



Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Rat VC in the CKD Model
2.3. VSMC Culture and VSMC Calcification Model
2.4. Biochemistry
2.5. Hematoxylin and Eosin Staining
2.6. Alizarin Red Staining
2.7. ROS Analysis
2.8. SOD Activity Assay
2.9. Mitochondrial Membrane Potential Measurements
2.10. Enzyme-Linked Immunosorbent Assay
2.11. Immunofluorescence Staining
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
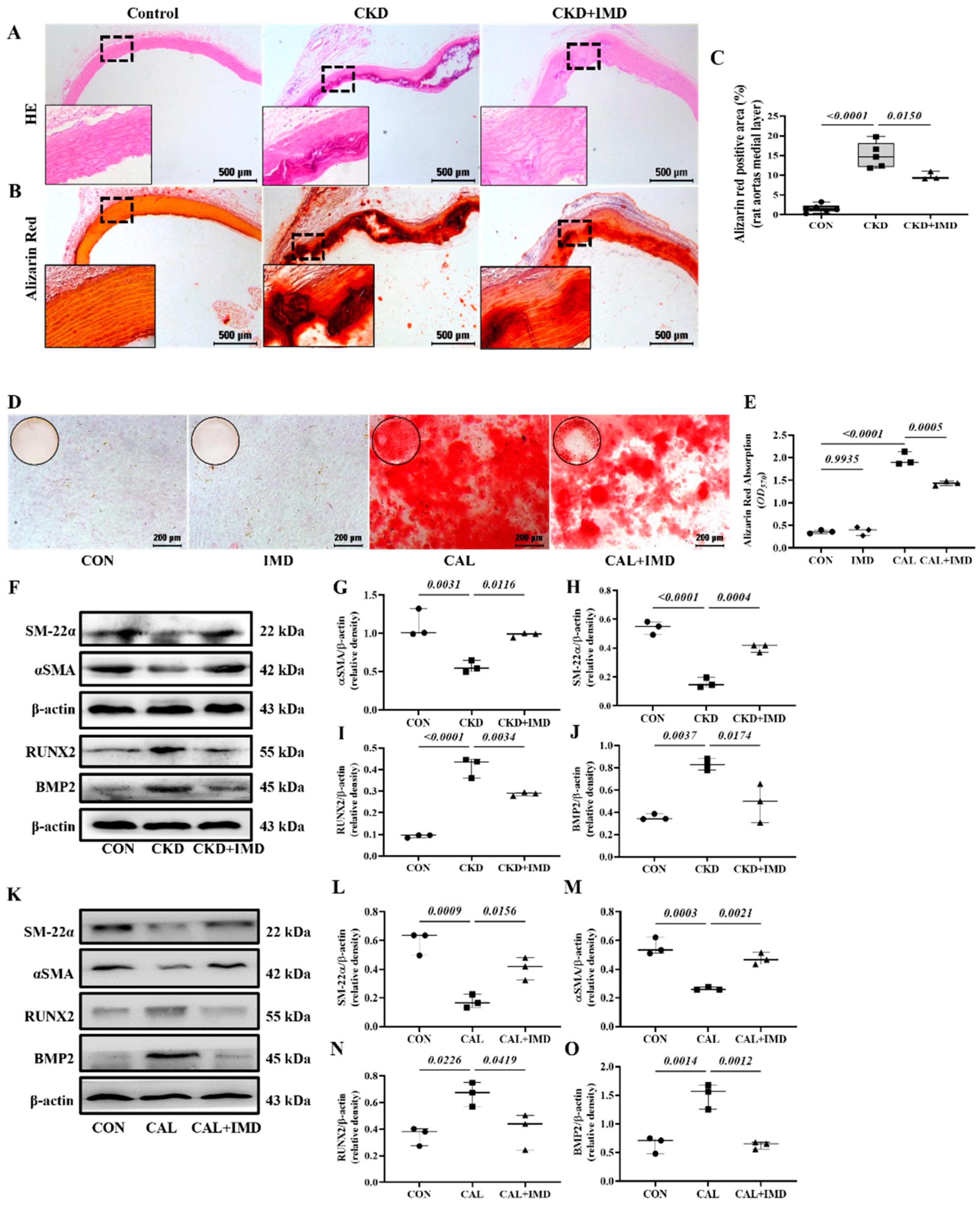
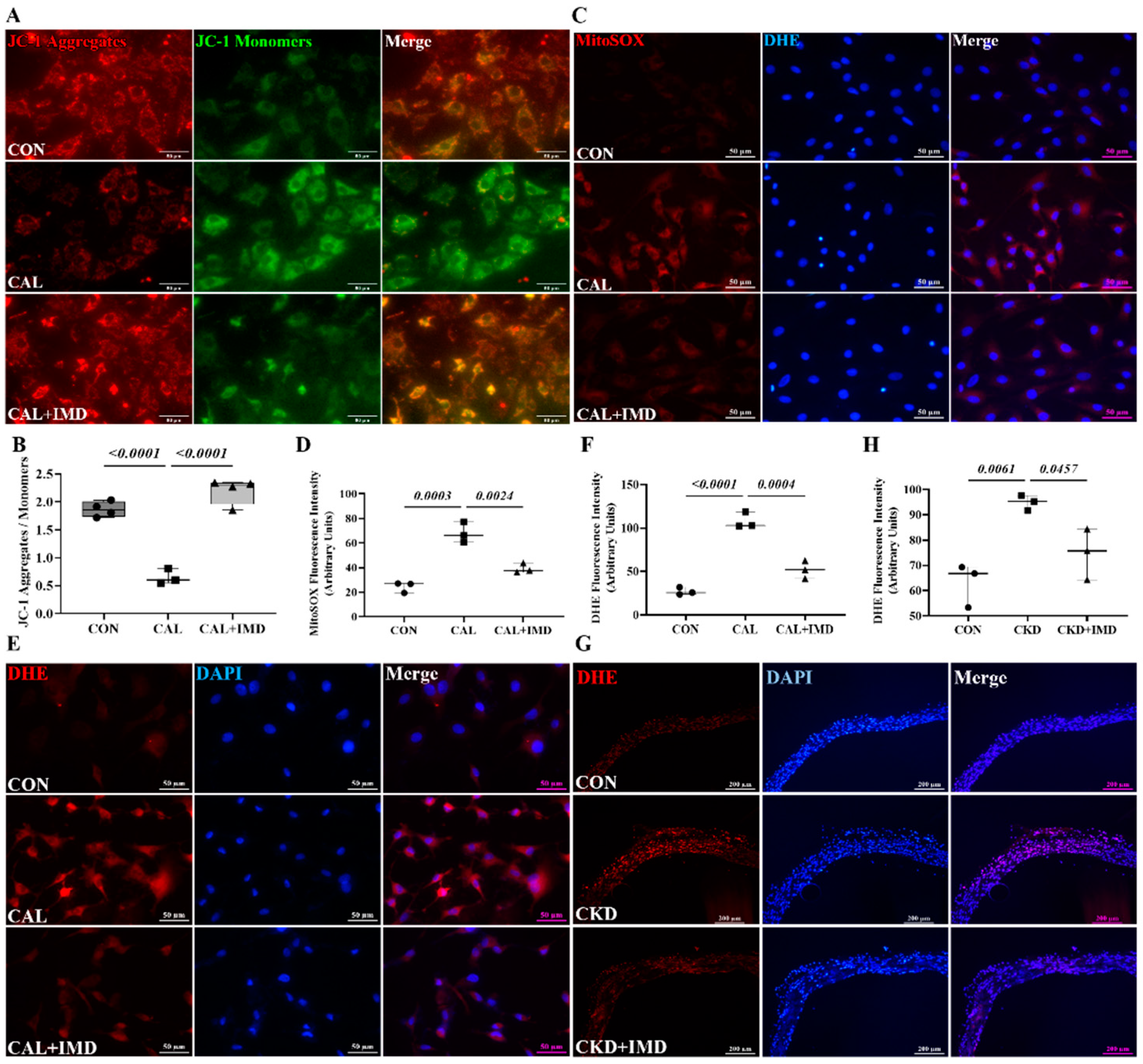
3.1. IMD Attenuated VC in CKD Rats by Improving Mitochondrial Function and Inhibiting Mitochondrial Oxidative Stress
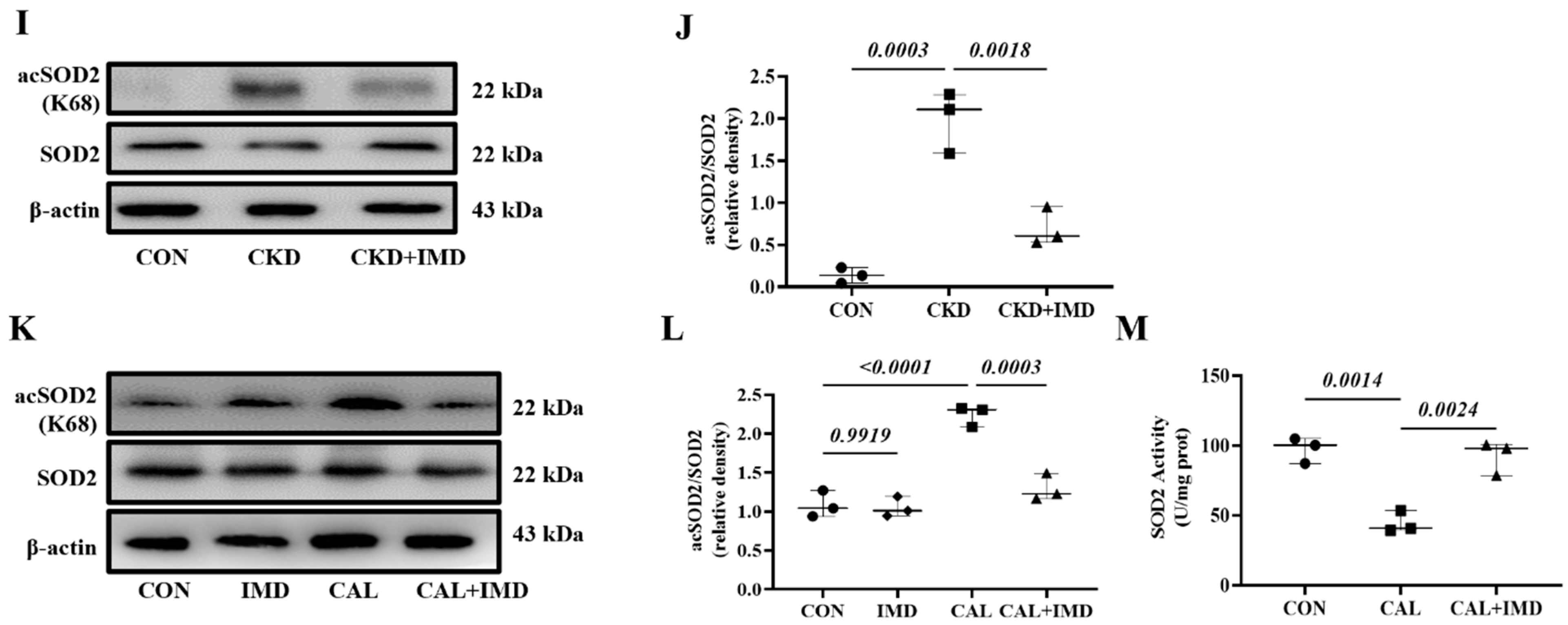
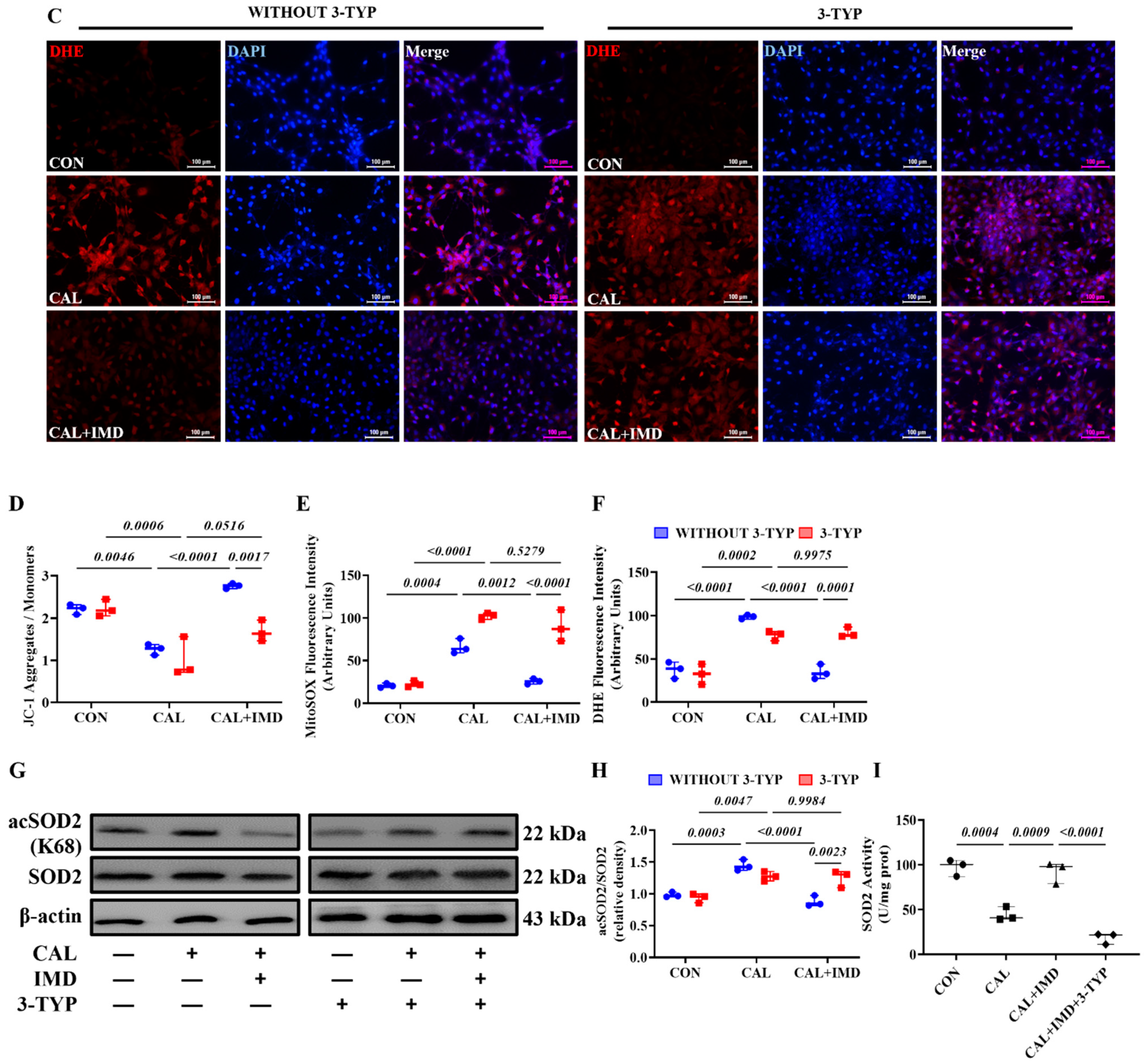
3.2. IMD Improved Mitochondrial Function and Inhibited Mitochondrial Oxidative Stress by Upregulating Sirt3
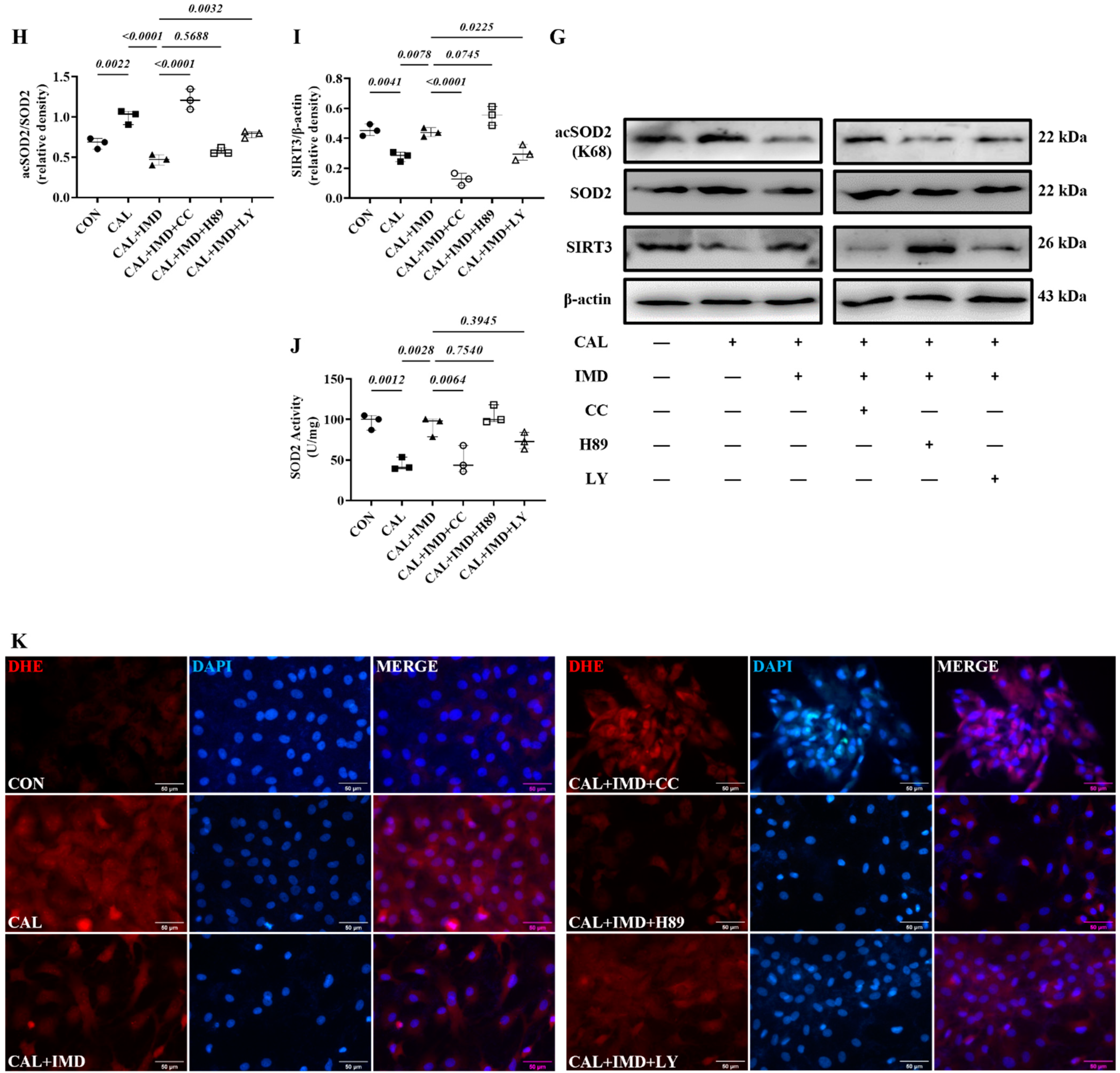
3.3. IMD Upregulated Sirt3 by Activating Its Receptor and the AMPK Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamura, K.; Suzuki, Y.; Hashiba, H.; Tamura, H.; Aizawa, S.; Kogo, H. Effect of Etidronate on Aortic Calcification and Bone Metabolism in Calcitoriol-Treated Rats with Subtotal Nephrectomy. J. Pharmacol. Sci. 2005, 99, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, L.S.; Ren, J.L.; Zhang, Y.R.; Wu, N.; Jia, M.Z.; Yu, Y.R.; Ning, Z.P.; Tang, C.S.; Qi, Y.F. Intermedin1-53 Attenuates Aging-Associated Vascular Calcification in Rats by Upregulating Sirtuin 1. Aging (Albany. NY) 2020, 12, 5651–5674. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.R.; Guo, J.; Wang, Y.; Hou, Y.L.; Lu, W.W.; Zhang, J.S.; Yu, Y.R.; Xu, M.J.; Liu, X.Y.; Wang, X.J.; et al. Intermedin1–53 Attenuates Vascular Calcification in Rats with Chronic Kidney Disease by Upregulation of α-Klotho. Kidney Int. 2016, 89, 586–600. [Google Scholar] [CrossRef]
- Balogh, E.; Tóth, A.; Méhes, G.; Trencsényi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)-Dependent and Reactive Oxygen Species-Dependent Manner. Arterioscler. Thromb. Vasc. Biol. 2019, 1, ATVBAHA119312509. [Google Scholar] [CrossRef]
- Lu, W.W.; Jia, L.X.; Ni, X.Q.; Zhao, L.; Chang, J.R.; Zhang, J.S.; Hou, Y.L.; Zhu, Y.; Guan, Y.F.; Yu, Y.R.; et al. Intermedin 1− 53 Attenuates Abdominal Aortic Aneurysm by Inhibiting Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2176–2190. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth Muscle Cell Phenotypic Transition Associated with Calcification: Upregulation of Cbfa1 and Downregulation of Smooth Muscle Lineage Markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Wang, P.W.; Pang, Q.; Zhou, T.; Song, X.Y.; Pan, Y.J.; Jia, L.P.; Zhang, A.H. Irisin Alleviates Vascular Calcification by Inhibiting VSMC Osteoblastic Transformation and Mitochondria Dysfunction via AMPK/Drp1 Signaling Pathway in Chronic Kidney Disease. Atherosclerosis 2022, 346, 36–45. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Yeh, H.-Y.; Chao, C.-T.; Chiang, C.-K. Superoxide Dismutase 2 (SOD2) in Vascular Calcification: A Focus on Vascular Smooth Muscle Cells, Calcification Pathogenesis, and Therapeutic Strategies. Oxid. Med. Cell. Longev. 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, J.Y.; Yu, B.; Cong, X.; Zhang, W.G.; Li, L.; Liu, L.M.; Zhou, Y.; Zhang, C.L.; Gu, P.L.; et al. Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Inhibits Vascular Calcification through Sirtuin 3-Mediated Reduction of Mitochondrial Oxidative Stress. Antioxid. Redox Signal. 2019, 31, 75–91. [Google Scholar] [CrossRef]
- Galli, U.; Mesenzani, O.; Coppo, C.; Sorba, G.; Canonico, P.L.; Tron, G.C.; Genazzani, A.A. Identification of a Sirtuin 3 Inhibitor That Displays Selectivity over Sirtuin 1 and 2. Eur. J. Med. Chem. 2012, 55, 58–66. [Google Scholar] [CrossRef]
- Ni, X.Q.; Zhang, J.S.; Tang, C.S.; Qi, Y.F. Intermedin/Adrenomedullin2: An Autocrine/Paracrine Factor in Vascular Homeostasis and Disease. Sci. China Life Sci. 2014, 57, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Xu, M.J.; Wang, X. Adrenomedullin 2/Intermedin: A Putative Drug Candidate for Treatment of Cardiometabolic Diseases. Br. J. Pharmacol. 2018, 175, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Chijiwa, T.; Mishima, A.; Hagiwara, M.; Sano, M.; Hayashi, K.; Inoue, T.; Naito, K.; Toshioka, T.; Hidaka, H. Inhibition of Forskolin-Induced Neurite Outgrowth and Protein Phosphorylation by a Newly Synthesized Selective Inhibitor of Cyclic AMP-Dependent Protein Kinase, N-[2-(p-Bromocinnamylamino)Ethyl]-5-Isoquinolinesulfonamide (H-89), of PC12D Pheochromocytoma. J. Biol. Chem. 1990, 265, 5267–5272. [Google Scholar] [CrossRef]
- Chaussade, C.; Rewcastle, G.W.; Kendall, J.D.; Denny, W.A.; Cho, K.; Grønning, L.M.; Chong, M.L.; Anagnostou, S.H.; Jackson, S.P.; Daniele, N.; et al. Evidence for Functional Redundancy of Class IA PI3K Isoforms in Insulin Signalling. Biochem. J. 2007, 404, 449–458. [Google Scholar] [CrossRef]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef]
- Cardús, A.; Panizo, S.; Parisi, E.; Fernandez, E.; Valdivielso, J.M. Differential Effects of Vitamin D Analogs on Vascular Calcification. J. Bone Miner. Res. 2007, 22, 860–866. [Google Scholar] [CrossRef]
- Gauthier-Bastien, A.; Ung, R.V.; Larivière, R.; Mac-Way, F.; Lebel, M.; Agharazii, M. Vascular Remodeling and Media Calcification Increases Arterial Stiffness in Chronic Kidney Disease. Clin. Exp. Hypertens. 2014, 36, 173–180. [Google Scholar] [CrossRef]
- Mendoza, F.J.; Lopez, I.; Montes De Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolic Acidosis Inhibits Soft Tissue Calcification in Uremic Rats. Kidney Int. 2008, 73, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; McDermott, B.J. Intermedin (Adrenomedullin-2): A Novel Counter-Regulatory Peptide in the Cardiovascular and Renal Systems. Br. J. Pharmacol. 2009, 153, S247–S262. [Google Scholar] [CrossRef] [PubMed]
- Disthabanchong, S.; Srisuwarn, P. Mechanisms of Vascular Calcification in Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial Reactive Oxygen Species Promote P65 Nuclear Translocation Mediating High-Phosphate-Induced Vascular Calcification in Vitro and in Vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef]
- Bi, X.; Du, C.; Wang, X.; Wang, X.Y.; Han, W.; Wang, Y.; Qiao, Y.; Zhu, Y.; Ran, L.; Liu, Y.; et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv. Sci. 2021, 8, 1–18. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin Attenuates Vascular Calcification by Inhibiting Oxidative Stress and Mitochondrial Fission. Vascul. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef]
- Byon, C.H.; Chen, Y. Molecular Mechanisms of Vascular Calcification in Chronic Kidney Disease: The Link between Bone and the Vasculature. Curr. Osteoporos. Rep. 2015, 13, 206–215. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone Morphogenetic Protein-2 Activates NADPH Oxidase to Increase Endoplasmic Reticulum Stress and Human Coronary Artery Smooth Muscle Cell Calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [CrossRef]
- Kang, Y.; Ding, L.; Dai, H.; Wang, F.; Zhou, H.; Gao, Q.; Xiong, X.; Zhang, F.; Song, T.; Yuan, Y.; et al. Intermedin in Paraventricular Nucleus Attenuates AngII-Induced Sympathoexcitation through the Inhibition of NADPH Oxidase-Dependent ROS Generation in Obese Rats with Hypertension. Int. J. Mol. Sci. 2019, 20, 4217. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A New Regulator of Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 7293861. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Bugger, H.; Kroemer, G.; Sedej, S. NAD + and Vascular Dysfunction: From Mechanisms to Therapeutic Opportunities. J. Lipid Atheroscler. 2022, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, H.; Chen, X.; Zou, Y.; Li, J.; Wang, L.; Wu, M.; Zang, J.; Yu, Y.; Zhuang, W.; et al. A Small Molecule Activator of SIRT3 Promotes Deacetylation and Activation of Manganese Superoxide Dismutase. Free Radic. Biol. Med. 2017, 112, 287–297. [Google Scholar] [CrossRef]
- Baldensperger, T.; Glomb, M.A. Pathways of Non-Enzymatic Lysine Acylation. Front. Cell Dev. Biol. 2021, 9, 664553. [Google Scholar] [CrossRef]
- He, W.; Huang, J.; Liu, Y.; Xie, C.; Zhang, K.; Zhu, X.; Chen, J.; Huang, H. Deletion of Soluble Epoxide Hydrolase Suppressed Chronic Kidney Disease-Related Vascular Calcification by Restoring Sirtuin 3 Expression. Cell Death Dis. 2021, 12, 992. [Google Scholar] [CrossRef]
- Byun, K.-A.; Oh, S.; Yang, J.Y.; Lee, S.Y.; Son, K.H.; Byun, K. Ecklonia Cava Extracts Decrease Hypertension-Related Vascular Calcification by Modulating PGC-1α and SOD2. Biomed. Pharmacother. 2022, 153, 113283. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-Derived ROS Activate AMP-Activated Protein Kinase (AMPK) Indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS Signaling under Metabolic Stress: Cross-Talk between AMPK and AKT Pathway. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol Blocks and Reverses Cardiac Hypertrophy in Mice by Activating Mitochondrial Sirt3. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD+ and Mitochondrial Sirtuins in Cardiac and Renal Diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zou, L.; Shi, D.; Liu, J.; Zhang, J.; Zhao, R.; Wang, G.; Zhang, L.; Ouyang, L.; Liu, B. Structure-Guided Design of a Small-Molecule Activator of Sirtuin-3 That Modulates Autophagy in Triple Negative Breast Cancer. J. Med. Chem. 2021, 64, 14192–14216. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Hay, D.L.; Quirion, R.; Poyner, D.R. The Pharmacology of Adrenomedullin 2/Intermedin. Br. J. Pharmacol. 2012, 166, 110–120. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-M.; Zhang, Y.-R.; Chen, Y.; Ji, D.-R.; Zhao, J.; Fu, S.; Jia, M.-Z.; Yu, Y.-R.; Tang, C.-S.; Huang, W.; et al. Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress. Pharmaceuticals 2022, 15, 1224. https://doi.org/10.3390/ph15101224
Liu S-M, Zhang Y-R, Chen Y, Ji D-R, Zhao J, Fu S, Jia M-Z, Yu Y-R, Tang C-S, Huang W, et al. Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress. Pharmaceuticals. 2022; 15(10):1224. https://doi.org/10.3390/ph15101224
Chicago/Turabian StyleLiu, Shi-Meng, Ya-Rong Zhang, Yao Chen, Deng-Ren Ji, Jie Zhao, Su Fu, Mo-Zhi Jia, Yan-Rong Yu, Chao-Shu Tang, Wei Huang, and et al. 2022. "Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress" Pharmaceuticals 15, no. 10: 1224. https://doi.org/10.3390/ph15101224
APA StyleLiu, S.-M., Zhang, Y.-R., Chen, Y., Ji, D.-R., Zhao, J., Fu, S., Jia, M.-Z., Yu, Y.-R., Tang, C.-S., Huang, W., Zhou, Y.-B., & Qi, Y.-F. (2022). Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress. Pharmaceuticals, 15(10), 1224. https://doi.org/10.3390/ph15101224