
Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors

 , ,
, ,  and
and 
Abstract
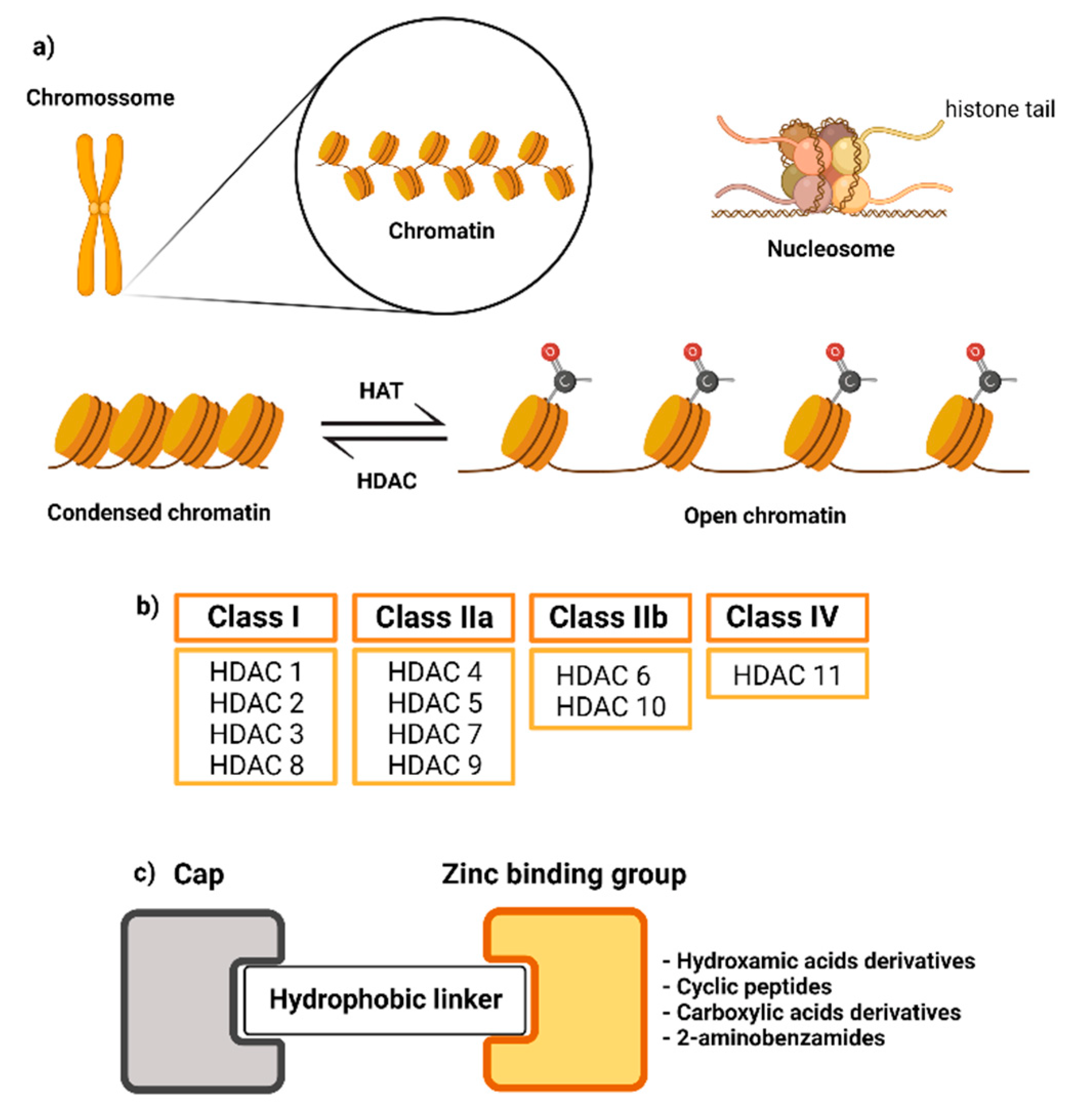
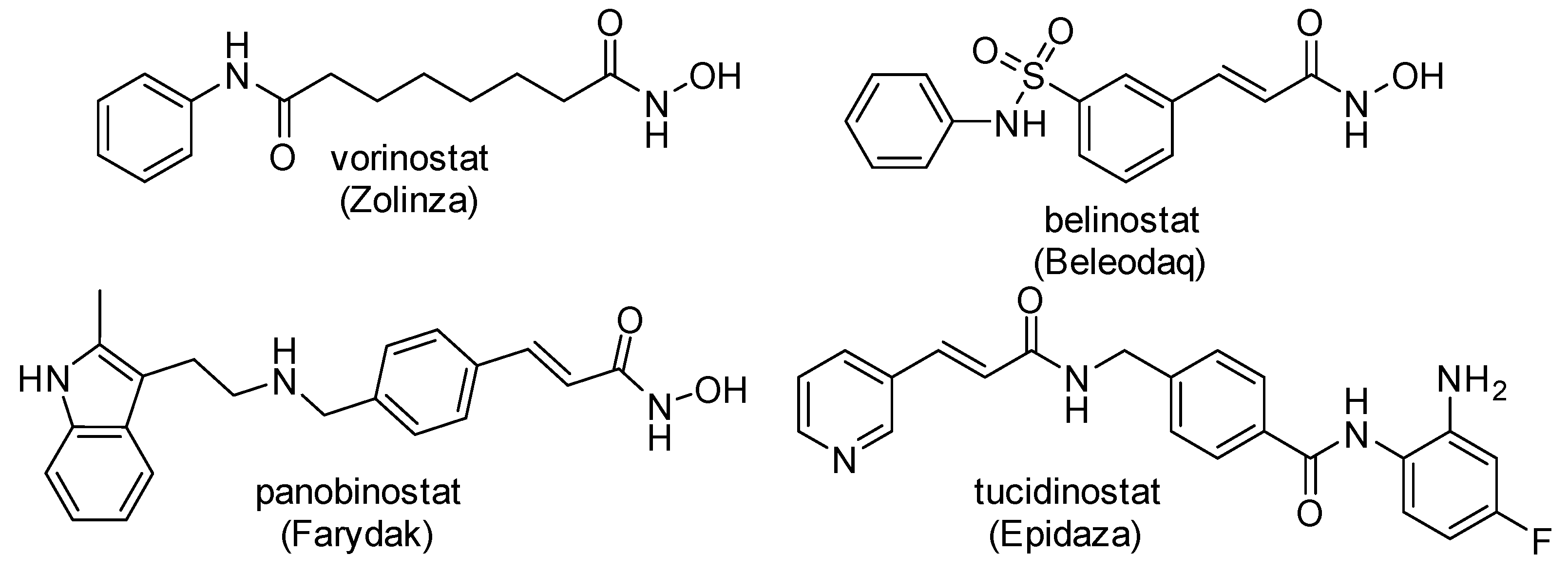
:1. Introduction
2. Results and Discussion
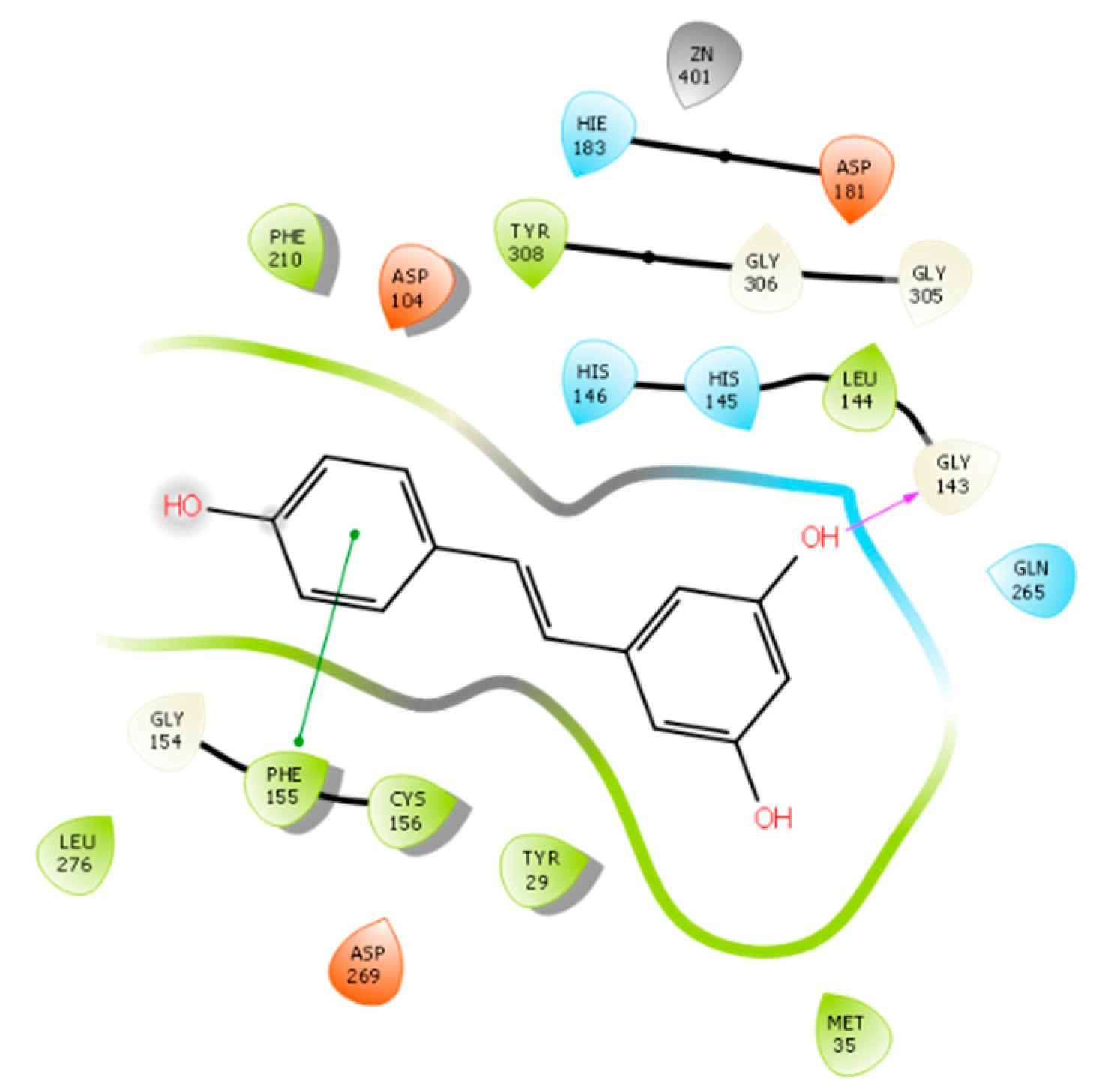
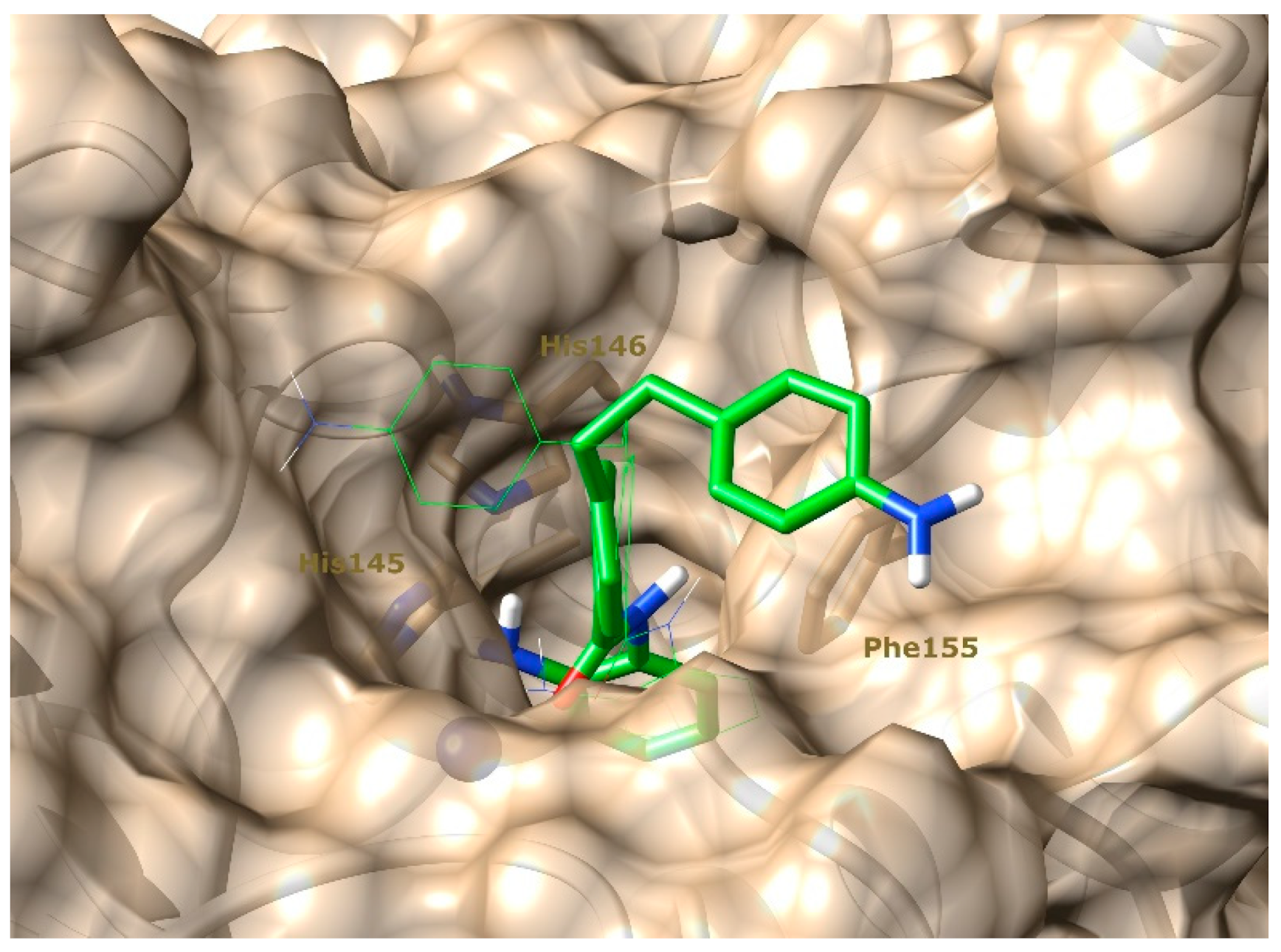
2.1. Molecular Docking Studies and In Silico Prediction of ADME Properties
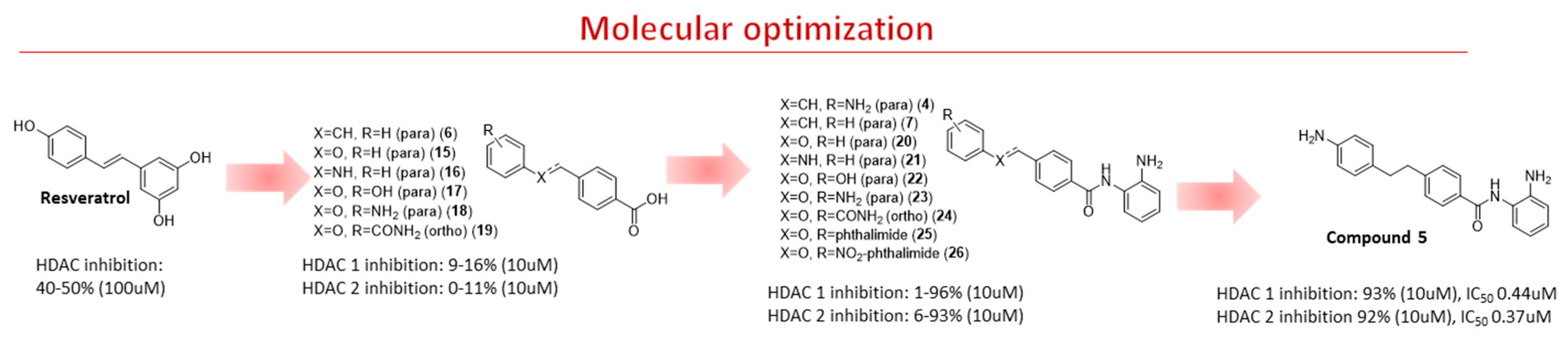
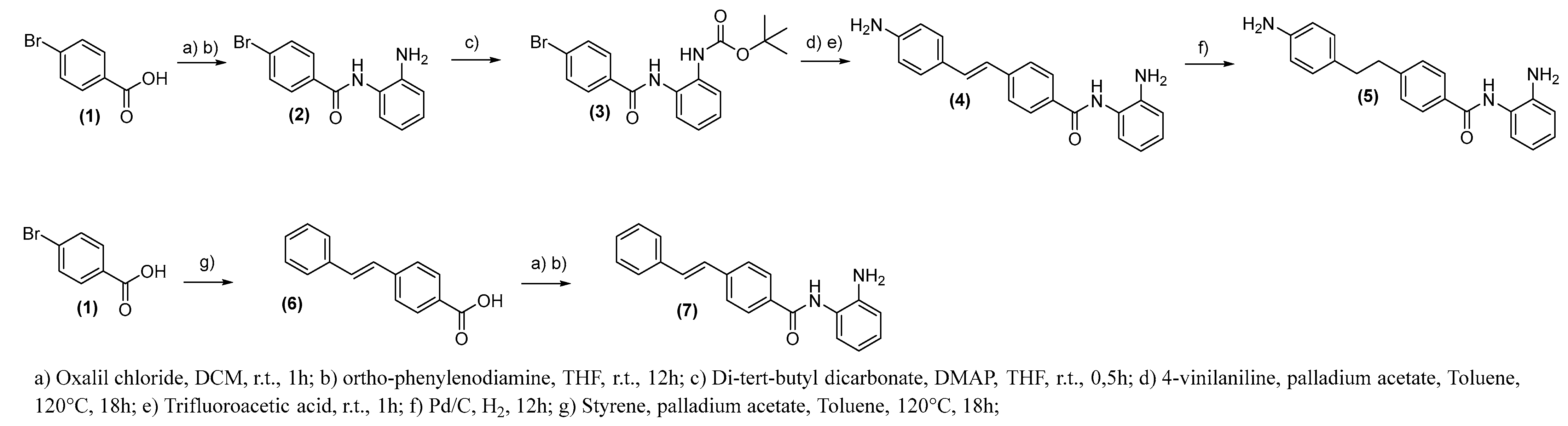
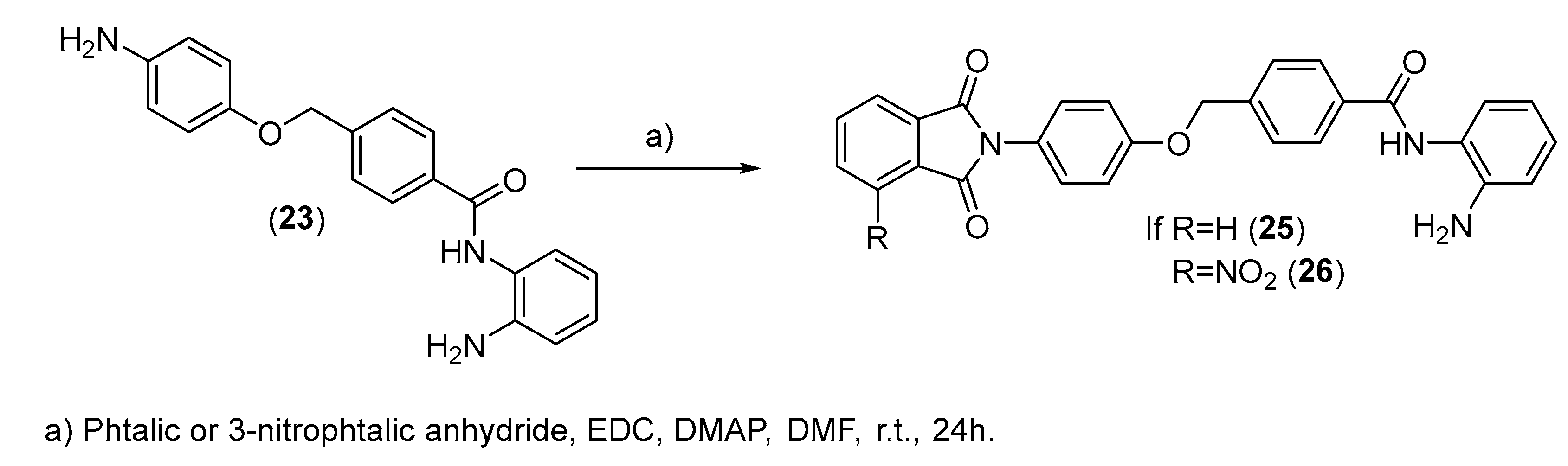
2.2. Chemistry
2.3. Enzymatic Evaluation
3. Materials and Methods
3.1. Computational Methods
3.1.1. Molecular Docking Studies
3.1.2. Molecular Dynamics Simulations
3.1.3. In Silico Prediction of ADME Properties
3.2. Chemistry
3.2.1. General Chemical Aspects
3.2.2. General Procedure for the Synthesis of Compound (2)
3.2.3. General Procedure for the Synthesis of Compound (4)
3.2.4. General Procedure for the Synthesis of Compound (5)
3.2.5. General Procedure for the Synthesis of Compounds (6)
3.2.6. General Procedure for the Synthesis of Compound (7)
3.2.7. General Procedure for the Synthesis of Compounds (10)–(13)
3.2.8. General Procedures for the Synthesis of Compounds (15)–(19)
3.2.9. General Procedure for the Synthesis of Compounds (20)–(24)
3.2.10. General Procedures for the Synthesis of Compounds (25) and (26)
3.3. Enzymatic Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficner, R. Novel Structural Insights into Class I and II Histone Deacetylases. Curr. Top. Med. Chem. 2009, 9, 235–240. [Google Scholar] [CrossRef]
- Pande, V. Understanding the Complexity of Epigenetic Target Space. J. Med. Chem. 2016, 59, 1299–1307. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical Phylogenetics of Histone Deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Smolewski, P.; Robak, T. The discovery and development of romidepsin for the treatment of T-cell lymphoma. Expert Opin. Drug Discov. 2017, 12, 859–873. [Google Scholar] [CrossRef]
- Campbell, P.; Thomas, C.M. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J. Oncol. Pharm. Pract. 2017, 23, 143–147. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Bondarev, A.; Attwood, M.; Jonsson, J.; Chubarev, V.; Tarasov, W.; Schiöth, H. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Liao, G.; Sun, P.; Yu, Z.; Chen, J. An Overview of HDAC Inhibitors and their Synthetic Routes. Curr. Topics Med. Chem. 2019, 19, 1005–1040. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Di Micco, S.; Chini, M.G.; Terracciano, S.; Bruno, I.; Riccio, R.; Bifulco, G. Structural basis for the design and synthesis of selective HDAC inhibitors. Bioorganic Med. Chem. 2013, 21, 3795–3807. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, H. Structure-based inhibitor discovery of class i histone deacetylases (HDACS). Int. J. Mol. Sci. 2020, 21, 8828. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavan, A.R.; Silva, G.D.; Jornada, D.H.; Chiba, D.H.; Fernandes, G.F.; Man Chin, C.; Dos Santos, J.L. Unraveling the Anticancer Effect of Curcumin and Resveratrol. Nutrients 2016, 8, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, B.I.; Ruiz-Ramos, M.; Pedraza-Chaverri, J.; Santiago-Osorio, E.; Mendoza-Núñez, V.M. Hypoglycemic effect of resveratrol: A systematic review and meta-analysis. Antioxidants 2021, 10, 69. [Google Scholar] [CrossRef]
- Islam, F.; Nafady, M.H.; Islam, M.R.; Saha, S.; Rashid, S.; Akter, A.; Harun-Or-Rashid, M.; Akhtar, M.F.; Perveen, A.; Ashraf, G.M.; et al. Resveratrol and neuroprotection: An insight into prospective therapeutic approaches against Alzheimer’s disease from bench to bedside. Mol. Neurobiol. 2022, 59, 4384–4404. [Google Scholar] [CrossRef]
- Venturelli, S.; Berger, A.; Böcker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a Pan-HDAC Inhibitor Alters the Acetylation Status of Jistone Proteins in Human-Derived Hepatoblastoma Cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, G.F.S.; Silva, G.D.B.; Pavan, A.R.; Chiba, D.E.; Chin, C.M.; Dos Santos, J.L. Epigenetic regulatory mechanisms induced by resveratrol. Nutrients 2017, 9, 1201. [Google Scholar] [CrossRef] [Green Version]
- Ingólfsson, H.I.; Thakur, P.; Herold, K.F.; Hobart, E.A.; Ramsey, N.B.; Periole, X.; de Jong, D.H.; Zwama, M.; Yilmaz, D.; Hall, K.; et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 2014, 9, 1788–1798. [Google Scholar] [CrossRef]
- Bosquesi, P.L.; Melchior, A.C.B.; Pavan, A.R.; Lanaro, C.; de Souza, C.M.; Rusinova, R.; Chelucci, R.C.; Barbieri, K.P.; Carlos, I.Z.; Andersen, O.S.; et al. Synthesis and evaluation of resveratrol derivatives as fetal hemoglobin inducers. Bioorganic Chem. 2020, 100, 103948. [Google Scholar] [CrossRef]
- Albert, S.; Horbach, R.; Deising, H.B.; Siewert, B.; Csuk, R. Synthesis and antimicrobial activity of (E) stilbene derivatives. Bioorganic Med. Chem. 2011, 19, 5155–5166. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Xeilari, M.; Yiotakis, A.; Grembecka, J.; Pawełczak, M.; Kafarski, P.; Mucha, A. A synthetic method for diversification of the P1′ substituent in phosphinic dipeptides as a tool for exploration of the specificity of the S1′ binding pockets of leucine aminopeptidases. Bioorganic Med. Chem. 2007, 15, 3187–3200. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Yao, Y.; Tang, P.; Chen, G.; Liu, X.; Yun, F.; Cheng, C.; Wu, X.; Yuan, Q. Design, synthesis and biological evaluation of novel hydroxamates and 2-aminobenzamides as potent histone deacetylase inhibitors and antitumor agents. Eur. J. Med. Chem. 2017, 134, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, F.; Zwinderman, M.R.H.; van Merkerk, R.; Ettema, P.E.; Quax, W.J.; Dekker, F.J. Inhibitory selectivity among class I HDACs has a major impact on inflammatory gene expression in macrophages. Eur. J. Med. Chem. 2019, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein—Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [Green Version]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [Green Version]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Lopes, J.R.; Prokopczyk, I.M.; Gerlack, M.; Chin, C.M.; Dos Santos, J.L. Design and synthesis of hybrid compounds as epigenetic modifiers. Pharmaceuticals 2021, 14, 1308. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Docking Score (DS) Values (Kcal/mol) | |
|---|---|---|
| HDAC-1 | HDAC-2 | |
| Resveratrol (RVT) | −5.565 | −6.453 |
| Tucidinostat | −7.606 | −11.436 |
| (4) | −4.462 | −10.967 |
| (5) | −8.724 | −11.707 |
| (7) | −5.331 | −10.997 |
| (20) | −8.542 | −11.399 |
| (21) | −8.760 | −11.621 |
| (22) | −5.068 | −11.839 |
| (23) | −7.268 | −12.234 |
| (24) | −10.763 | −12.780 |
| (25) | −8.764 | −11.128 |
| (26) | −8.437 | −10.971 |
| Compound (10 µM) | HDAC-1 (%) | HDAC- 2 (%) |
|---|---|---|
| (4) | 45 ± 1.2 | 28 ± 1.0 |
| (5) | 93 ± 1.0 | 92 ± 0.8 |
| (6) | 9 ± 1.0 | 0 |
| (7) | 42 ± 0.7 | 32 ± 0.7 |
| (15) | 16 ± 0.6 | 10 ± 0.5 |
| (17) | 12 ± 0.7 | 11 ± 0.5 |
| (19) | 13 ± 0.9 | 8 ± 0.6 |
| (20) | 87 ± 1.1 | 85 ± 1.1 |
| (21) | 96 ± 1.3 | 93 ± 1.2 |
| (22) | 96 ± 1.2 | 92 ± 0.7 |
| (23) | 72 ± 1.1 | 52 ± 0.8 |
| (24) | 86 ± 1.0 | 81 ± 1.1 |
| (25) | 14 ± 0.5 | 21 ± 0.8 |
| (26) | 1 ± 0.5 | 6 ± 0.6 |
| Compound | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|
| (5) | 82 ± 1.2 | 5 ± 0.4 | 5 ± 0.5 | 3 ± 0.4 | 15 ± 0.7 | 13 ± 1.0 | 7 ± 0.4 | 60 ± 1.4 | 15 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urias, B.S.; Pavan, A.R.; Albuquerque, G.R.; Prokopczyk, I.M.; Alves, T.M.F.; de Melo, T.R.F.; Sartori, G.R.; da Silva, J.H.M.; Chin, C.M.; Santos, J.L.D. Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors. Pharmaceuticals 2022, 15, 1260. https://doi.org/10.3390/ph15101260
Urias BS, Pavan AR, Albuquerque GR, Prokopczyk IM, Alves TMF, de Melo TRF, Sartori GR, da Silva JHM, Chin CM, Santos JLD. Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors. Pharmaceuticals. 2022; 15(10):1260. https://doi.org/10.3390/ph15101260
Chicago/Turabian StyleUrias, Beatriz Silva, Aline Renata Pavan, Gabriela Ribeiro Albuquerque, Igor Muccilo Prokopczyk, Tânia Mara Ferreira Alves, Thais Regina Ferreira de Melo, Geraldo Rodrigues Sartori, João Hermínio Martins da Silva, Chung Man Chin, and Jean Leandro Dos Santos. 2022. "Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors" Pharmaceuticals 15, no. 10: 1260. https://doi.org/10.3390/ph15101260
APA StyleUrias, B. S., Pavan, A. R., Albuquerque, G. R., Prokopczyk, I. M., Alves, T. M. F., de Melo, T. R. F., Sartori, G. R., da Silva, J. H. M., Chin, C. M., & Santos, J. L. D. (2022). Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors. Pharmaceuticals, 15(10), 1260. https://doi.org/10.3390/ph15101260