
Rational Design and Synthesis of New Selective COX-2 Inhibitors with In Vivo PGE2-Lowering Activity by Tethering Benzenesulfonamide and 1,2,3-Triazole Pharmacophores to Some NSAIDs

 and
and 
Abstract
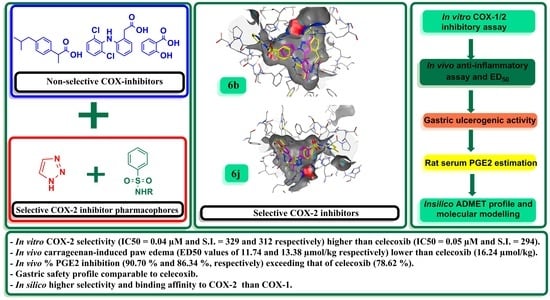
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Human COX-1 and Human COX-2 Enzymatic Inhibitory Activities
2.2.2. In Vivo Carrageenan-Induced Paw Edema in Mice
2.2.3. In Vivo Estimation of Rat Serum Prostaglandin E2 (PGE2)
2.2.4. Ulcerogenic Effects
2.3. Docking Studies of the Potential Selective COX-2 Target Inhibitors vs. the Weaker Selective Inhibitors
2.4. In Silico Prediction of the Physicochemical Properties, Drug-Likeness Score, Pharmacokinetics, Toxicity Profile and Ligand Efficiency Metrics
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of 4-Substituted Phenyl azides (1a–d)
- 4-Azido-N-(pyrimidin-2-yl)benzenesulfonamide (1c)
- 4-Azido-N-(4-methylpyrimidin-2-yl)benzenesulfonamide (1d)
3.1.2. General Procedure for Synthesis of 1-(1-(4-Substituted phenyl)-5-methyl-1H-1,2,3-triazol-4-yl) ethan-1-ones (4a–d)
- 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (4c)
- 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-(4-methylpyrimidin-2-yl)benzenesulfonamide (4d)
3.1.3. General Procedure for Synthesis of Compounds 6a–j
- 4-(4-(1-(2-(2-(4-Isobutylphenyl)propanoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6a)
- 4-(4-(1-(2-(2-(4-Isobutylphenyl)propanoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(thiazol-2-yl)benzenesulfonamide (6b)
- 4-(4-(1-(2-(2-(4-Isobutylphenyl)propanoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (6c)
- 4-(4-(1-(2-(2-(2-((2,6-Dichlorophenyl)amino)phenyl)acetyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6d)
- 4-(4-(1-(2-(2-(2-((2,6-Dichlorophenyl)amino)phenyl)acetyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(thiazol-2-yl)benzenesulfonamide (6e)
- 4-(4-(1-(2-(2-(2-((2,6-Dichlorophenyl)amino)phenyl)acetyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (6f)
- 4-(4-(1-(2-(2-Hydroxybenzoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6g)
- 4-(4-(1-(2-(2-Hydroxybenzoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(thiazol-2-yl)benzenesulfonamide (6h)
- 4-(4-(1-(2-(2-Hydroxybenzoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (6i)
- 4-(4-(1-(2-(2-Hydroxybenzoyl)hydrazineylidene)ethyl)-5-methyl-1H-1,2,3-triazol-1-yl)-N-(4-methylpyrimidin-2-yl)benzenesulfonamide (6j)
3.2. Biological Screening
3.2.1. In Vitro Human COX-1 and COX-2 Enzymatic Inhibitory Activities
3.2.2. Carrageenan-Induced Paw Edema in Mice
3.2.3. Determination of ED50
3.2.4. Estimation of Rat Serum Prostaglandin E2 (PGE2)
3.2.5. Ulcerogenic Effects
3.3. Molecular Docking Studies
3.4. In Silico Prediction of the Physicochemical Properties, Drug-Likeness Score, Pharmacokinetics, Toxicity Profile and Ligand Efficiency Metrics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghlichloo, I.; Gerriets, V. Nonsteroidal Anti-inflammatory Drugs (NSAIDs). In StatPears [Internet]; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Osafo, N. Mechanism of Action of Nonsteroidal Anti-Inflammatory Drugs. In Nonsteroidal Anti-Inflammatory Drugs; Agyare, C., Ed.; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef]
- Kam, P.C.A.; See, A.U.-L. Cyclo-oxygenase isoenzymes: Physiological and pharmacological role. Anaesthesia 2000, 55, 442–449. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.; Henry, D. Current problems with non-specific COX inhibitors. Curr. Pharm. Des. 2000, 6, 1693–1724. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.-H.; So, S.-P.; Ruan, K.-H. Inducible COX-2 dominates over COX-1 in prostacyclin biosynthesis: Mechanisms of COX-2 inhibitor risk to heart disease. Life Sci. 2011, 88, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Alajbegovic, A.; Gomes, A.V. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2015, 2015, 536962. [Google Scholar] [CrossRef]
- Gunter, B.R.; Butler, K.A.; Wallace, R.L.; Smith, S.M.; Harirforoosh, S. Non-steroidal anti-inflammatory drug-induced cardiovascular adverse events: A meta-analysis. J. Clin. Pharm. Ther. 2017, 42, 27–38. [Google Scholar] [CrossRef]
- Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar]
- Kassab, S.E.; Khedr, M.A.; Ali, H.I.; Abdalla, M.M. Discovery of new indomethacin-based analogs with potentially selective cyclooxygenase-2 inhibition and observed diminishing to PGE2 activities. Eur. J. Med. Chem. 2017, 141, 306–321. [Google Scholar] [CrossRef]
- Dwivedi, A.K.; Gurjar, V.; Kumar, S.; Singh, N. Molecular basis for nonspecificity of nonsteroidal anti-inflammatory drugs (NSAIDs). Drug Discov. Today 2015, 20, 863–873. [Google Scholar] [CrossRef]
- Bhattacharyya, D.K.; Lecomte, M.; Rieke, C.J.; Garavito, M.; Smith, W.L. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 1996, 271, 2179–2184. [Google Scholar] [CrossRef]
- Solomon, D.H. Selective cyclooxygenase 2 inhibitors and cardiovascular events. Arthritis Rheum. 2005, 52, 1968–1978. [Google Scholar] [CrossRef]
- Agouram, N.; el Hadrami, E.M.; Bentama, A. 1,2,3-Triazoles as Biomimetics in Peptide Science. Molecules 2021, 26, 2937. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule, V.D. 1,2,3-Triazoles of 8-Hydroxyquinoline and HBT: Synthesis and Studies (DNA Binding, Antimicrobial, Molecular Docking, ADME, and DFT). ACS Omega 2021, 6, 27089–27100. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef]
- Assali, M.; Abualhasan, M.; Sawaftah, H.; Hawash, M.; Mousa, A. Synthesis, Biological Activity, and Molecular Modeling Studies of Pyrazole and Triazole Derivatives as Selective COX-2 Inhibitors. J. Chem. 2020, 2020, 6393428. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Carbonic Anhydrase Inhibitors: Inhibition of Isozymes I, II, and IX with Triazole-Linked O-Glycosides of Benzene Sulfonamides. J. Med. Chem. 2007, 50, 1651–1657. [Google Scholar] [CrossRef]
- Elzahhar, P.A.; el Wahab, S.M.A.; Elagawany, M.; Daabees, H.; Belal, A.S.F.; L-Yazbi, A.F.E.; Eid, A.H.; Alaaeddine, R.; Hegazy, R.R.; Allam, R.M.; et al. Expanding the anticancer potential of 1,2,3-triazoles via simultaneously targeting Cyclooxygenase-2, 15-lipoxygenase and tumor-associated carbonic anhydrases. Eur. J. Med. Chem. 2020, 200, 112439. [Google Scholar] [CrossRef]
- Baena, Y.A.; Manzo, R.H.; D’León, L.F.Q.P. Preparation and physicochemical characterization of some polyelectrolyte-diclofenac complexes. Vitae 2011, 18, 305–311. [Google Scholar]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Inc.: New York, NY, USA, 1989; pp. 1076–1080. ISBN 0-582-46236-3. [Google Scholar] [CrossRef]
- Al-Ajely, M.; Yaseen, A. Synthesis and Characterization of Some New Hydrazides and Their Derivatives, Ibn Al-Haitham. J. Pure Appl. Sci. 2015, 28, 103–112. [Google Scholar]
- Gedawy, E.M.; Kassab, A.E.; el Kerdawy, A.M. Design, synthesis and biological evaluation of novel pyrazole sulfonamide derivatives as dual COX-2/5-LOX inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef]
- El-Dershaby, N.H.; El-Hawash, S.A.; Kassab, S.E.; Dabees, H.G.; Moneim, A.E.A.; Wahab, I.A.A.; Abd-Alhaseeb, M.M.; El-Miligy, M.M.M. Rational design of biodegradable sulphonamide candidates treating septicaemia by synergistic dual inhibition of COX-2/PGE2 axis and DHPS enzyme. J. Enzyme Inhib. Med. Chem. 2022, 37, 1737–1751. [Google Scholar] [CrossRef]
- El-Miligy, M.M.M.; Al-Kubeisi, A.K.; El-Zemity, S.R.; Nassra, R.A.; Abu-Serie, M.M.; Hazzaa, A.A. Discovery of small molecule acting as multitarget inhibitor of colorectal cancer by simultaneous blocking of the key COX-2, 5-LOX and PIM-1 kinase enzymes. Bioorg. Chem. 2021, 115, 105171. [Google Scholar] [CrossRef] [PubMed]
- El-Dash, Y.; Khalil, N.A.; Ahmed, E.M.; Hassan, M.S.A. Synthesis and biological evaluation of new nicotinate derivatives as potential anti-inflammatory agents targeting COX-2 enzyme. Bioorg. Chem. 2021, 107, 104610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.B.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Stallings, Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Molinspiration, (n.d.). Available online: https://www.molinspiration.com/ (accessed on 4 March 2022).
- Pre-ADMET, (n.d.). Available online: https://preadmet.qsarhub.com/ (accessed on 14 February 2022).
- Osiris Property Explorer, (n.d.). Available online: https://www.organic-chemistry.org/prog/peo/ (accessed on 14 February 2022).
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Govindasamy, J.; Khalilullah, H.; Mohan, G.; Stables, J.P.; Pannecouque, C.; de Clercq, E. POMA analyses as new efficient bioinformatics’ platform to predict and optimise bioactivity of synthesized 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg. Med. Chem. Lett. 2012, 22, 7029–7035. [Google Scholar] [CrossRef]
- Hassan, N.W.; Saudi, M.N.; Abdel-Ghany, Y.S.; Ismail, A.; Elzahhar, P.A.; Sriram, D.; Nassra, R.; Abdel-Aziz, M.M.; El-Hawash, S.A. Novel pyrazine based anti-tubercular agents: Design, synthesis, biological evaluation and in silico studies. Bioorg. Chem. 2020, 96, 103610. [Google Scholar] [CrossRef]
- Ali, M.R.; Kumar, S.; Afzal, O.; Shalmali, N.; Ali, W.; Sharma, M.; Bawa, S. 2-Benzamido-4-methylthiazole-5-carboxylic Acid Derivatives as Potential Xanthine Oxidase Inhibitors and Free Radical Scavengers. Arch. Pharm. 2017, 350, 1600313. [Google Scholar] [CrossRef]
- Kenny, P.W.; Leitão, A.; Montanari, C.A. Ligand efficiency metrics considered harmful. J. Comput. Aided Mol. Des. 2014, 28, 699–710. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- El-Dershaby, N.H.; El-Hawash, S.A.; Kassab, S.E.; Daabees, H.G.; El-Miligy, M.M.M.; Moneim, A.E.A. Design, Synthesis and Biological Evaluation of Some Novel Sulfonamide Derivatives as Dual Antiinflammatory and Antibacterial Agents; Approval No. HU2021/Z/AE0721-01; Institutional Animal Care and Use Committee (HU-IACUC), Faculty of Science, Helwan University: Cairo, Egypt, 2021. [Google Scholar]
- Shibaike, Y.; Gotoh, M.; Ogawa, C.; Nakajima, S.; Yoshikawa, K.; Kobayashi, T.; Murakami-Murofushi, K. 2-Carba cyclic phosphatidic acid inhibits lipopolysaccharide-induced prostaglandin E2 production in a human macrophage cell line. Biochem. Biophys. Rep. 2019, 19, 100668. [Google Scholar] [CrossRef]
- Abouzeit-Har, M.S.; Verimer, T.; Long, J.P. Effect of long term estrogen and lithium treatment on restraint induced gastric erosion in intact and ovariectomized rats. Pharmazie 1982, 37, 593–595. [Google Scholar]
- El-Miligy, M.M.M.; Hazzaa, A.A.; El-Messmary, H.; Nassra, R.A.; El-Hawash, S.A.M. New benzothiophene derivatives as dual COX-1/2 and 5-LOX inhibitors: Synthesis, biological evaluation and docking study. Future Med. Chem. 2017, 9, 443–468. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound NO. | COX-1 | COX-2 | SI c |
|---|---|---|---|
| IC50 (µM) a | IC50 (µM) b | COX-1/COX-2 | |
| Celecoxib | 14.7 ± 0.0577 | 0.05 ± 0.0003 | 294 |
| Indomethacin | 0.1 ± 0.0033 | 0.48 ± 0.0058 | 0.21 |
| Diclofenac sodium | 3.8 ± 0.0333 | 0.84 ± 0.0033 | 4.52 |
| 1c | 5.93 ± 0.0882 | 0.09 ± 0.0006 | 65.89 |
| 1d | 4.6 ± 0.1155 | 0.1 ± 0.0033 | 46 |
| 4c | 8.23 ± 0.0667 | 0.09 ± 0.0003 | 91.44 |
| 4d | 9.27 ± 0.0667 | 0.11 ± 0.0057 | 84.27 |
| 6a | 9.13 ± 0.0882 | 0.08 ± 0.0006 | 114.13 |
| 6b | 13.17 ± 0.1202 | 0.04 ± 0.0007 | 329.25 |
| 6c | 11.17 ± 0.0333 | 0.07 ± 0.0006 | 159.57 |
| 6d | 8.4 ± 0.0577 | 0.08 ± 0.0009 | 105 |
| 6e | 10.23 ± 0.0333 | 0.05 ± 0.0006 | 204.6 |
| 6f | 12.67 ± 0.0333 | 0.08 ± 0.0007 | 158.38 |
| 6g | 6.67 ± 0.0667 | 0.08 ± 0.0006 | 83.38 |
| 6h | 8.27 ± 0.0667 | 0.09 ± 0.0006 | 91.89 |
| 6i | 10.23 ± 0.0882 | 0.07 ± 0.0006 | 146.14 |
| 6j | 12.5 ± 0.0577 | 0.04 ± 0.0006 | 312.5 |
| Compound No. a | Thickness of Edema (mm) b | ED50 f (µmol/kg) (95% Confidence Level) | ||||
|---|---|---|---|---|---|---|
| 0 h | 2 h | 4 h | 6 h | 8 h | ||
| Control (carrageenan) | 2.13 ± 0.006 | 3.64 ± 0.007 d | 3.77 ± 0.006 d | 3.86 ± 0.007 d | 3.85 ± 0.009 d | |
| Celecoxib | 2.10 ± 0.005 | 2.54 ± 0.007 c (70.86%) e | 2.44 ± 0.011 c (79.27%) | 2.32 ± 0.010 c (87.28%) | 2.30 ± 0.009 c (89.01%) | 16.24 (14.09–18.60) |
| Diclofenac | 2.08 ± 0.006 | 2.96 ± 0.009 c,d (41.72%) | 2.81 ± 0.009 c,d (55.49%) | 2.64 ± 0.01 c,d (67.63%) | 2.58 ± 0.01 c,d (72.53%) | 18.35 (16.22–20.63) |
| 6b | 2.14 ± 0.006 | 2.34 ± 0.009 c,d (86.75%) | 2.23 ± 0.009 c,d (94.51%) | 2.18 ± 0.009 c,d (97.69%) | 2.15 ± 0.007 c,d (99.45%) | 11.74 (10.46–13.16) |
| 6e | 2.12 ± 0.007 | 2.75 ± 0.008 c,d (58.28%) | 2.63 ± 0.010 c,d (68.90%) | 2.54 ± 0.009 c,d (75.72%) | 2.52 ± 0.006 c,d (78.02%) | 18.23 (16.61–19.92) |
| 6j | 2.08 ± 0.007 | 2.48 ± 0.005 c,d (73.51%) | 2.37 ± 0.009 c,d (82.32%) | 2.28 ± 0.011 c,d (88.44%) | 2.24 ± 0.009 c,d (91.21%) | 13.38 (12.04–14.84) |
| Compound NO. a | PGE2 Serum Conc. (pg/mL) b | % Inhibition |
|---|---|---|
| Control (pre-carrageenan) | 116.450 ± 5.47 c,d | - |
| Control 1 (post-carrageenan) | 735.470 ± 8.03 d | - |
| Celecoxib | 157.245 ± 7.33 c,d | 78.62% |
| Diclofenac | 203.973 ± 6.35 c,d | 72.27% |
| 6b | 68.388 ± 7.13 c,d | 90.70% |
| 6e | 185.329 ± 6.79 c,d | 74.80% |
| 6j | 100.435 ± 5.66 c,d | 86.34% |
| 6b | 6j | Celecoxib | Diclofenac | |
|---|---|---|---|---|
| Physicochemical Parameters and Drug-Likeness | ||||
| Log Pa | 4.67 | 2.53 | 3.61 | 4.57 |
| M.Wtb | 565.73 | 506.55 | 381.38 | 296.15 |
| HBAc | 10 | 12 | 5 | 3 |
| HBDd | 2 | 3 | 2 | 2 |
| Lipinski’s violation e | 1 | 2 | 0 | 0 |
| NROTBf | 10 | 7 | 4 | 4 |
| TPSAg | 131.24 | 164.36 | 77.99 | 49.33 |
| %ABSh | 63.72 | 52.30 | 82.09 | 91.98 |
| Volumei | 493.62 | 423.19 | 298.65 | 238.73 |
| Sj | 0.011 | 4.81 | 2.08 | 6.17 |
| Drug-likeness score k | 0.16 | 0.25 | 0.37 | 0.38 |
| Pharmacokinetics (ADME) | ||||
| Caco-2l | 11.54 | 0.56 | 0.49 | 24.53 |
| MDCKm | 0.052 | 2.704 | 45.05 | 51.46 |
| HIAn | 96.59 | 91.53 | 96.69 | 95.96 |
| BBBo | 0.125 | 0.049 | 0.027 | 1.39 |
| PPBp | 87.67 | 97.87 | 91.08 | 91.96 |
| Ligand efficiency metrics | ||||
| LE (COX-2)q | 0.26 | 0.28 | 0.38 | 0.44 |
| LLEr | 2.73 | 4.87 | 2.87 | 1.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Dershaby, N.H.; El-Hawash, S.A.; Kassab, S.E.; Daabees, H.G.; Abdel Moneim, A.E.; El-Miligy, M.M.M. Rational Design and Synthesis of New Selective COX-2 Inhibitors with In Vivo PGE2-Lowering Activity by Tethering Benzenesulfonamide and 1,2,3-Triazole Pharmacophores to Some NSAIDs. Pharmaceuticals 2022, 15, 1165. https://doi.org/10.3390/ph15101165
El-Dershaby NH, El-Hawash SA, Kassab SE, Daabees HG, Abdel Moneim AE, El-Miligy MMM. Rational Design and Synthesis of New Selective COX-2 Inhibitors with In Vivo PGE2-Lowering Activity by Tethering Benzenesulfonamide and 1,2,3-Triazole Pharmacophores to Some NSAIDs. Pharmaceuticals. 2022; 15(10):1165. https://doi.org/10.3390/ph15101165
Chicago/Turabian StyleEl-Dershaby, Nada H., Soad A. El-Hawash, Shaymaa E. Kassab, Hoda G. Daabees, Ahmed E. Abdel Moneim, and Mostafa M. M. El-Miligy. 2022. "Rational Design and Synthesis of New Selective COX-2 Inhibitors with In Vivo PGE2-Lowering Activity by Tethering Benzenesulfonamide and 1,2,3-Triazole Pharmacophores to Some NSAIDs" Pharmaceuticals 15, no. 10: 1165. https://doi.org/10.3390/ph15101165
APA StyleEl-Dershaby, N. H., El-Hawash, S. A., Kassab, S. E., Daabees, H. G., Abdel Moneim, A. E., & El-Miligy, M. M. M. (2022). Rational Design and Synthesis of New Selective COX-2 Inhibitors with In Vivo PGE2-Lowering Activity by Tethering Benzenesulfonamide and 1,2,3-Triazole Pharmacophores to Some NSAIDs. Pharmaceuticals, 15(10), 1165. https://doi.org/10.3390/ph15101165