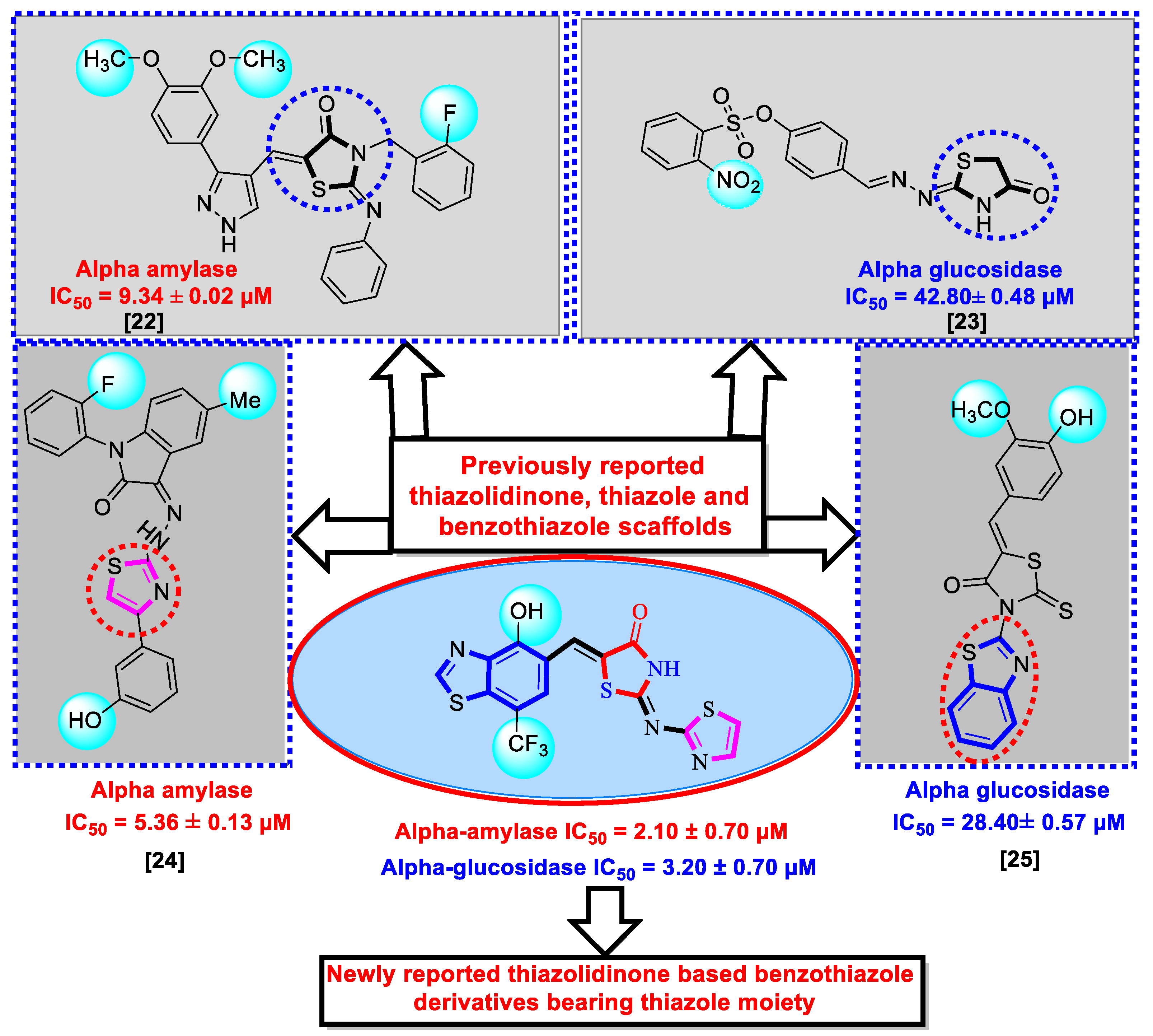
2. Results and Discussion
2.1. Chemistry
Spectroscopic Characterization and Elucidation of Derivative-1
2.2. In Vitro α-Amylase and α-Glucosidase Inhibitory Activities
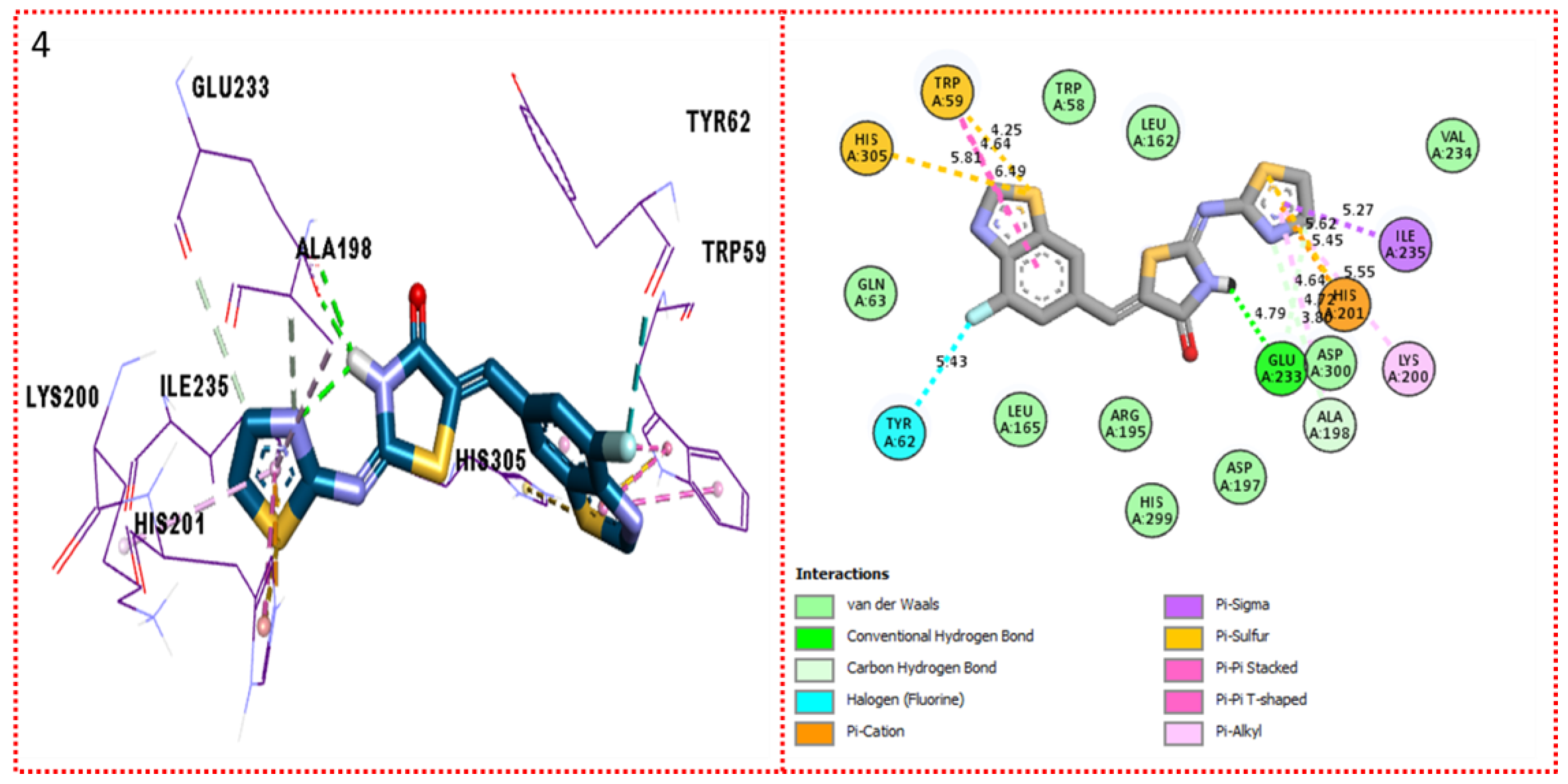
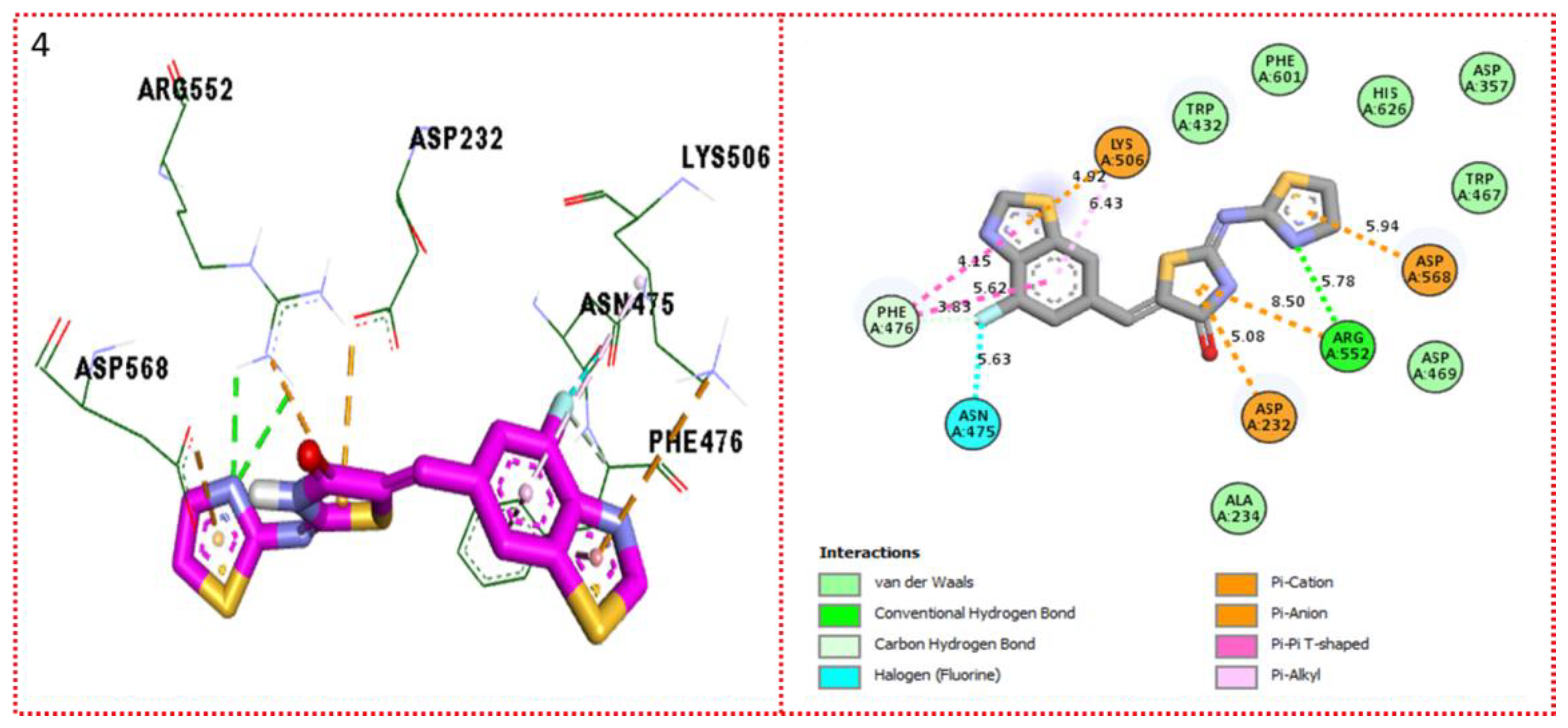
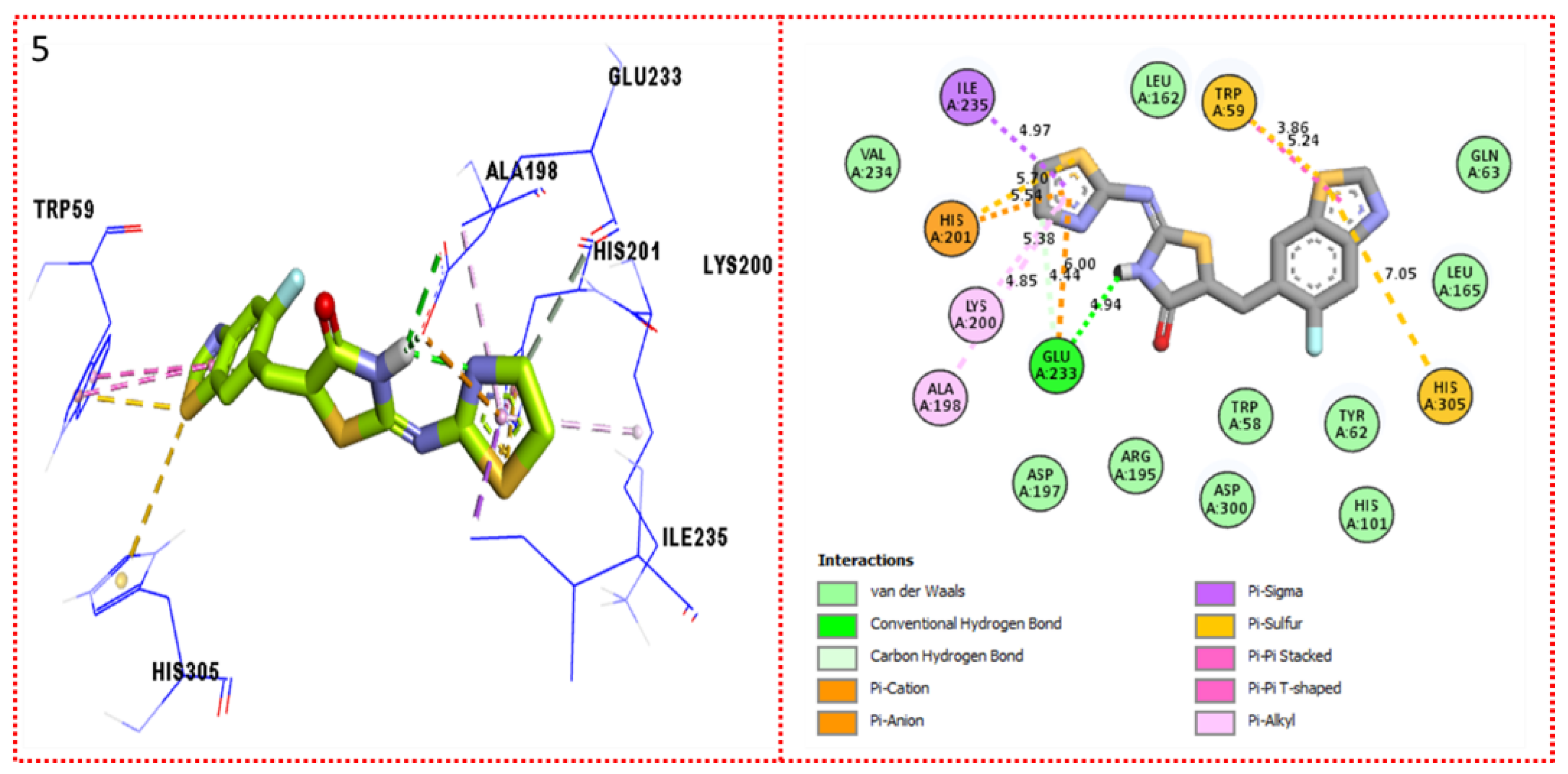
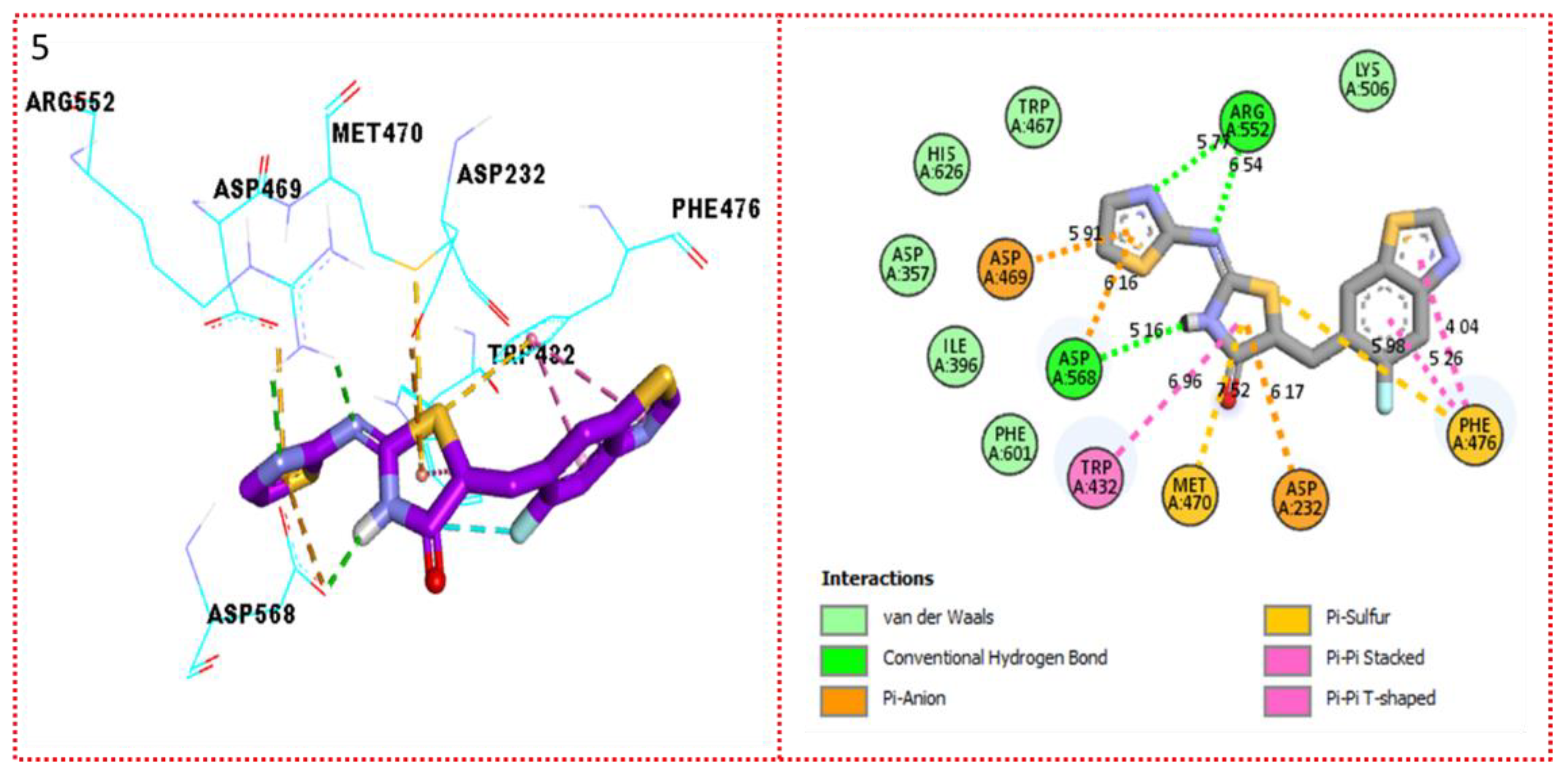
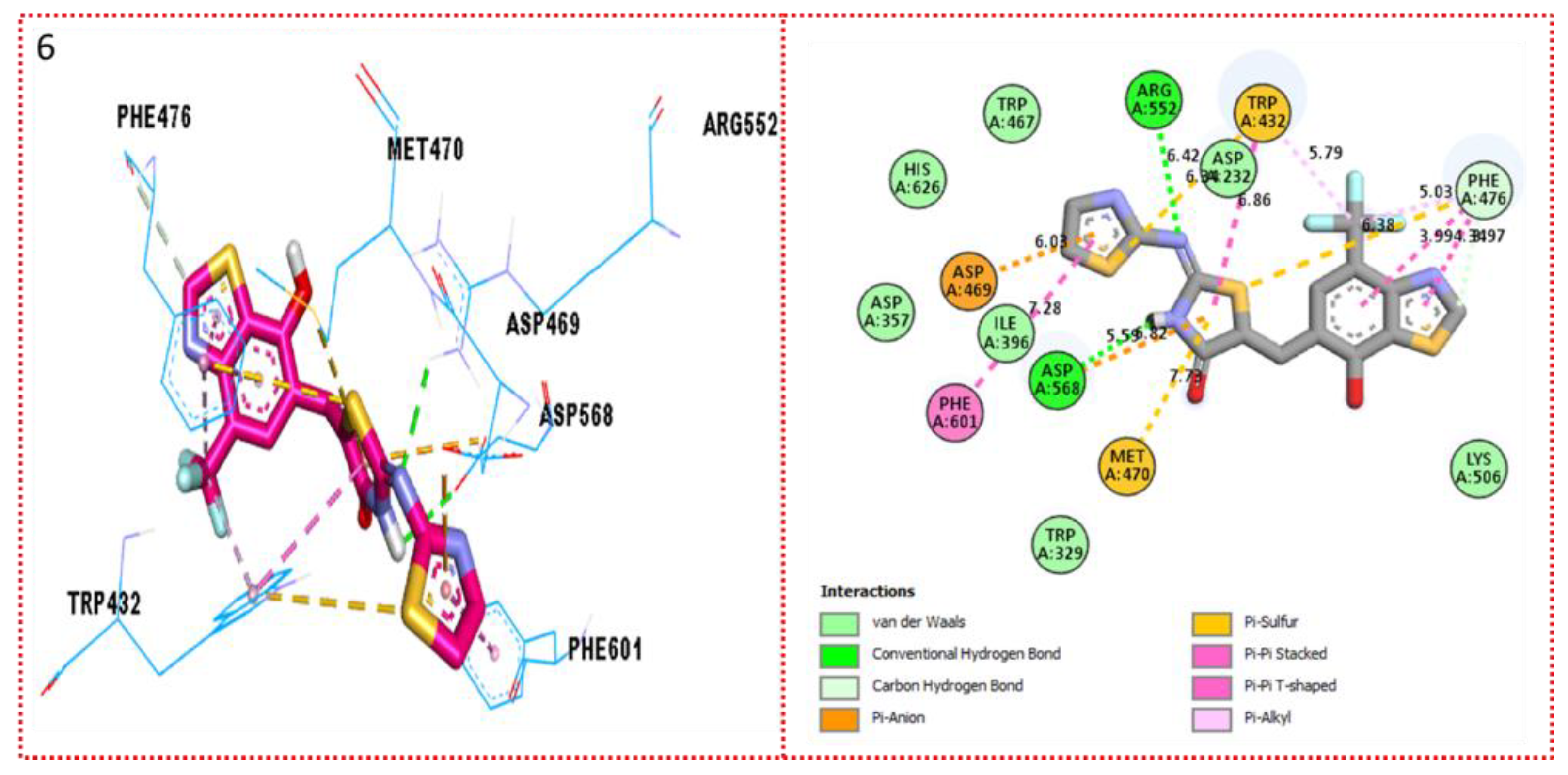
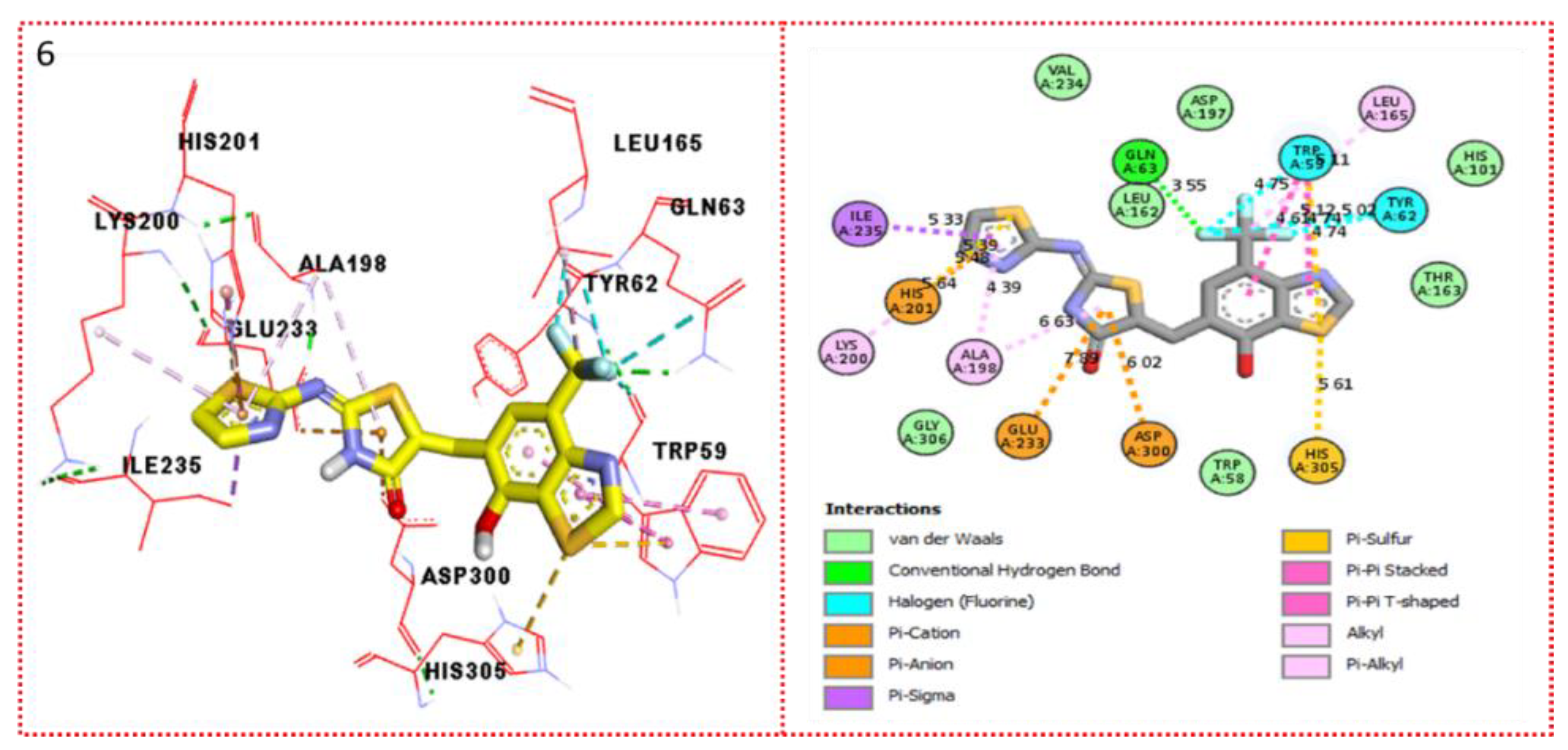
2.3. Molecular Docking
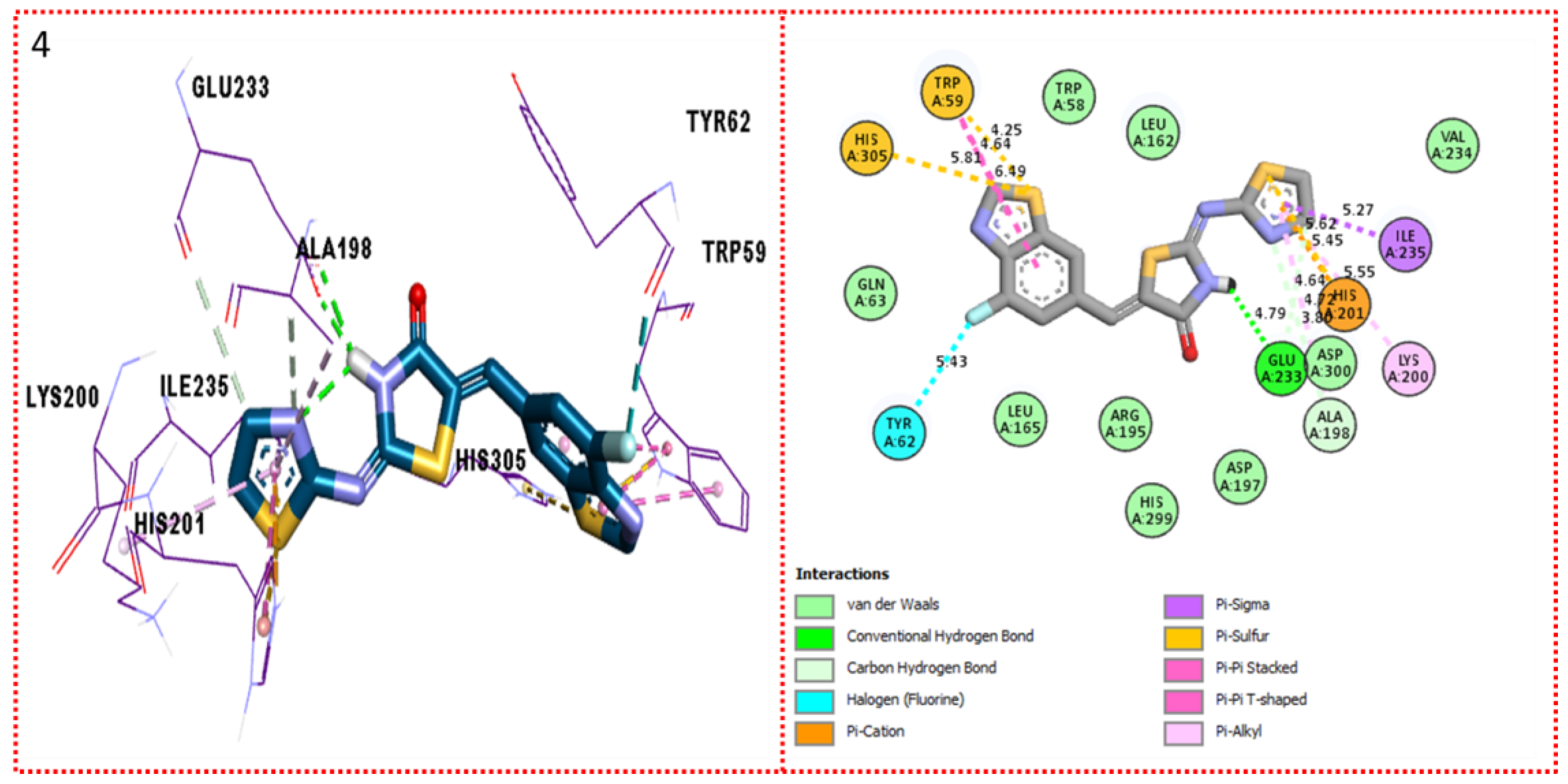
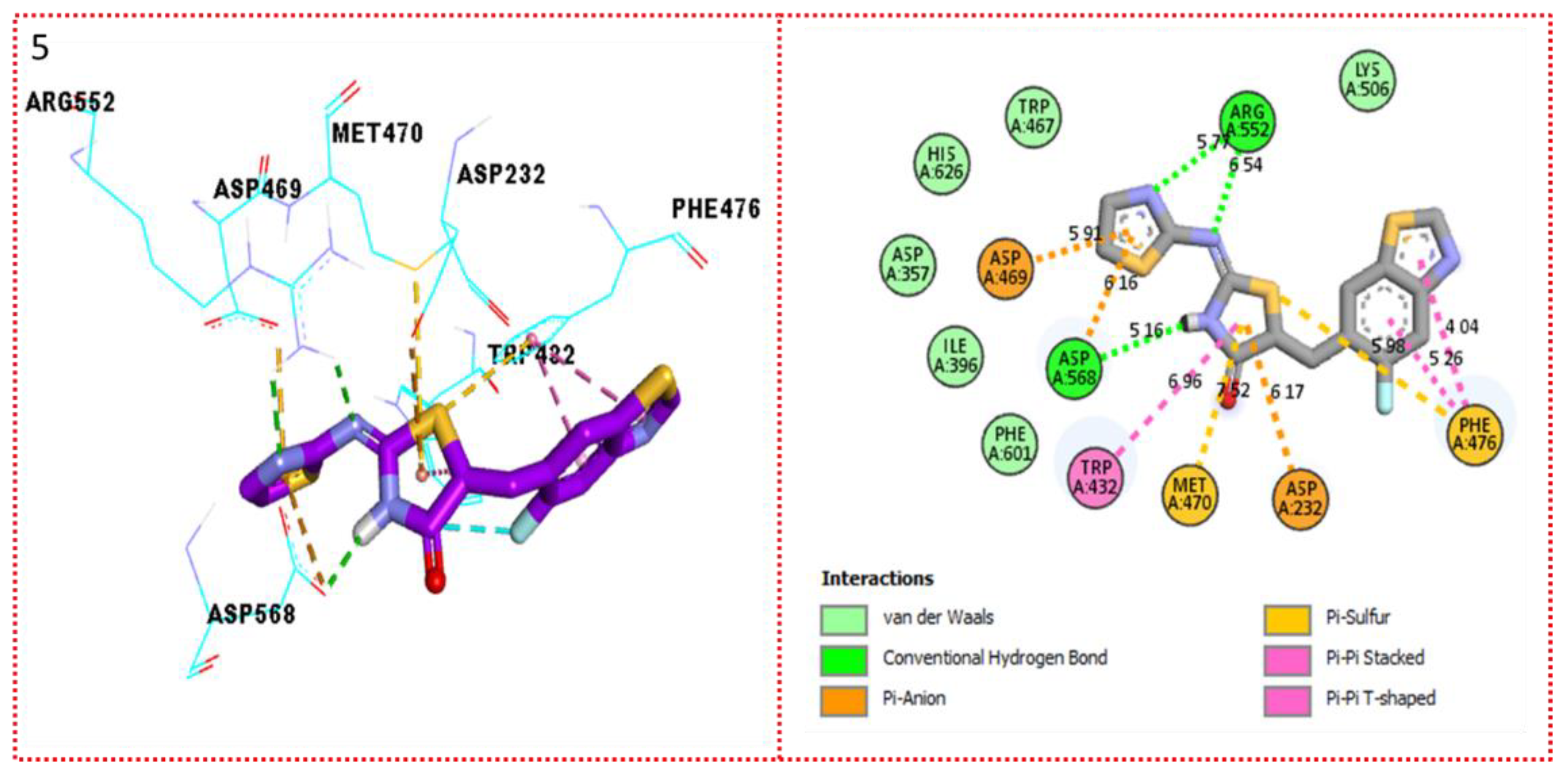
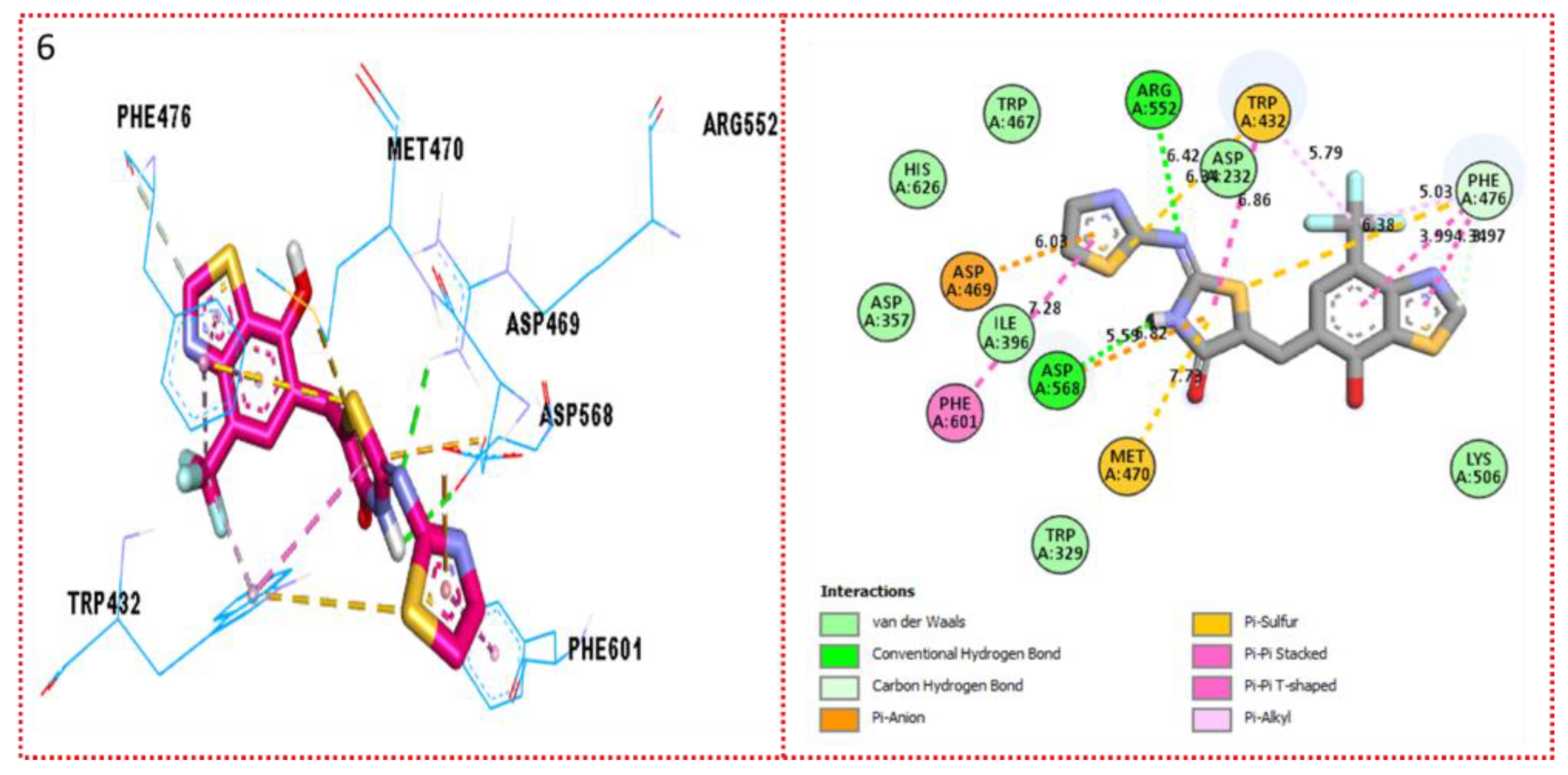
Docking Results
3. Materials and Methods
3.1. General Information
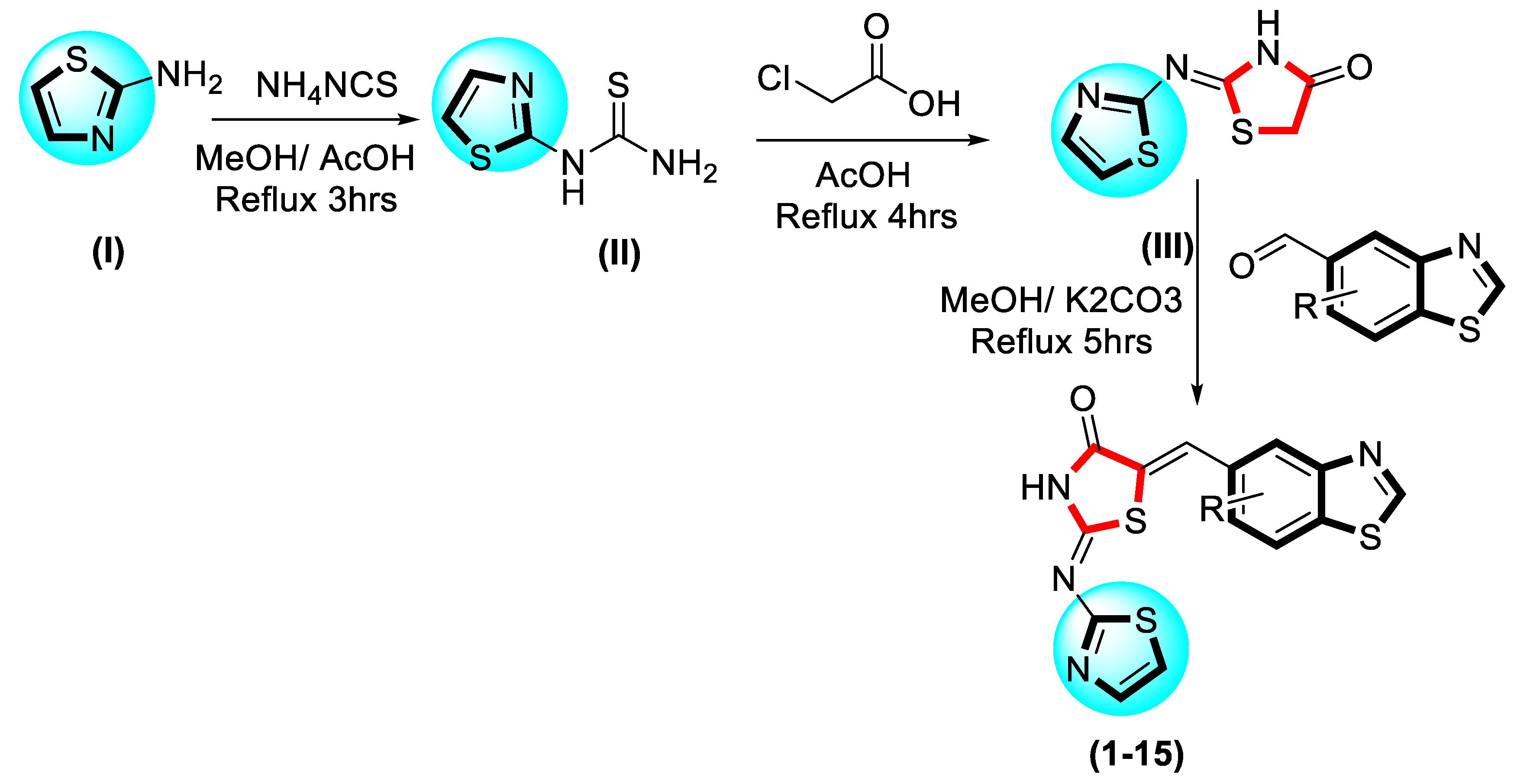
3.2. General Procedure for the Synthesis of Thiazole-Based Thiazolidinone Derivatives 1–15
3.3. Spectral Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atlas, D. International diabetes federation. In IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; p. 33. [Google Scholar]
- Gin, H.; Rigalleau, V. Post-prandial hyperglycemia. post-prandial hyperglycemia and diabetes. Diabetes Metab. 2000, 26, 265–272. [Google Scholar]
- Lordan, S.; Smyth, T.J.; Soler-Vila, A.; Stanton, C.; Ross, R.P. The α-amylase and α glucosidase inhibitory effects of Irish seaweed extracts. Food Chem. 2013, 141, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. α-glucosidase inhibitors. Endocrinol. Metab. Clin. N. Am. 1997, 26, 539–551. [Google Scholar] [CrossRef]
- Van de Laar, F.A. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; de la Garza, A.L.; Campión, J.; Martínez, J.A.; Milagro, F.I. Antidiabetic effects of natural plant extracts via inhibition of carbohydrate hydrolysis enzymes with emphasis on pancreatic alpha amylase. Expert Opin. Ther. Targets 2012, 16, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.A.D.; Souza, E.C.F.D.; EchazúBöschemeier, A.G.; Costa, C.C.M.D.; Bezerra, H.S.; Feitosa, E.E.L.C. Diagnosis of diabetes mellitus and living with a chronic condition: Participatory study. BMC Public Health 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fatmawati, S.; Shimizu, K.; Kondo, R.; Ganoderol, B. A potent α-glucosidase inhibitor isolated from the fruiting body of ganodermalucidum. Phytomedicine 2011, 18, 1053–1055. [Google Scholar] [CrossRef]
- Kawamura-Konishi, Y.; Watanabe, N.; Saito, M.; Nakajima, N.; Sakaki, T.; Katayama, T.; Enomoto, T. Isolation of a new phlorotannin, a potent inhibitor of carbohydrate-hydrolyzing enzymes, from the brown alga sargassum patens. J. Agric. Food Chem. 2012, 60, 5565–5570. [Google Scholar] [CrossRef] [PubMed]
- Orhan, N.; Aslan, M.; Şüküroğlu, M.; Orhan, D.D. In vivo and in vitro antidiabetic effect of Cistuslaurifolius L. and detection of major phenolic compounds by UPLC–TOF-MS analysis. J. Ethnopharmacol. 2013, 146, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Panwar, H.; Calderwood, D.; Grant, I.R.; Grover, S.; Green, B.D. Lactobacillus strains isolated from infant faeces possess potent inhibitory activity against intestinal alpha-and beta-glucosidases suggesting anti-diabetic potential. Eur. J. Nutr. 2014, 53, 1465–1474. [Google Scholar] [CrossRef]
- Ali, R.B.; Atangwho, I.J.; Kuar, N.; Ahmad, M.; Mahmud, R.; Asmawi, M.Z. In vitro and in vivo effects of standardized extract and fractions of phaleriamacrocarpa fruits pericarp on lead carbohydrate digesting enzymes. BMC Complement. Altern. Med. 2013, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.T.; Rioux, L.E.; Turgeon, S.L. Alpha-amylase and alpha-glucosidase inhibition is differentially modulated by fucoidan obtained from fucusvesiculosus and ascophyllumnodosum. Phytochemistry 2014, 98, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.H.; Siddiqui, M.J.A.; Ang, L.F.; Sadikun, A.; Chan, S.H.; Tan, S.C.; Yam, M.F. Potent α-glucosidase and α-amylase inhibitory activities of standardized 50% ethanolic extracts and sinensetin from orthosiphonsta-mineusbenth as anti-diabetic mechanism. BMC Complement. Altern. Med. 2012, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gutierrez, R.M.; Damian-Guzman, M. Meliacinolin: A potent α-glucosidase and α-amylase inhibitor isolated from azadirachtaindica leaves and in vivo antidiabetic property in streptozotocin-nicotinamide-induced type 2 diabetes in mice. Biol. Pharm. Bull. 2012, 35, 1516–1524. [Google Scholar] [CrossRef]
- Patel, D.; Kumari, P.; Patel, N. Synthesis and characterization of some new thiazolidinones containing coumarin moiety and their antimicrobial study. Arch. Appl. Sci. Res. 2010, 2, 68–75. [Google Scholar]
- Mishra, R.; Tomar, I.; Singhal, S.; Jha, K.K. Facile synthesis of thiazolidinones bearing thiophene nucleus as antimicrobial agents. Der. Pharm. Chem. 2012, 4, 489–496. [Google Scholar]
- Rao, A.; Carbone, A.; Chimirri, A.; De Clercq, E.; Monforte, A.M.; Monforte, P.; Zappalà, M. Synthesis and anti-HIV activity of 2, 3-diaryl-1, 3-thiazolidin-4-ones. Il Farm. 2003, 58, 115–120. [Google Scholar] [CrossRef]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Oruç, E.E.; Rollas, S.; Şahin, F.; Özbek, A. Synthesis, characterisation and biological activity of novel 4-thiazolidinones, 1,3,4-oxadiazoles and some related compounds. Eur. J. Med. Chem. 2002, 37, 197–206. [Google Scholar] [CrossRef]
- Ullah, H.; Uddin, I.; Rahim, F.; Khan, F.; Taha, M.; Khan, M.U.; Hussain, J. In vitro α-glucosidase and α-amylase inhibitory potential and molecular docking studies of benzohydrazide based imines and thiazolidine-4-one derivatives. J. Mol. Struct. 2022, 1251, 132058. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Chen, M.; Peng, Y.; Li, L.; Deng, B.; Li, W. Synthesis, biological evaluation, and molecular docking studies of novel isatin-thiazole derivatives as-glucosidase inhibitors. Molecules 2017, 22, 659. [Google Scholar]
- Srinivasa, M.G.; Aggarwal, N.N.; Gatpoh, B.F.D.; Shankar, M.K.; Byadarahalli Ravindranath, K.; Gurubasavaraj Veeranna, P.; Dixit, S.; Mandal, S.P.; Bommenahally Ravanappa, P.K.; Khanal, P.; et al. Identification of benzothiazole-rhodanine derivatives as α-amylase and α-glucosidase inhibitors: Design, synthesis, in silico, and in vitro analysis. J. Mol. Recognit. 2022, 35, e2959. [Google Scholar] [CrossRef] [PubMed]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Shivachandra, J.C. In vitro and in silico studies of fluorinated 2, 3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2022, 355, 2100342. [Google Scholar] [CrossRef]
- Kaur, R.; Kumar, R.; Dogra, N.; Yadav, A.K. Design, synthesis, biological evaluations and in silico studies of sulfonate ester derivatives of 2-(2-benzylidenehydrazono) thiazolidin-4-one as potential α-glucosidase inhibitors. J. Mol. Struct. 2022, 1247, 131266. [Google Scholar] [CrossRef]
- Kharb, M.; Jat, R.K.; Parjapati, G.; Gupta, A. Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Technol. 2012, 2, 189–197. [Google Scholar]
- Li, Z.; Gu, J.; Zhuang, H.; Kang, L.; Zhao, X.; Guo, Q. Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- Rao, C.M.M.P.; Naidu, N.; Priya, J.; Rao, K.P.C.; Ranjith, K.; Shobha, S.; Siddiraju, S. Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 2021, 17, 404. [Google Scholar] [PubMed]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
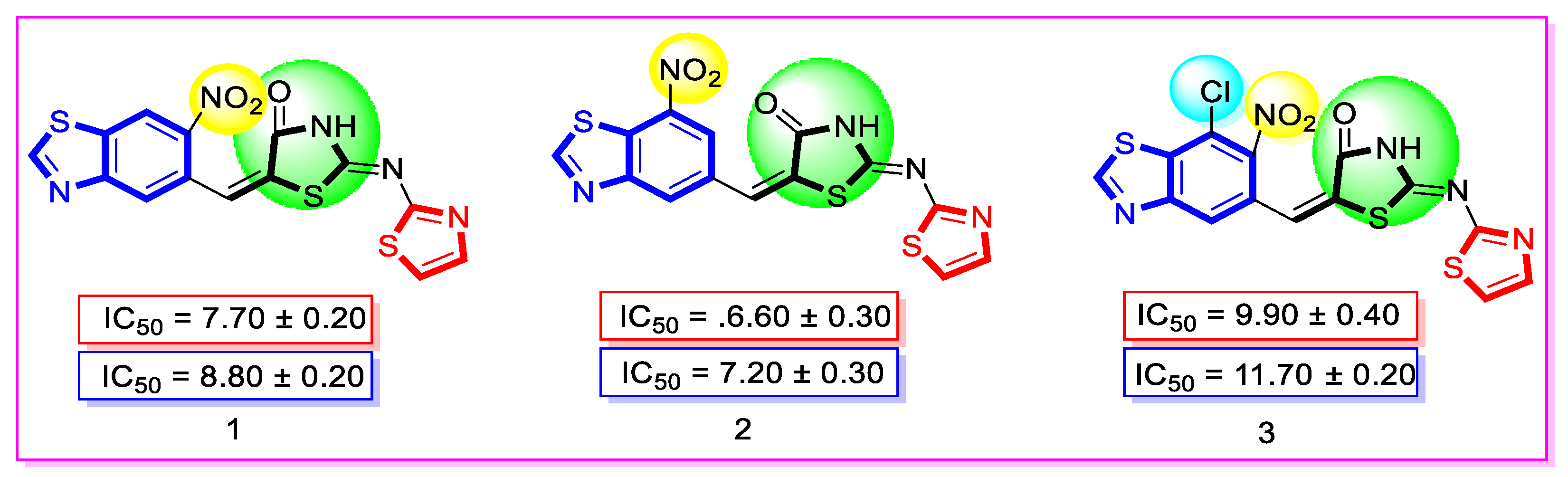
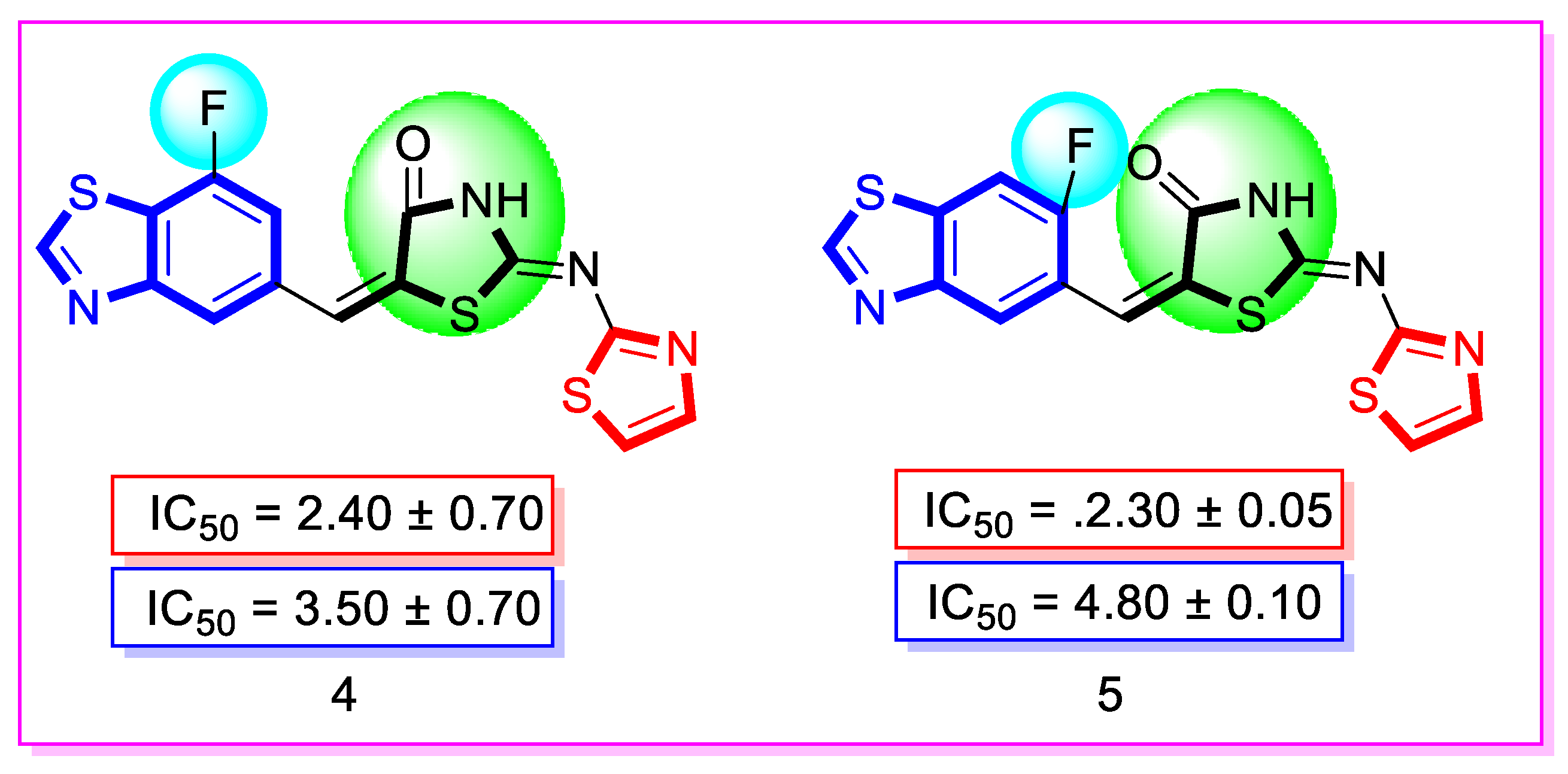
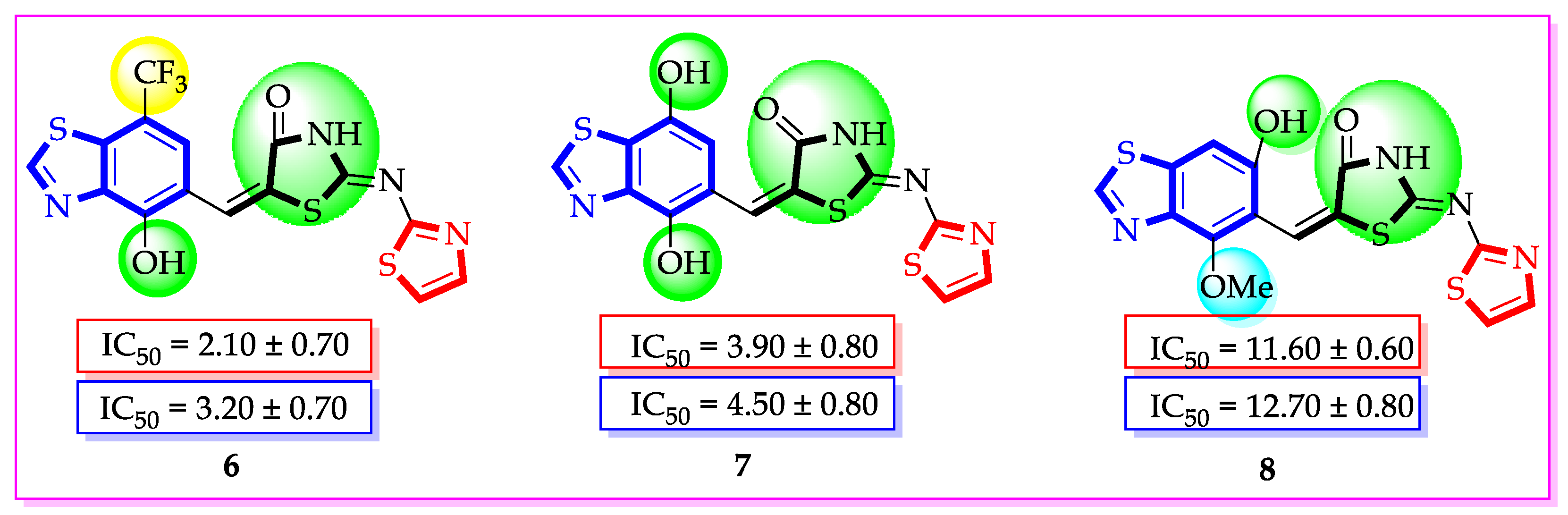
| Synthesized Compounds | R | α-Amylase IC50 (µM) | α-Glucosidase IC50 (µM) |
|---|---|---|---|
| 1 | 6-Nitro | 7.70 ± 0.20 | 8.80 ± 0.20 |
| 2 | 7-Nitro | 6.60 ± 0.30 | 7.20 ± 0.30 |
| 3 | 7-Chloro-6-nitro | 9.90 ± 0.40 | 11.70 ± 0.20 |
| 4 | 7-Fluoro | 2.40 ± 0.70 | 3.50 ± 0.70 |
| 5 | 7-Fluoro | 2.30 ± 0.05 | 4.80 ± 0.10 |
| 6 | 7-Trifluoromethyl-4-hydroxy | 2.10 ± 0.70 | 3.20 ± 0.70 |
| 7 | 4,7-dihydroxy | 3.90 ± 0.80 | 4.50 ± 0.80 |
| 8 | 4-hydroxy-6-methoxy | 11.60 ± 0.60 | 12.70 ± 0.80 |
| 9 | 7-(2-chloro-3-nitrophenyl) | 28.50 ± 0.30 | 29.20 ± 0.40 |
| 10 | 4-dimethylamino | 31.20 ± 0.10 | 33.60 ± 0.20 |
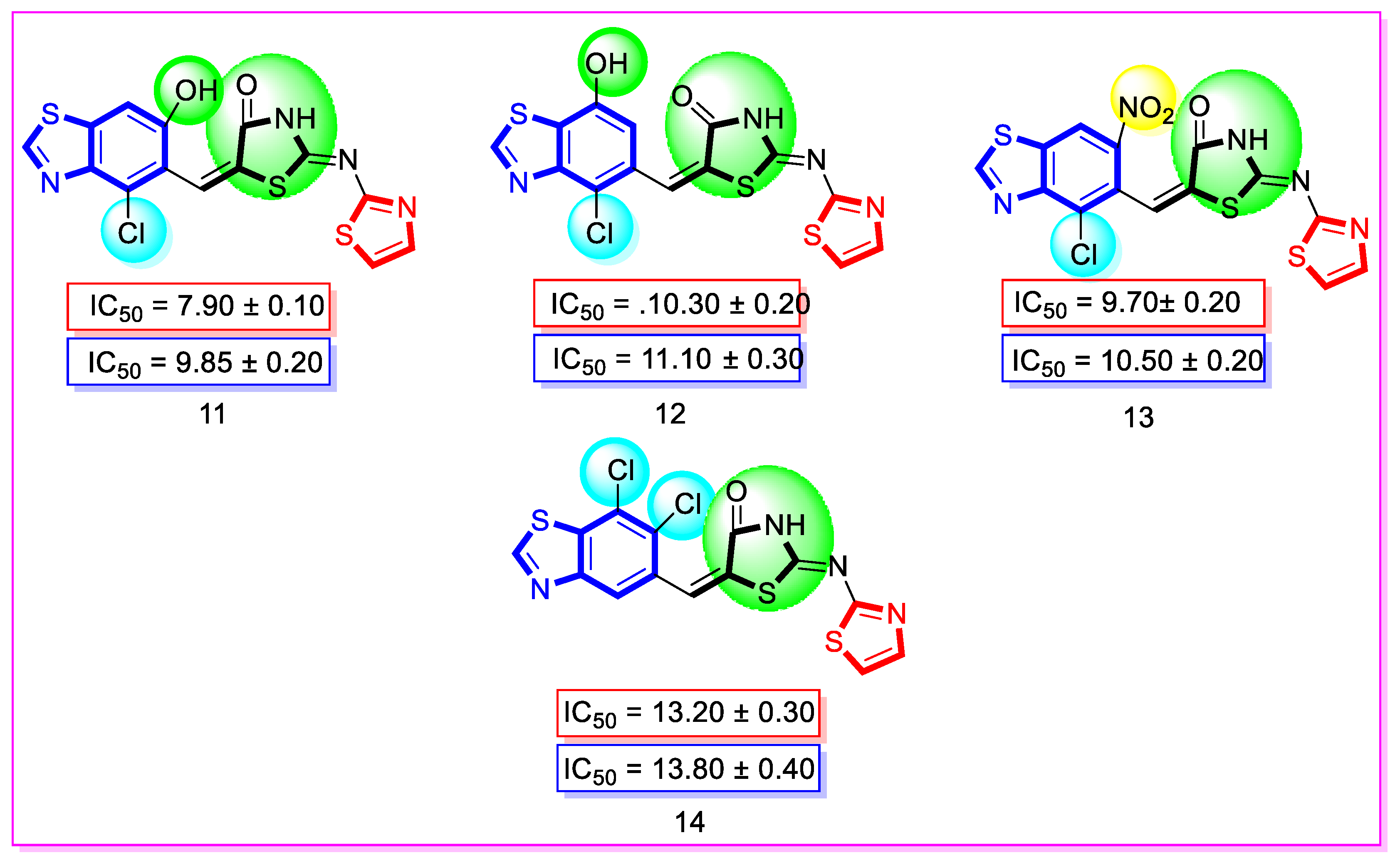
| 11 | 4-Chloro-6-hydroxy | 7.90 ± 0.10 | 9.85 ± 0.20 |
| 12 | 4-Chloro-7-hydroxy | 10.30 ± 0.20 | 11.10 ± 0.30 |
| 13 | 4-Chloro | 9.70 ± 0.20 | 10.50 ± 0.20 |
| 14 | 6,7-dichloro | 13.20 ± 0.30 | 13.80 ± 0.40 |
| 15 | 7-(3-cyanophenyl) | 37.50 ± 0.70 | 39.40 ± 0.80 |
| Standard drug acarbose | 9.10 ± 0.10 | 10.70 ± 0.10 | |
| Derivative-4 against α-amylase | Mode | Affinity Kcal/mol | Dist from rmsdl.b | Best Mode rmsdu.b |
| 1 | −8.7 | 0.000 | 0.000 | |
| 2 | −8.2 | 3.509 | 9.326 | |
| 3 | −8.1 | 3.524 | 9.035 | |
| 4 | −7.7 | 2.064 | 2.537 | |
| 5 | −7.7 | 3.222 | 4.078 | |
| 6 | −7.5 | 2.491 | 3.578 | |
| 7 | −7.4 | 3.997 | 9.249 | |
| 8 | −7.2 | 3.265 | 4.589 | |
| 9 | −7.2 | 4.618 | 6.871 | |
| Derivative-4 against α-glucosidase | 1 | −7.2 | 0.000 | 0.000 |
| 2 | −7.1 | 4.386 | 8.557 | |
| 3 | −7.1 | 2.493 | 2.825 | |
| 4 | −7.1 | 5.907 | 8.803 | |
| 5 | −6.5 | 4.612 | 5.490 | |
| 6 | −6.2 | 3.062 | 8.751 | |
| 7 | −6.1 | 4.812 | 8.361 | |
| 8 | −5.9 | 4.444 | 8.781 | |
| 9 | −5.9 | 6.901 | 8.970 |
| Derivative-5 against α-amylase | Mode | Affinity Kcal/mol | Dist from rmsdl.b | Best Mode rmsdu.b |
| 1 | −8.1 | 0.000 | 0.000 | |
| 2 | −7.7 | 2.690 | 3.719 | |
| 3 | −7.7 | 4.164 | 9.216 | |
| 4 | −7.3 | 1.867 | 2.573 | |
| 5 | −7.2 | 3.246 | 5.204 | |
| 6 | −7.2 | 3.340 | 9.046 | |
| 7 | −7.1 | 2.511 | 8.599 | |
| 8 | −6.6 | 12.800 | 14.844 | |
| 9 | −6.5 | 2.796 | 8.422 | |
| Derivative-5 against α-glucosidase | 1 | −7.3 | 0.000 | 0.000 |
| 2 | −7.2 | 4.474 | 8.247 | |
| 3 | −7.0 | 2.535 | 3.033 | |
| 4 | −6.5 | 6.261 | 8.114 | |
| 5 | −6.3 | 5.148 | 5.843 | |
| 6 | −6.2 | 3.705 | 4.420 | |
| 7 | −6.0 | 6.046 | 9.057 | |
| 8 | −6.0 | 16.081 | 17.997 | |
| 9 | −5.9 | 6.508 | 9.537 |
| Derivative-6 against α-amylase | Mode | Affinity Kcal/mol | Dist from rmsdl.b | Best Mode rmsdu.b |
| 1 | −8.6 | 0.000 | 0.000 | |
| 2 | −8.1 | 3.324 | 8.460 | |
| 3 | −8.0 | 3.376 | 8.475 | |
| 4 | −7.6 | 4.032 | 8.861 | |
| 5 | −7.5 | 2.904 | 8.532 | |
| 6 | −7.5 | 3.546 | 4.849 | |
| 7 | −7.3 | 3.664 | 5.155 | |
| 8 | −7.3 | 4.675 | 9.102 | |
| 9 | −7.3 | 3.260 | 3.961 | |
| Derivative-6 against α-glucosidase | 1 | −7.3 | 0.000 | 0.000 |
| 2 | −6.8 | 3.771 | 4.374 | |
| 3 | −6.6 | 5.743 | 8.923 | |
| 4 | −6.4 | 7.483 | 8.685 | |
| 5 | −6.1 | 5.822 | 8.279 | |
| 6 | −6.1 | 6.616 | 9.305 | |
| 7 | −6.0 | 15.492 | 16.488 | |
| 8 | −5.9 | 13.940 | 18.195 | |
| 9 | −5.9 | 6.522 | 10.003 |
| Acarbose against α-amylase | Mode | Affinity Kcal/mol | Dist from rmsdl.b | Best Mode rmsdu.b |
| 1 | −8.0 | 0.000 | 0.000 | |
| 2 | −7.8 | 3.267 | 11.500 | |
| 3 | −7.8 | 3.228 | 10.552 | |
| 4 | −7.7 | 2.163 | 9.832 | |
| 5 | −7.5 | 4.169 | 7.220 | |
| 6 | −7.5 | 5.320 | 9.443 | |
| 7 | −7.4 | 4.477 | 8.490 | |
| 8 | −7.3 | 3.580 | 11.779 | |
| 9 | −7.5 | 5.259 | 9.041 | |
| Acarbose against α-glucosidase | 1 | −7.3 | 0.000 | 0.000 |
| 2 | −7.3 | 3.562 | 11.099 | |
| 3 | −7.3 | 1.489 | 2.269 | |
| 4 | −6.9 | 3817 | 10.685 | |
| 5 | −6.9 | 3,838 | 10.852 | |
| 6 | −6.7 | 7.042 | 13.565 | |
| 7 | −6.7 | 6.866 | 11.671 | |
| 8 | −6.6 | 2.089 | 11.269 | |
| 9 | −6.6 | 4.540 | 11.478 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Iqbal, S.; Khan, M.; Rehman, W.; Shah, M.; Hussain, R.; Rasheed, L.; Khan, Y.; Dera, A.A.; Pashameah, R.A.; et al. Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase. Pharmaceuticals 2022, 15, 1164. https://doi.org/10.3390/ph15101164
Khan S, Iqbal S, Khan M, Rehman W, Shah M, Hussain R, Rasheed L, Khan Y, Dera AA, Pashameah RA, et al. Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase. Pharmaceuticals. 2022; 15(10):1164. https://doi.org/10.3390/ph15101164
Chicago/Turabian StyleKhan, Shoaib, Shahid Iqbal, Marwa Khan, Wajid Rehman, Mazloom Shah, Rafaqat Hussain, Liaqat Rasheed, Yousaf Khan, Ayed A. Dera, Rami Adel Pashameah, and et al. 2022. "Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase" Pharmaceuticals 15, no. 10: 1164. https://doi.org/10.3390/ph15101164
APA StyleKhan, S., Iqbal, S., Khan, M., Rehman, W., Shah, M., Hussain, R., Rasheed, L., Khan, Y., Dera, A. A., Pashameah, R. A., Alzahrani, E., & Farouk, A.-E. (2022). Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase. Pharmaceuticals, 15(10), 1164. https://doi.org/10.3390/ph15101164