
Identification of Novel Cyanopyridones and Pyrido[2,3-d]Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
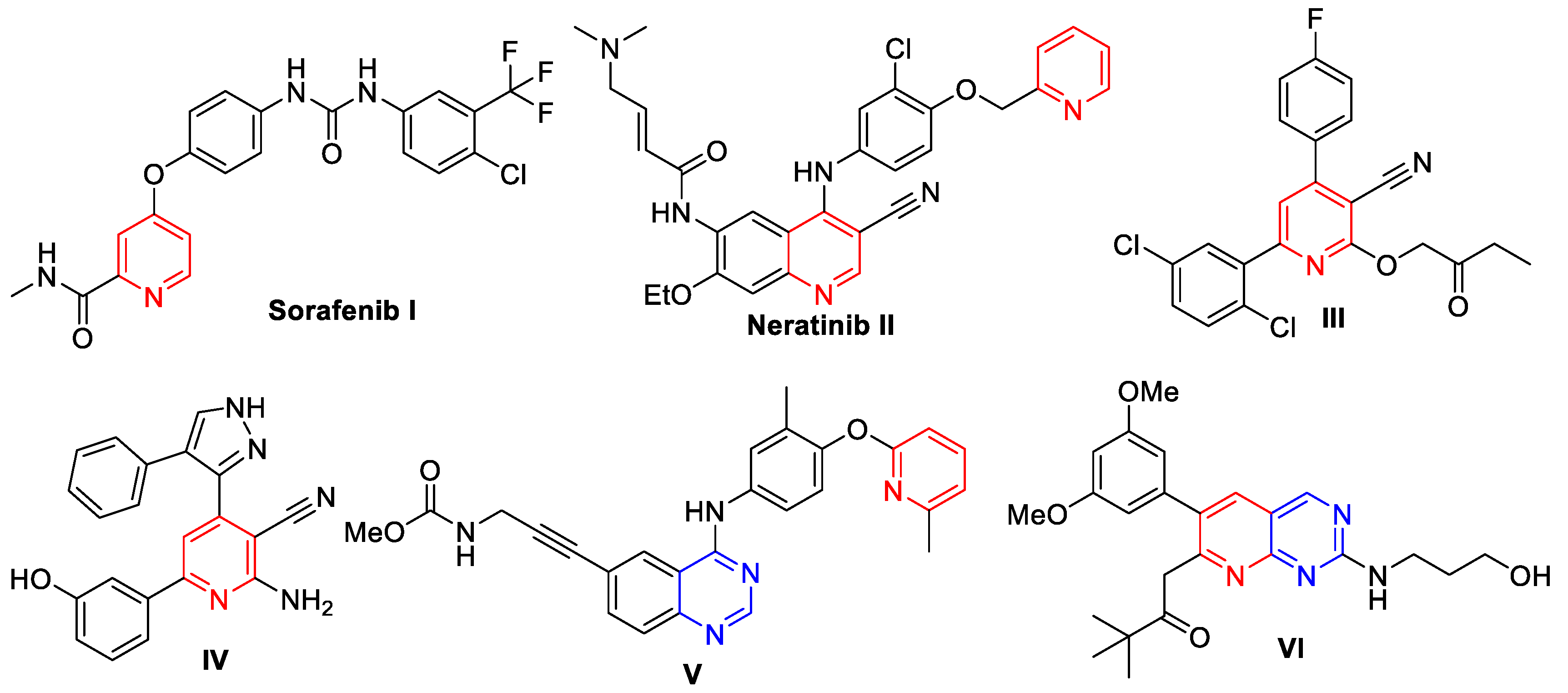
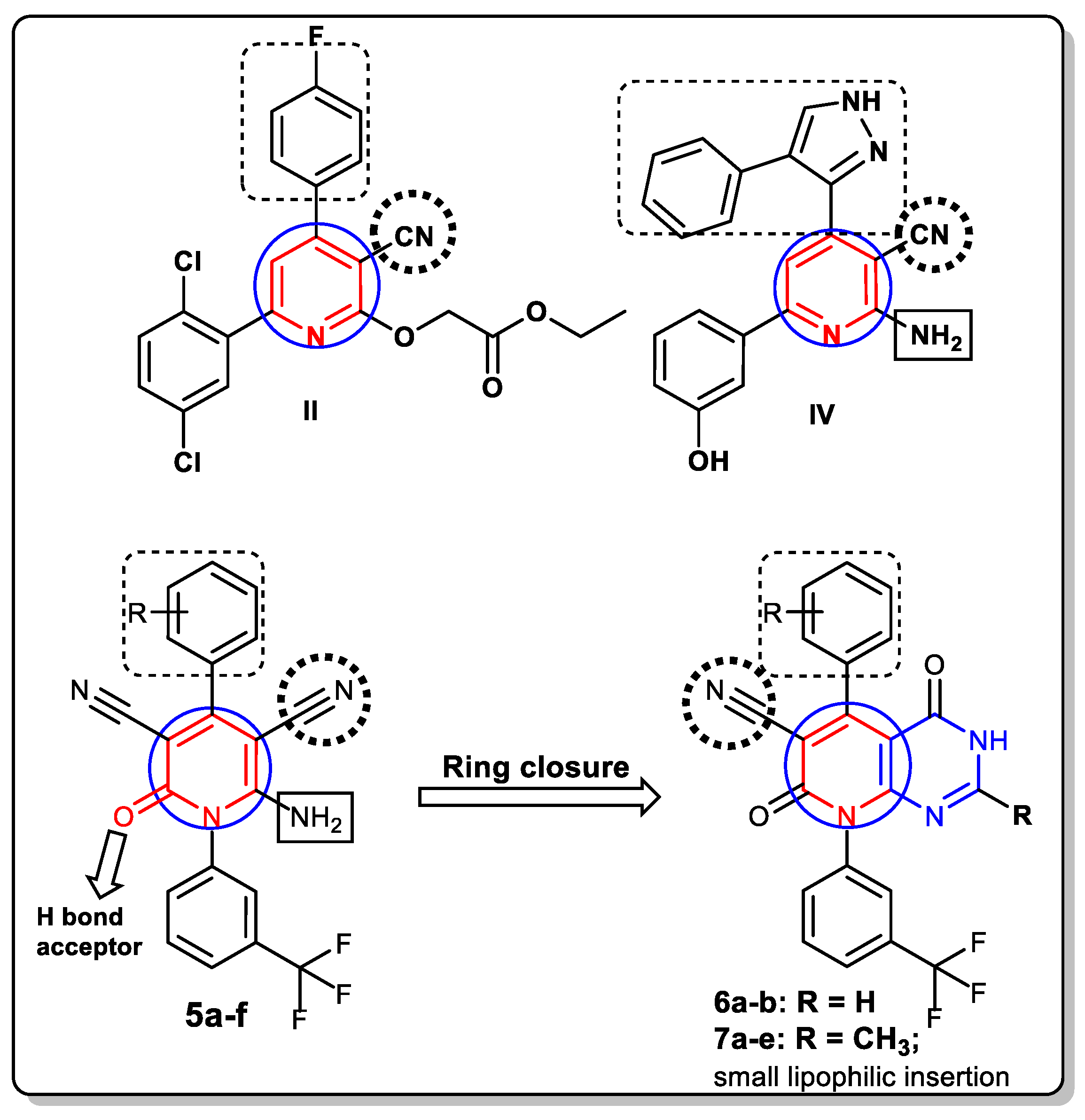
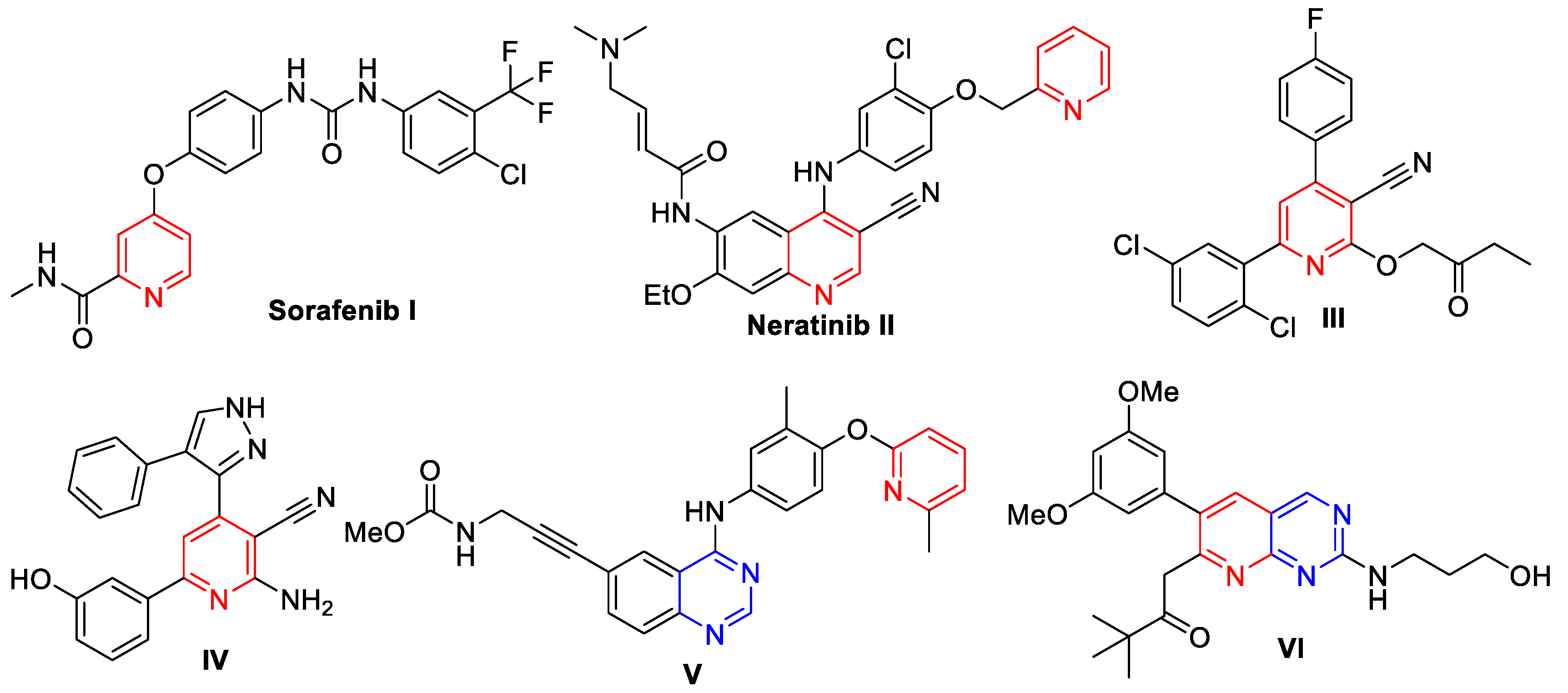
2. Rationale and Design
3. Results and Discussion
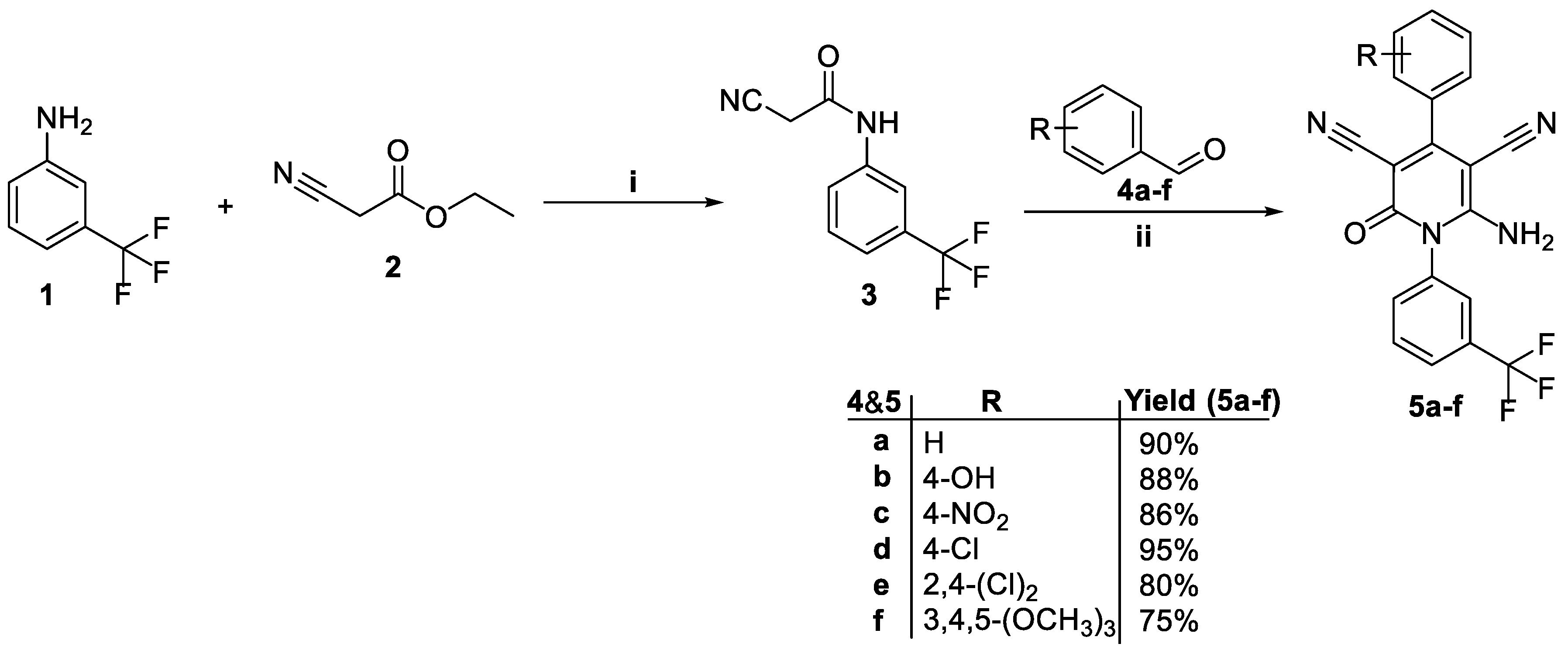
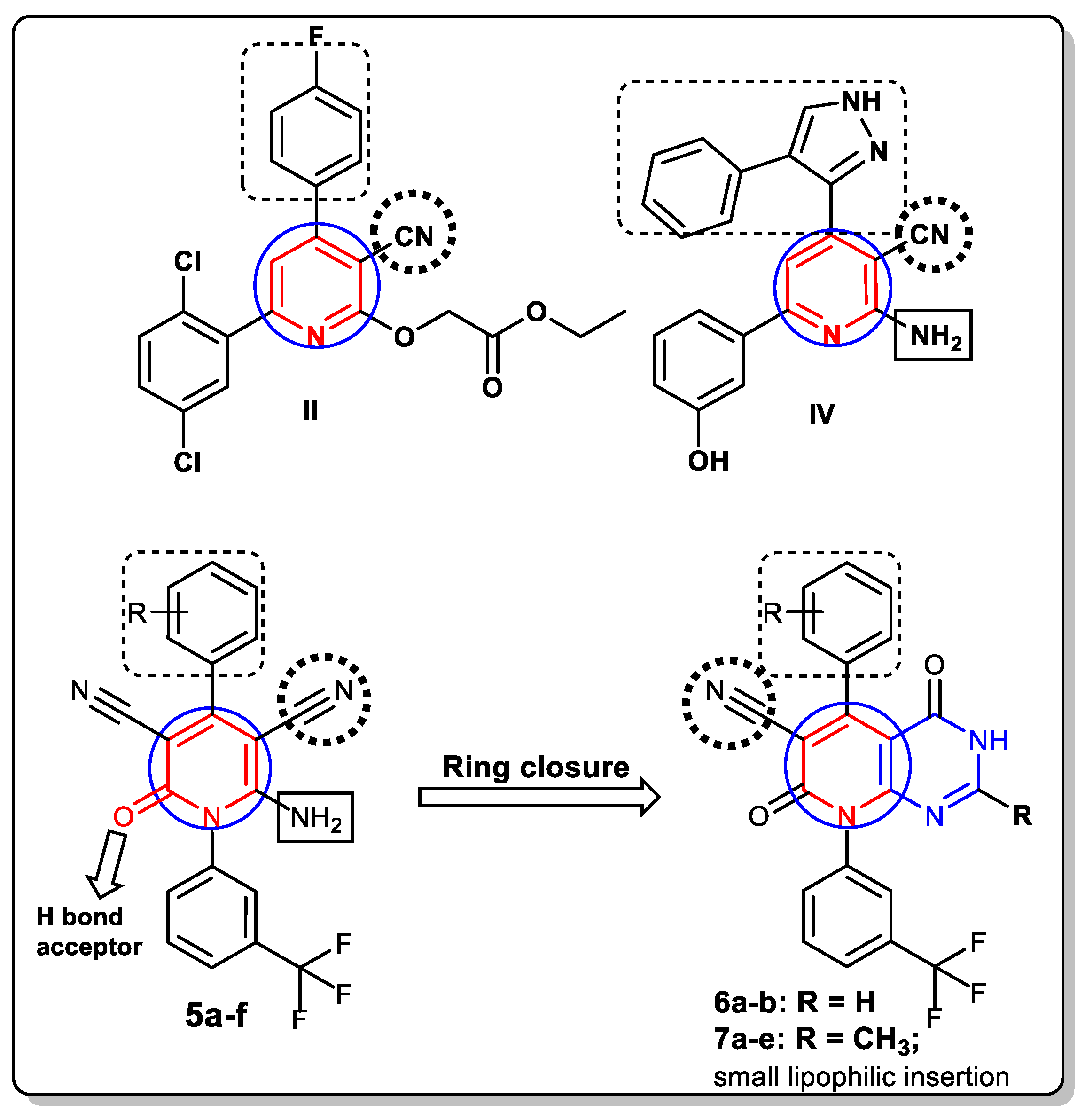
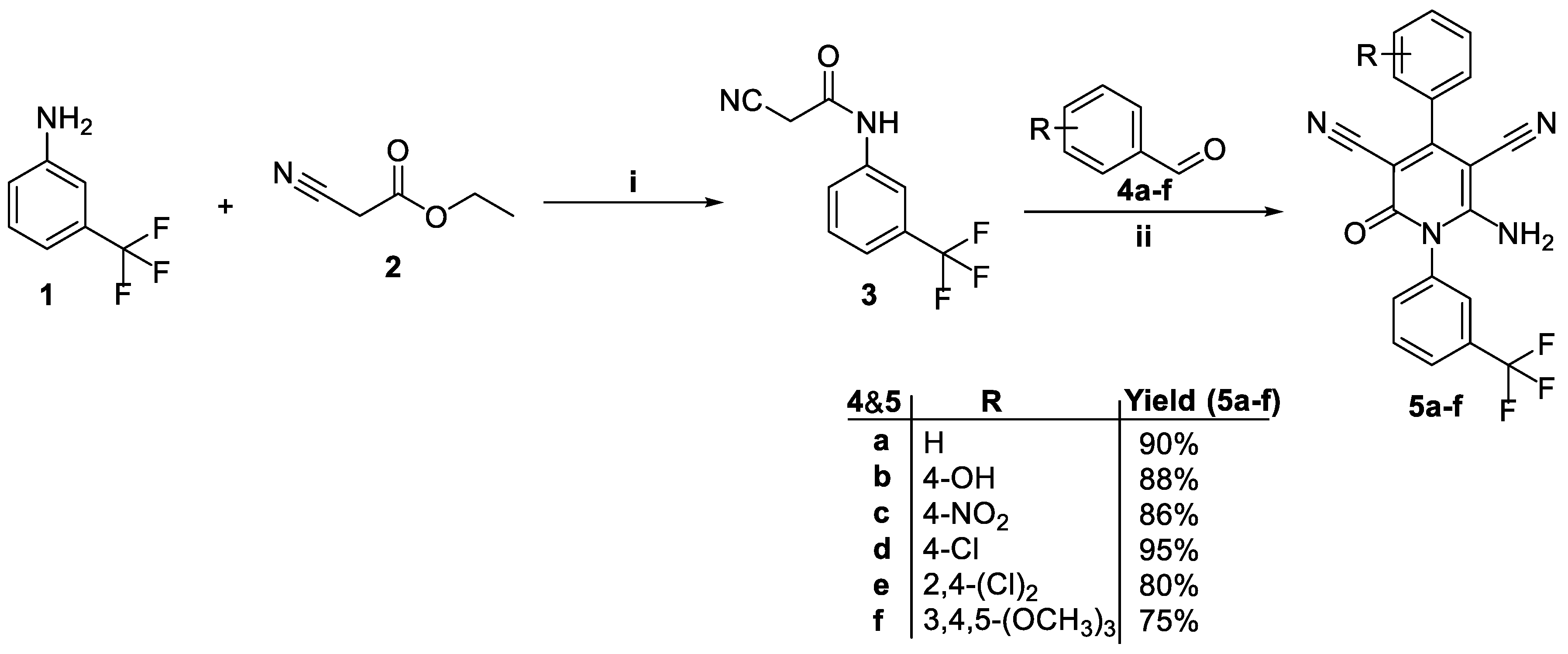
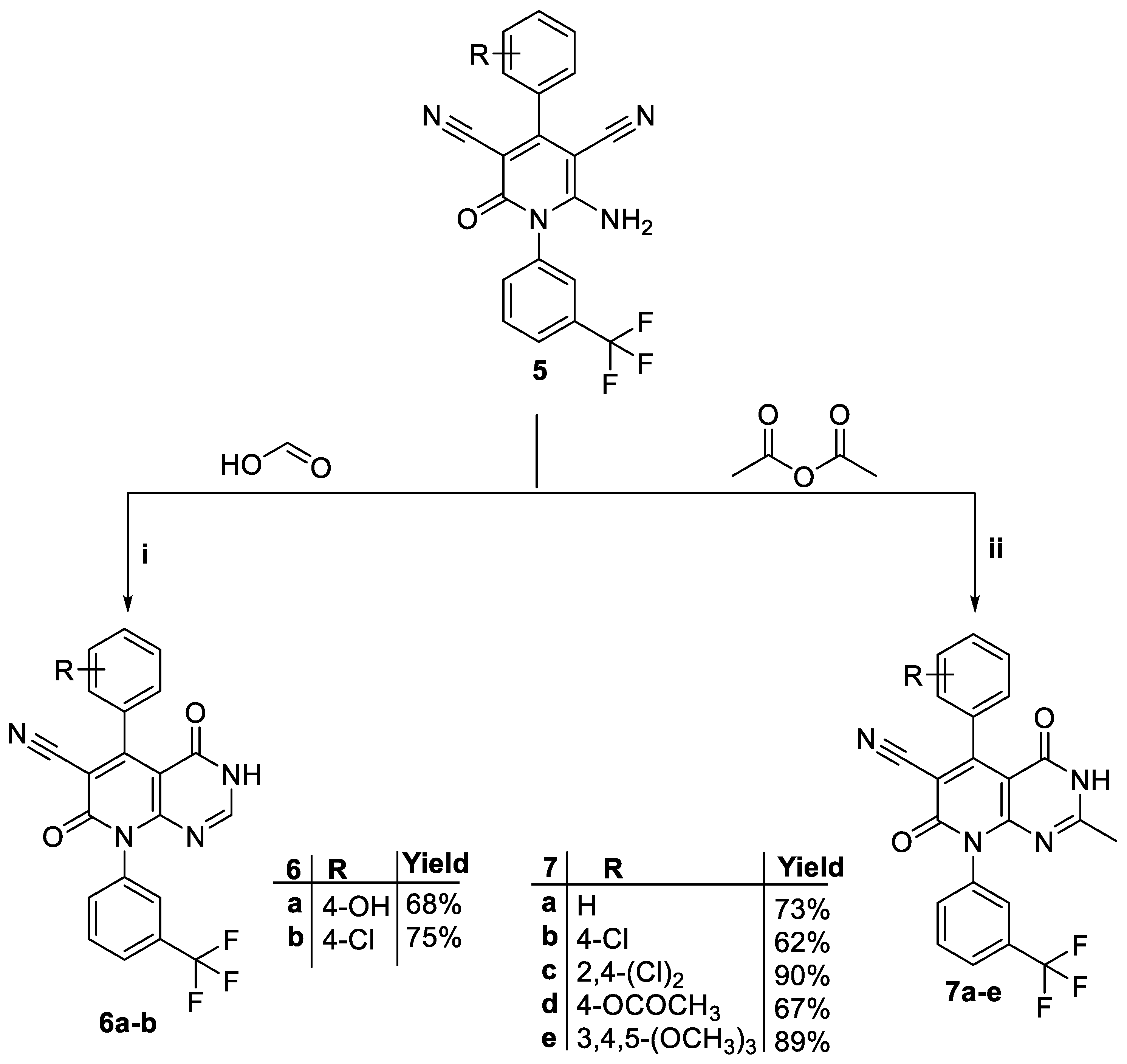
3.1. Chemistry
3.2. Biological Evaluations
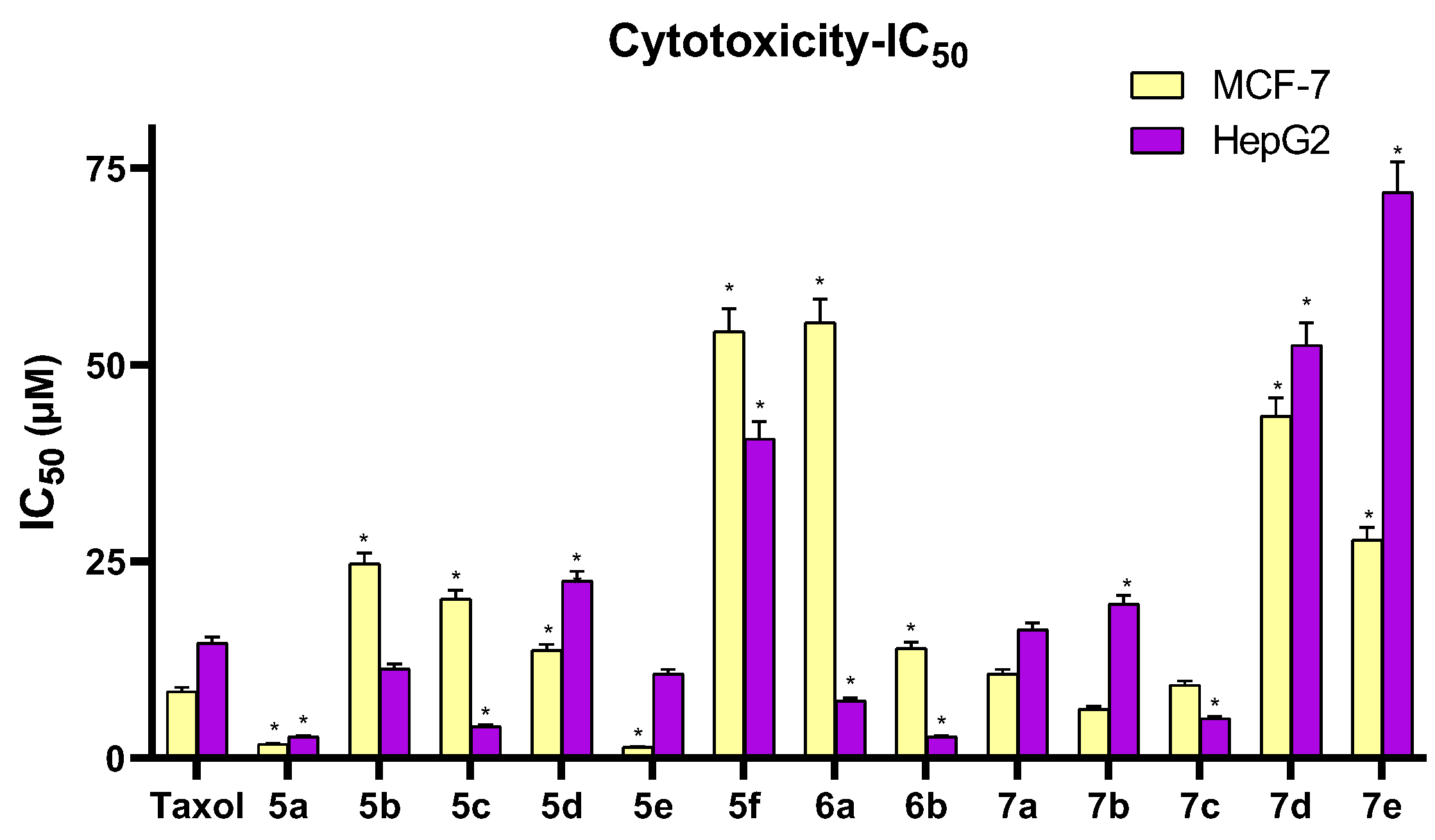
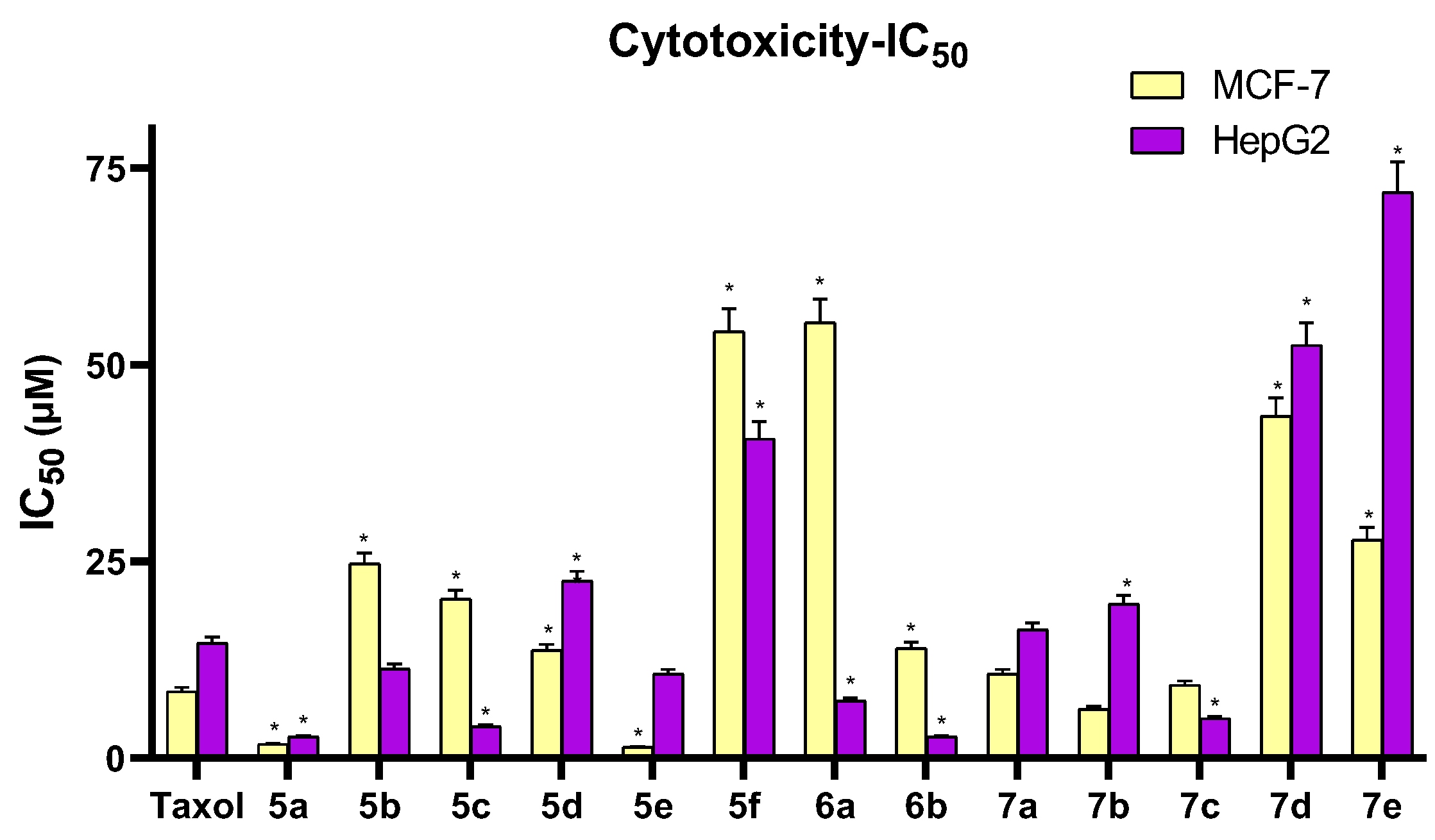
3.2.1. Cytotoxicity Assay
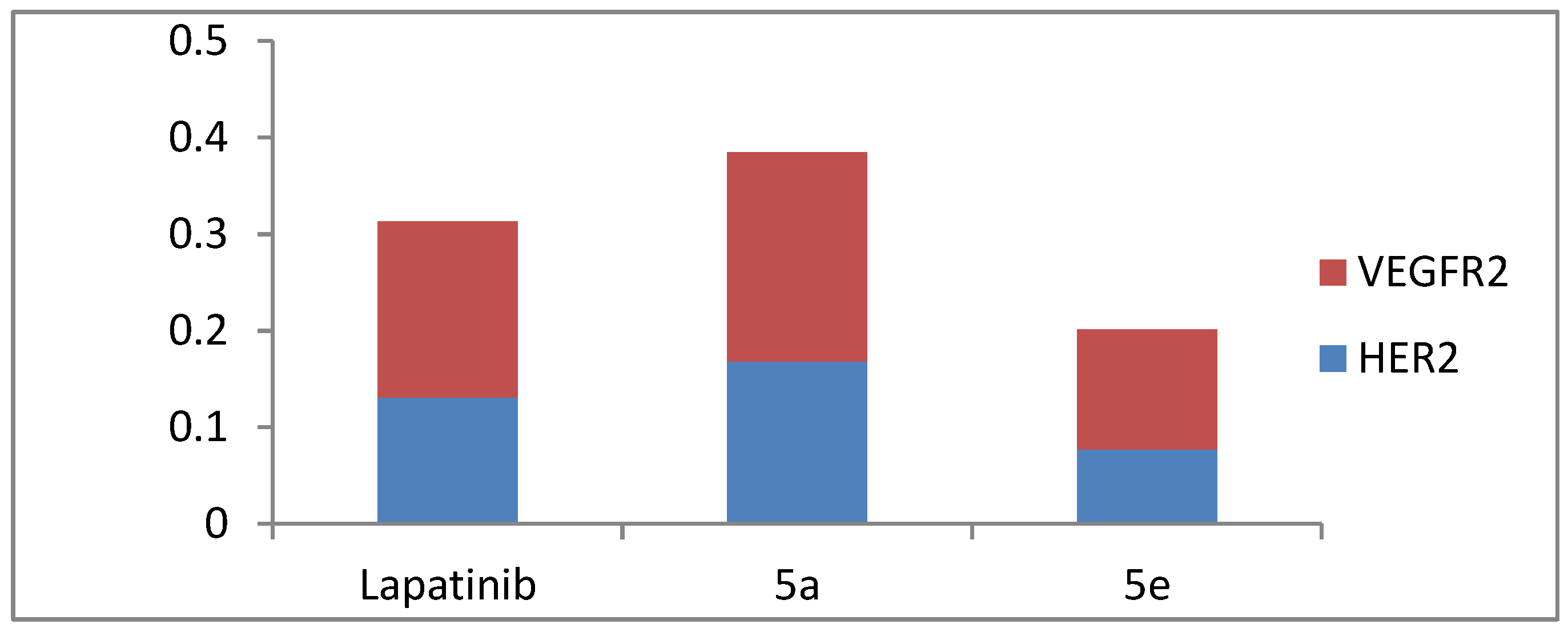
3.2.2. VEGFR-2 and HER-2 Kinase Assay
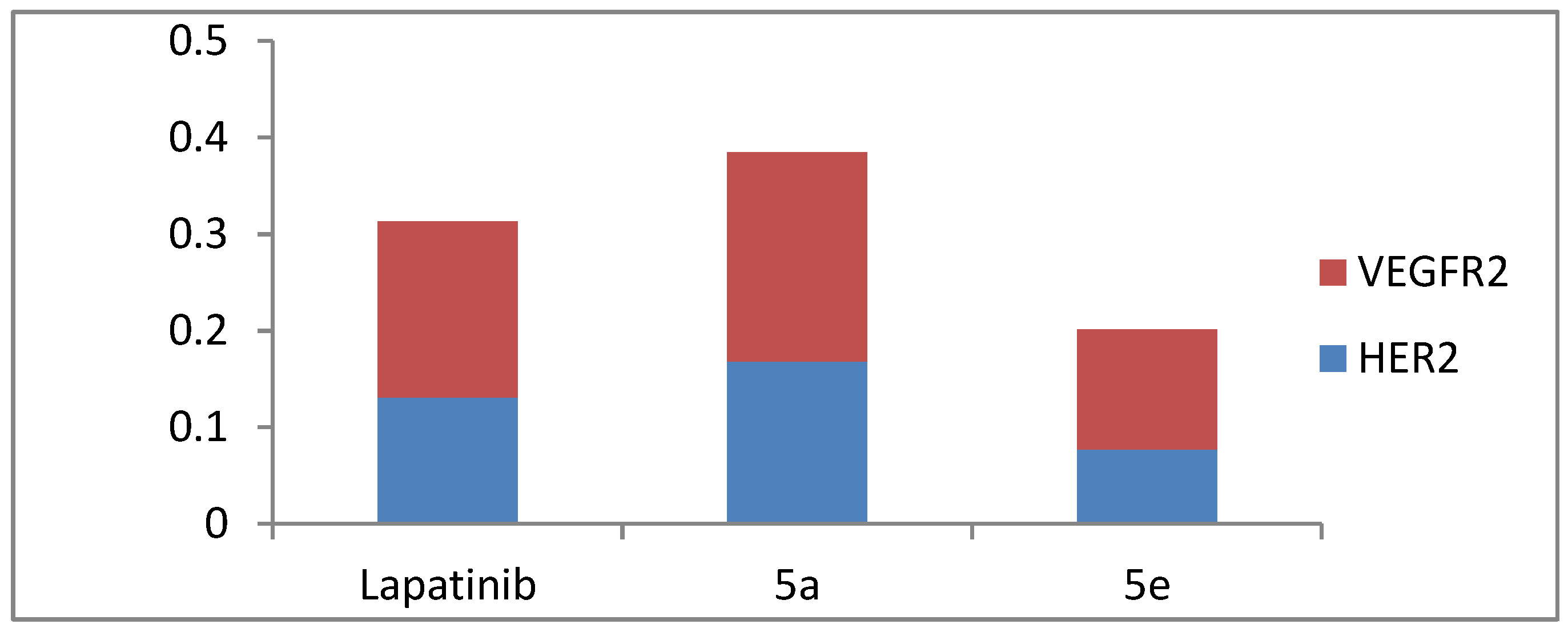
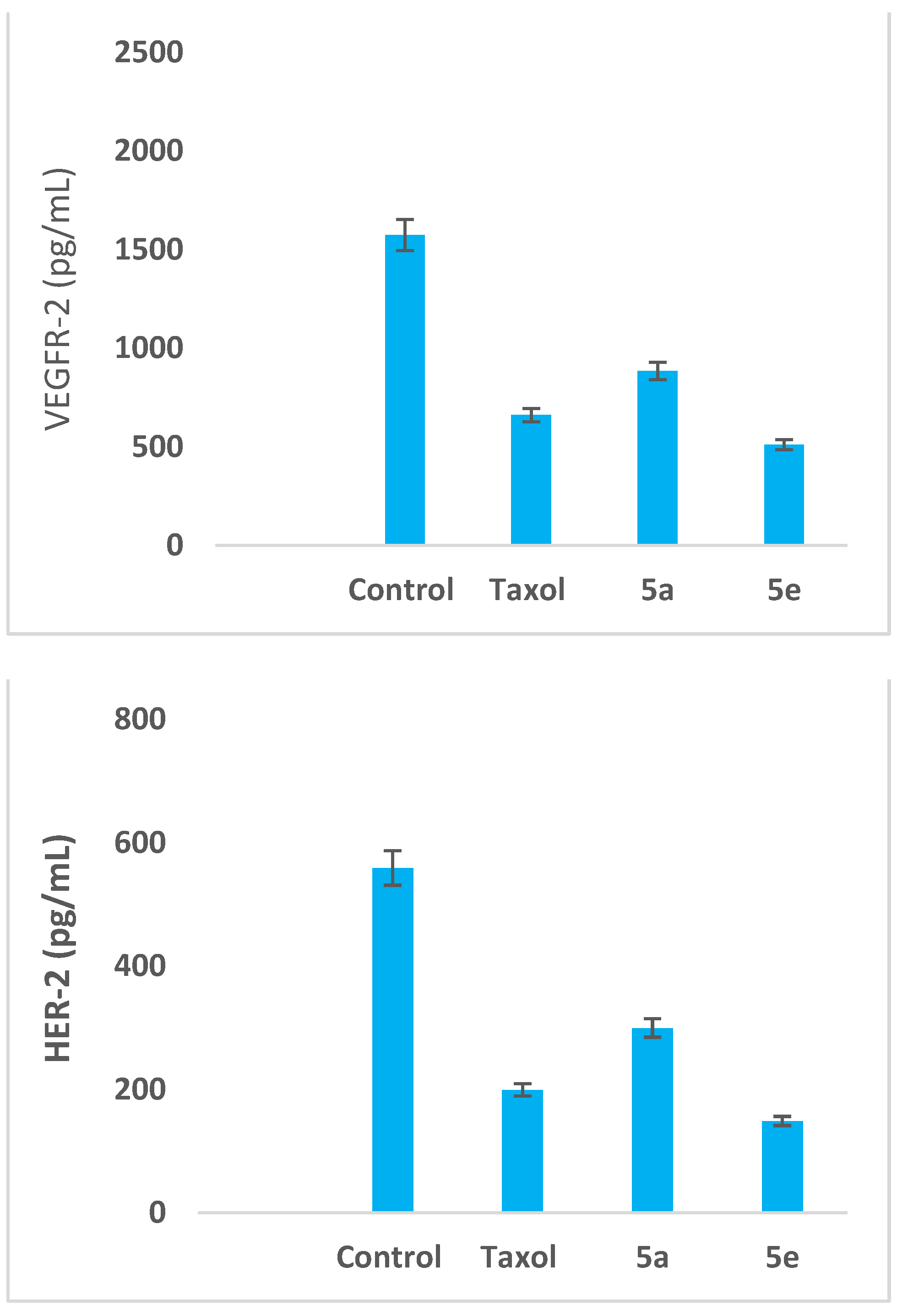
3.2.3. Impact on the Expression Levels of VEGFR-2 and HER-2 in MCF-7 Cells
3.3. Molecular Docking Studies
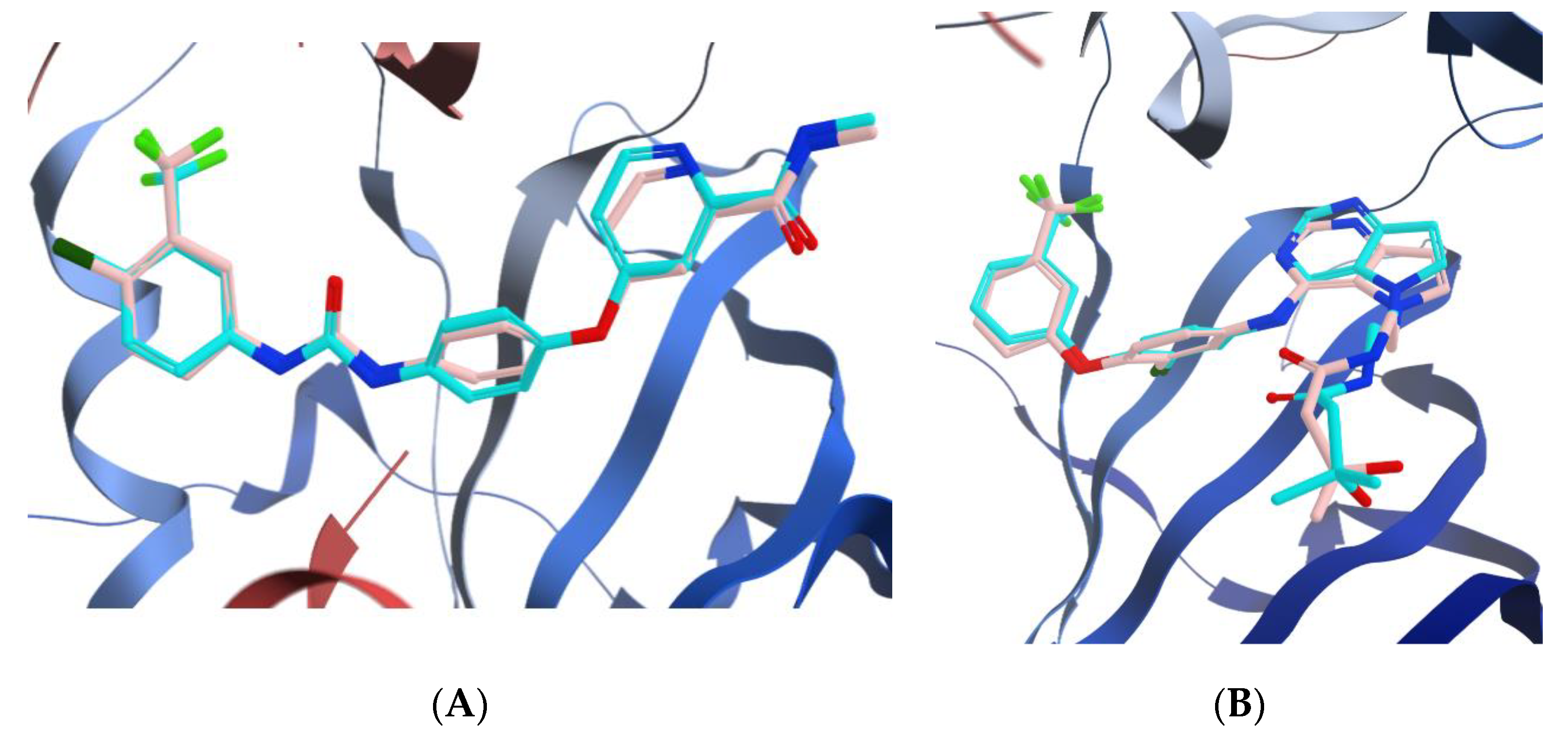
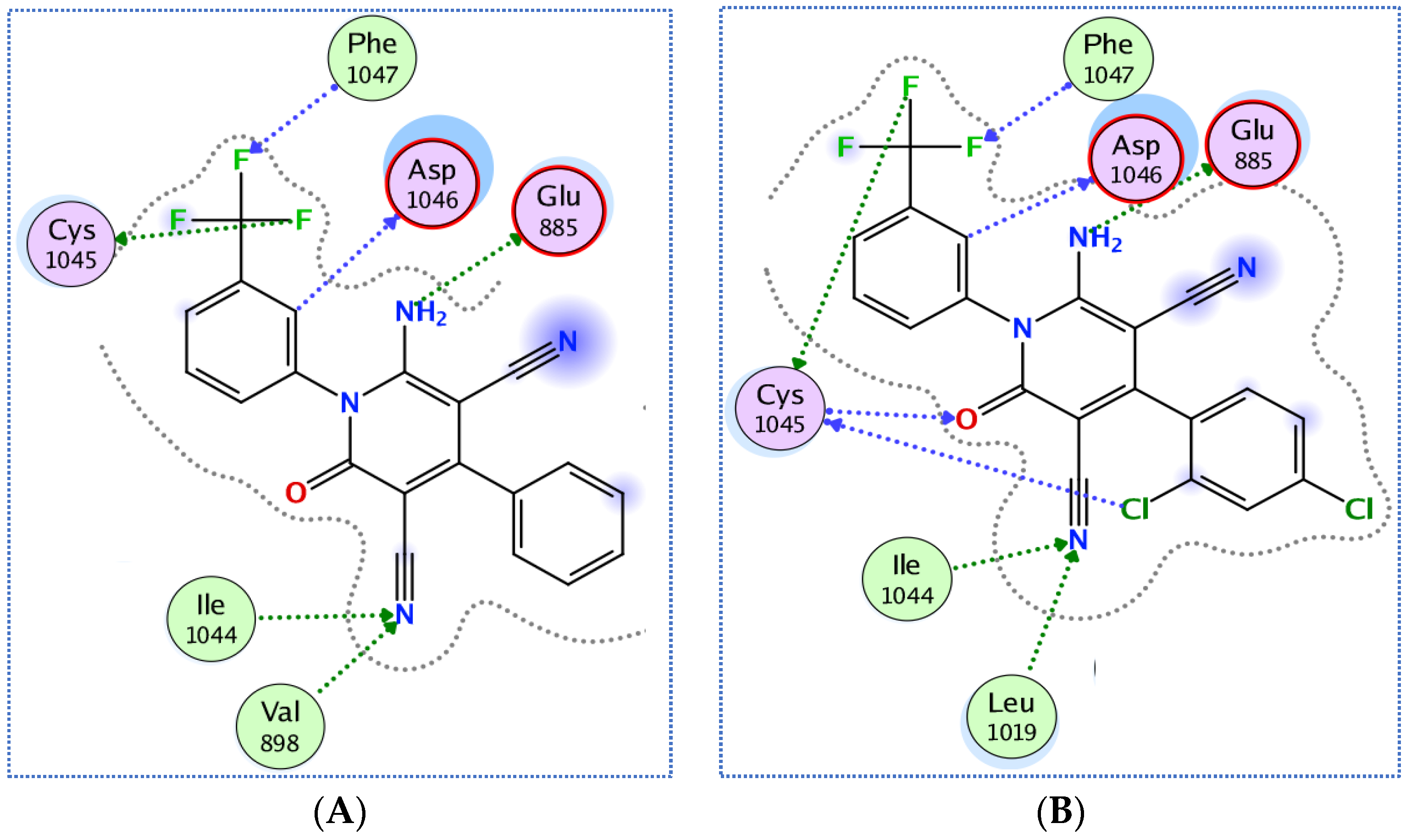
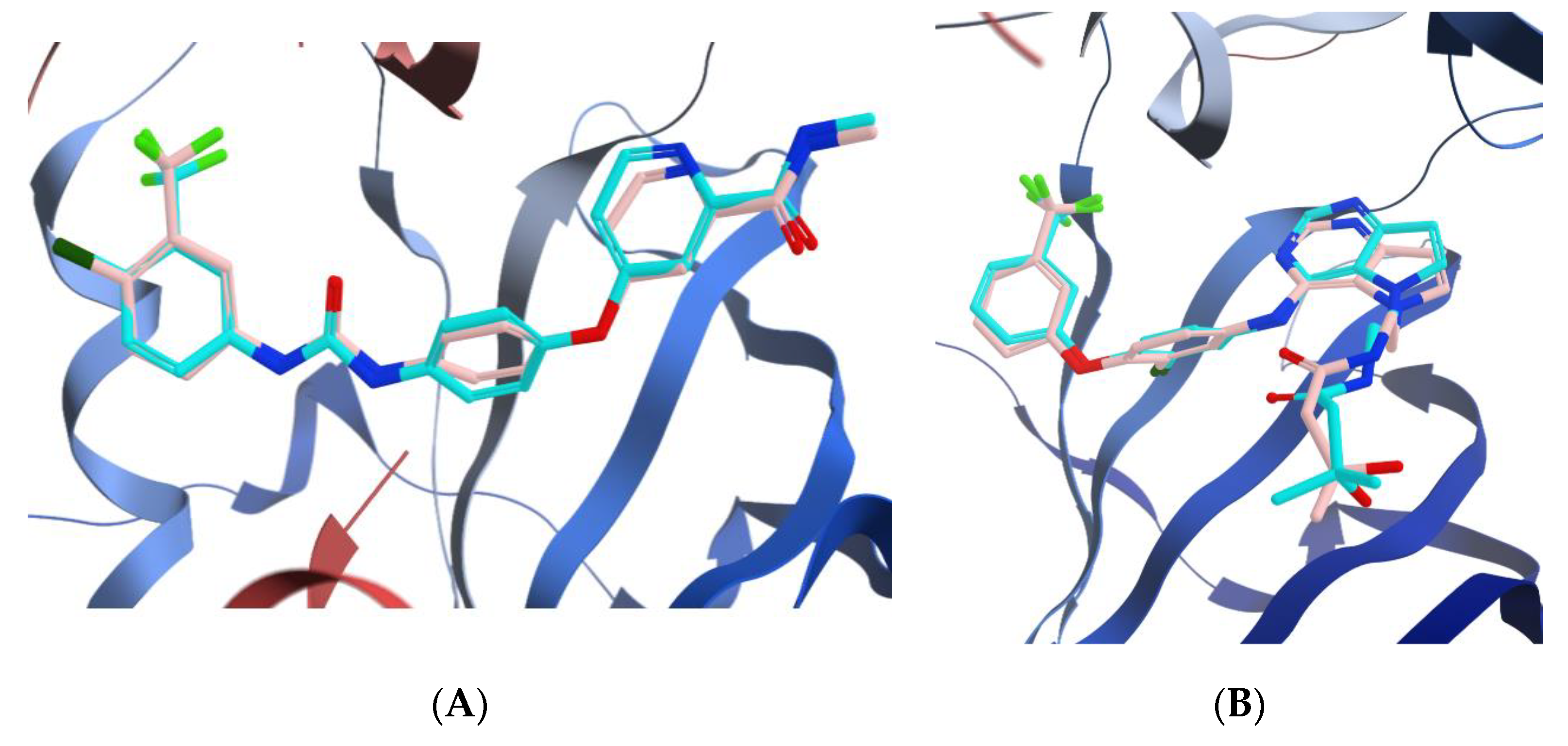
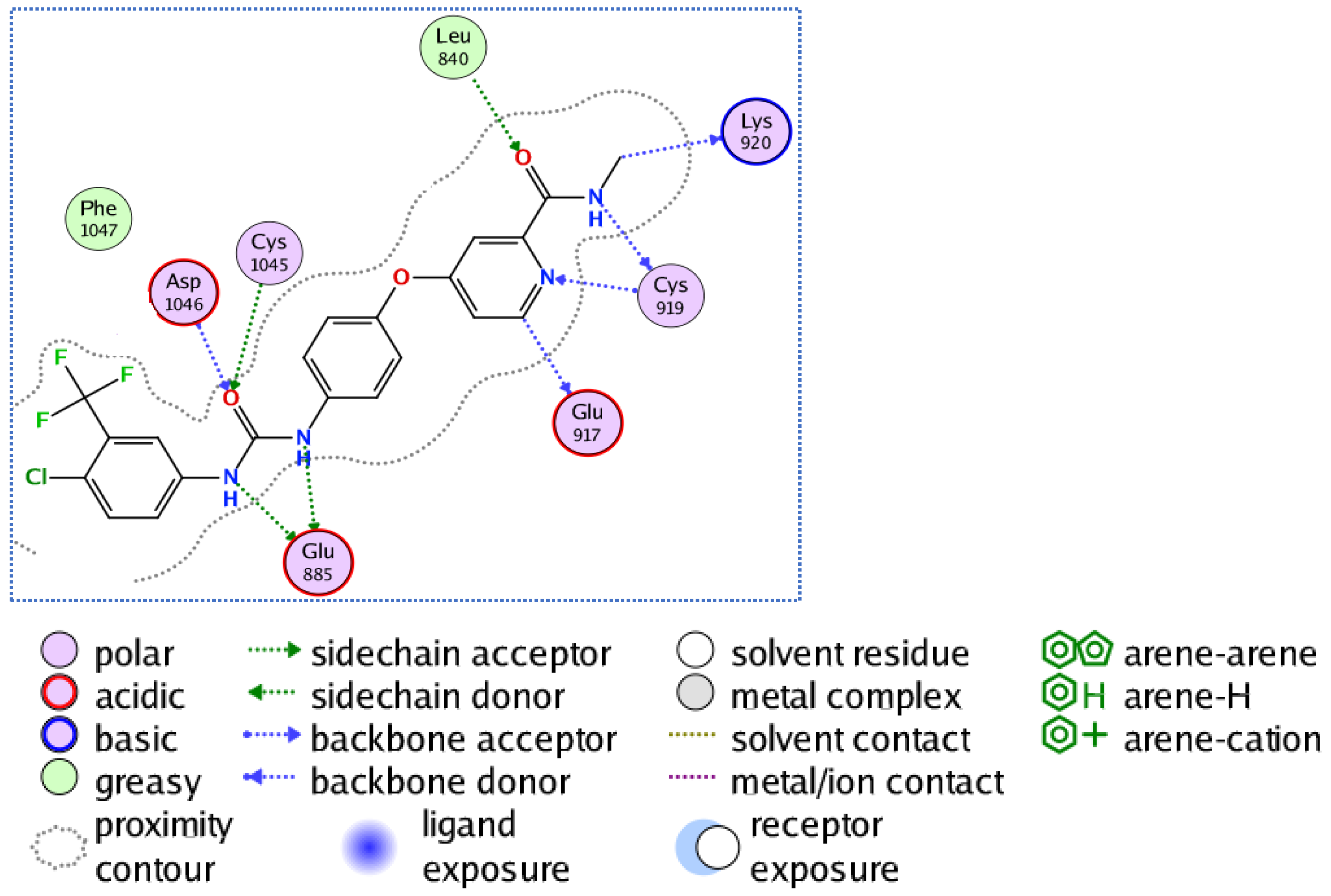
3.3.1. Docking of Compounds 5a and 5e into VEGFR-2 Active Site
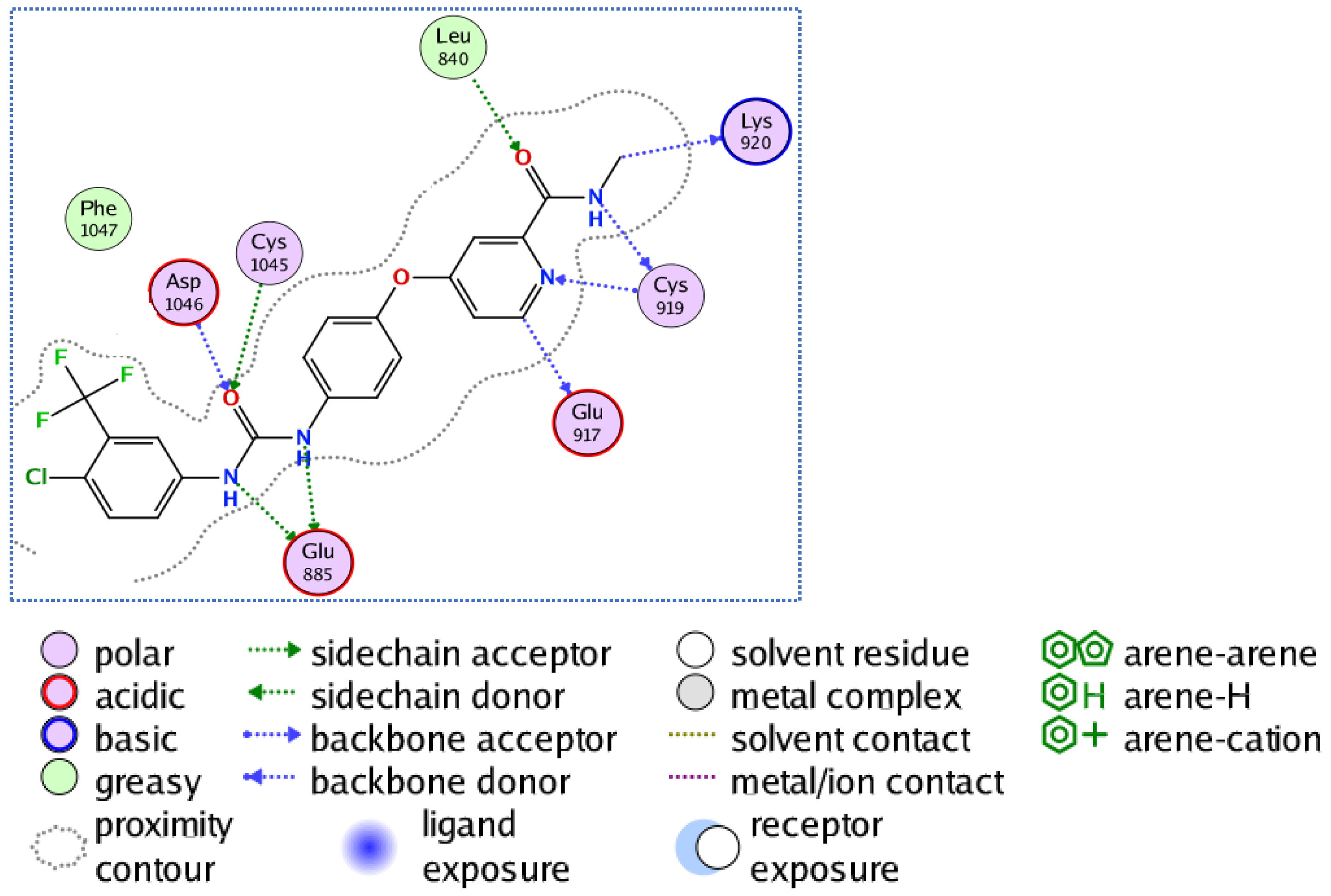
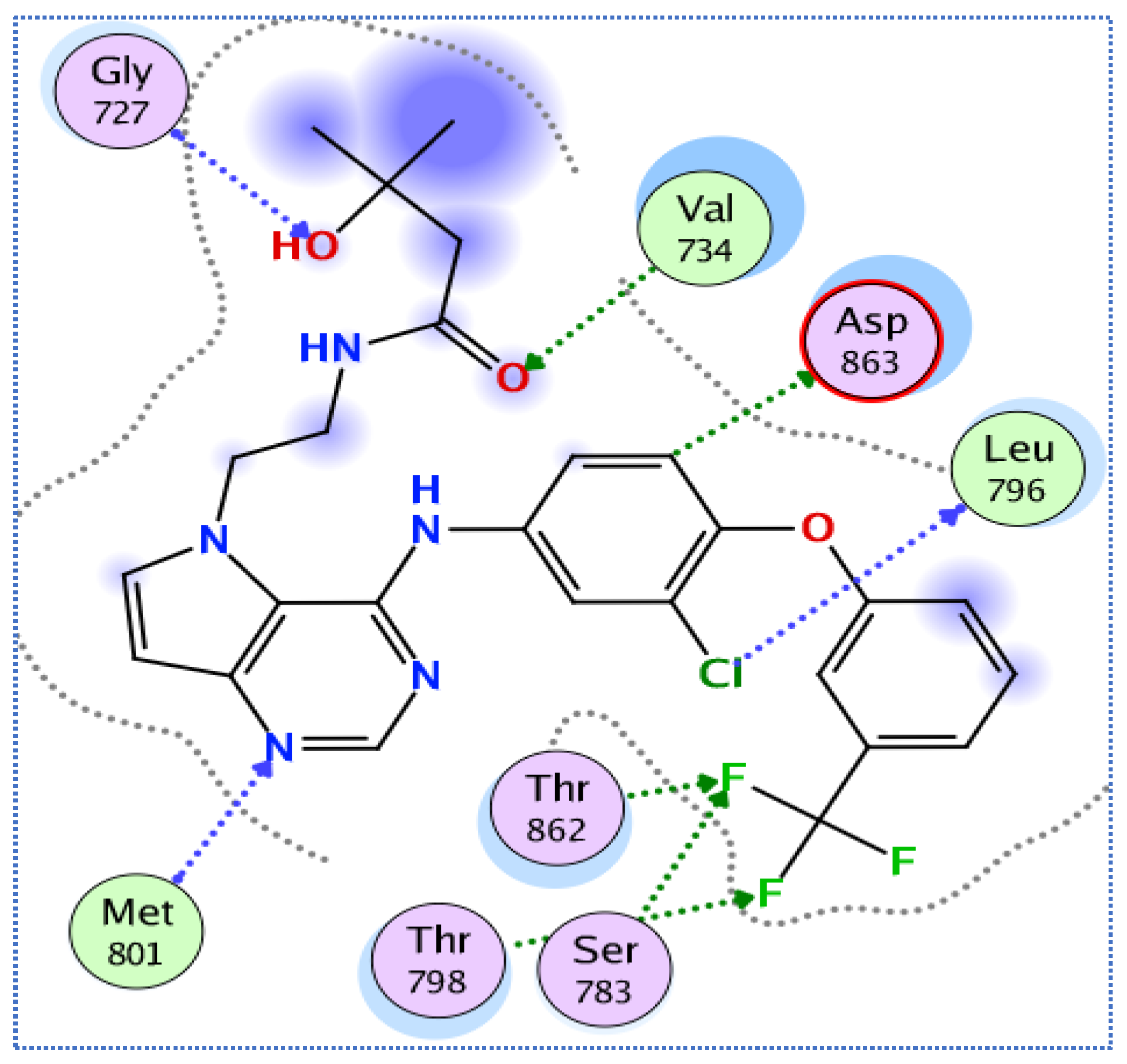
3.3.2. Molecular Docking Studies of Compounds 5a and 5e to Explain Their in Vitro Enzymatic Activity with Her-2 Active Site
3.4. Molecular Dynamics
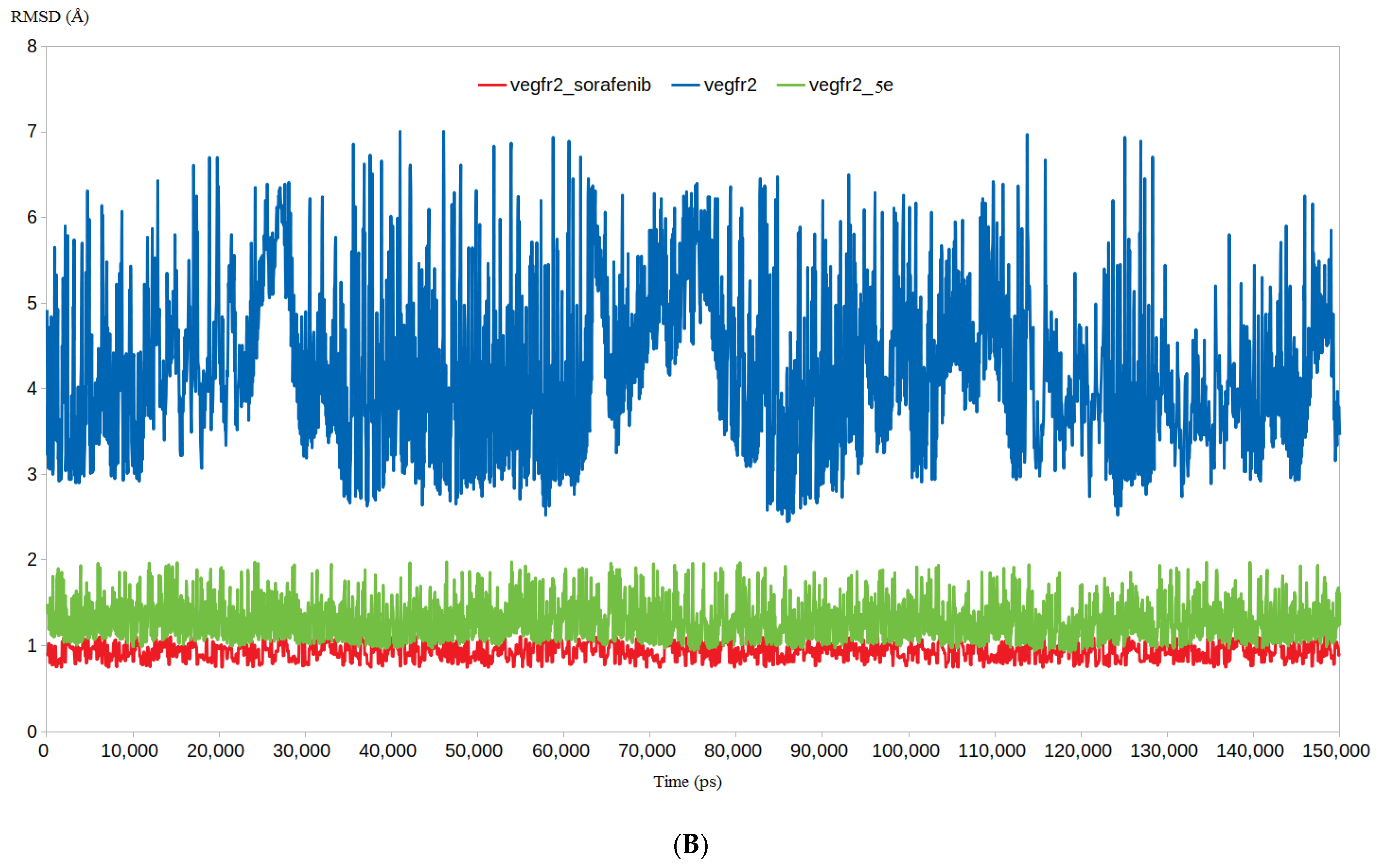
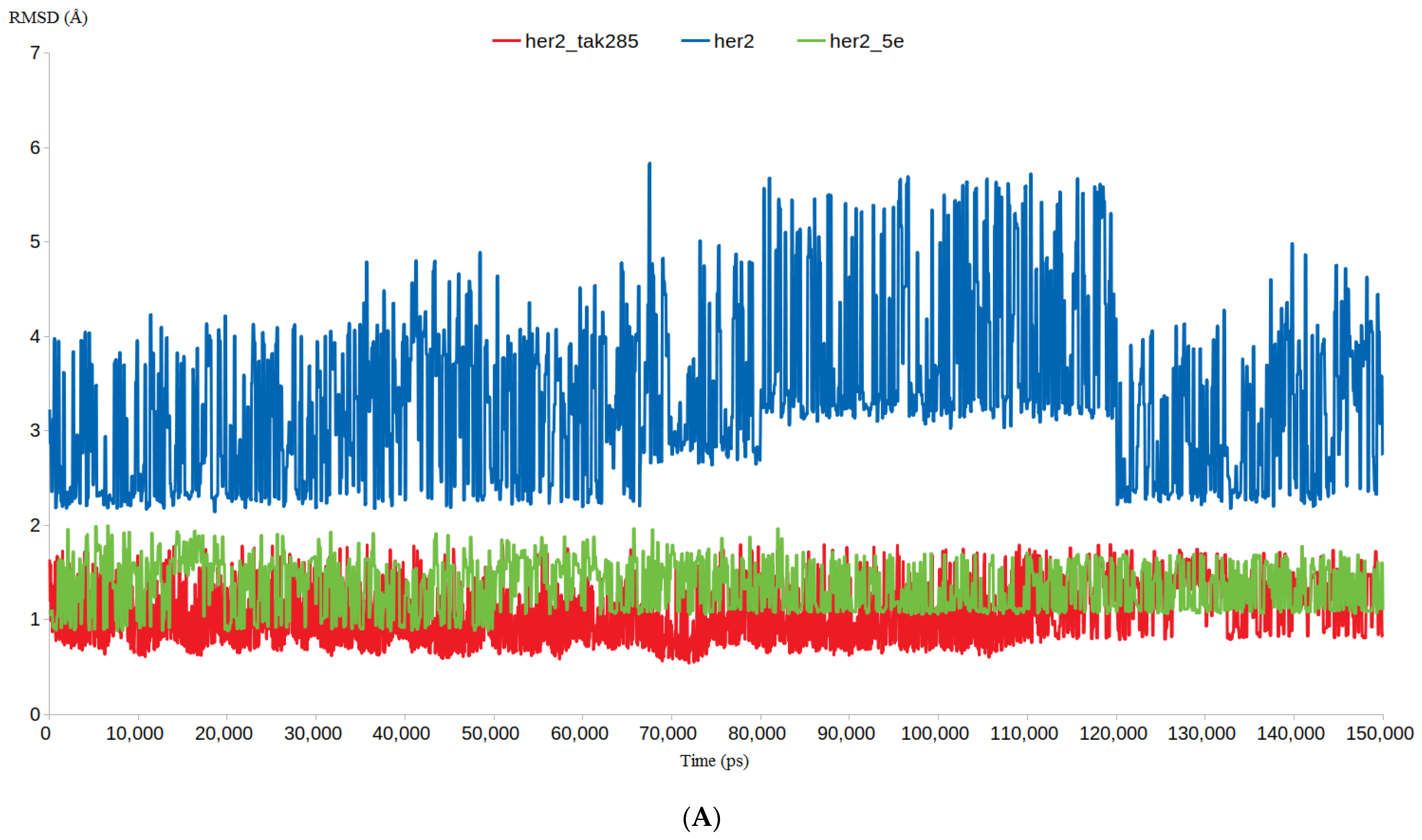
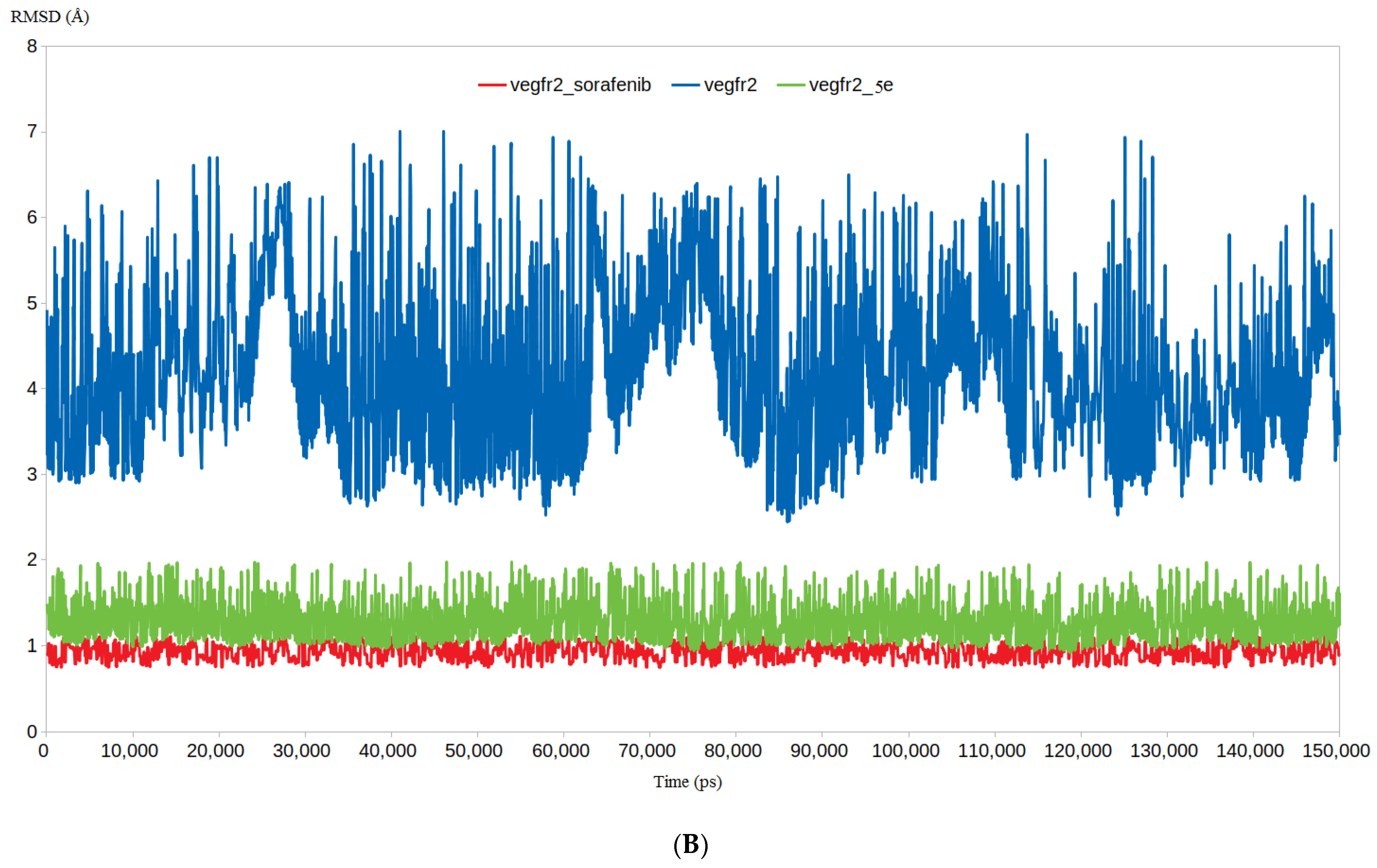
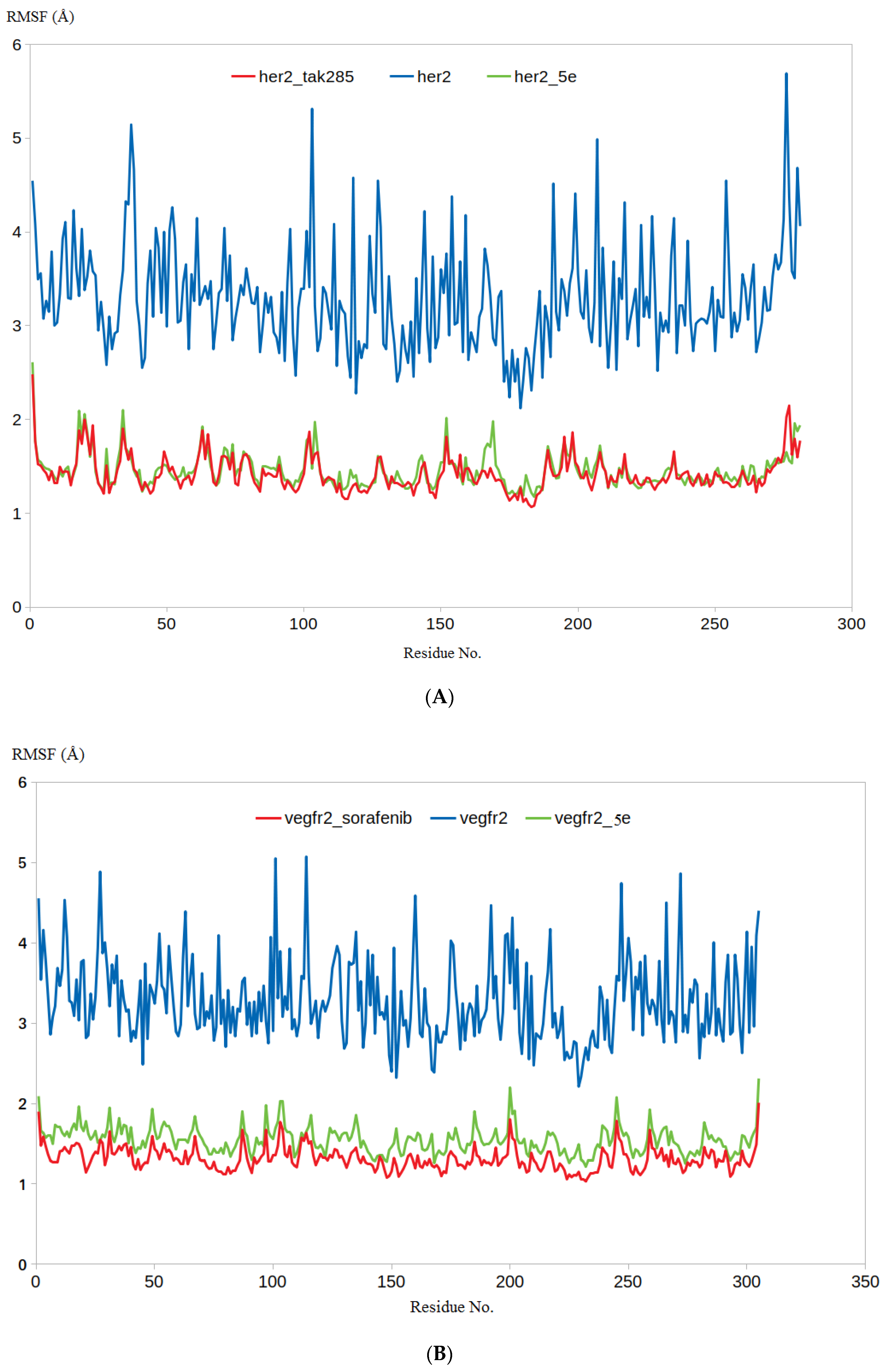
3.4.1. RMSD and RMSF Analysis
3.4.2. Binding Free Energy Calculations Using MM-PBSA Approach
4. Experimental
4.1. Chemistry
4.1.1. General Procedures for the Synthesis of 2-cyano-N-(3-(trifluoromethyl)phenyl) acetamide (3)
4.1.2. General Procedure for the Synthesis of 6-amino-4-(aryl)-2-oxo-1-(3-(trifluoromethyl)phenyl)-1,2-dihydropyridine-3,5-dicarbonitriles (5a-f)
4.1.3. General Procedure for the Synthesis of 4,7-dioxo-5-(aryl)-8-(3-(trifluoromethyl)phenyl)-3,4,7,8-tetrahydropyrido[2,3-d]pyrimidine-6-carbonitriles (6a-b)
4.1.4. General Procedure for the Synthesis of 5-(4-(aryl)-2-methyl-4,7-dioxo-8-(3-(trifluoromethyl)phenyl)-3,4,7,8-tetrahydropyrido[2,3-d]pyrimidine-6-carbonitriles (7a-e)
4.2. Cytotoxicity Assay
4.2.1. Chemicals and Reagents
4.2.2. Cell Culture
4.2.3. Cell Viability Assay
4.3. In Vitro VEGFR-2 and HER-2 Inhibition Assays
4.4. Molecular Docking Studies
4.5. Molecular Dynamics
MM-PBSA Calculation and Per-Residue Contribution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Rastogi, T.; Hildesheim, A.; Sinha, R. Opportunities for cancer epidemiology in developing countries. Nat. Rev. Cancer 2004, 4, 909–917. [Google Scholar] [CrossRef]
- Brunelleschi, S.; Penengo, L.; Santoro, M.M.; Gaudino, G. Receptor tyrosine kinases as target for anti-cancer therapy. Curr. Pharm. Des. 2002, 8, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Nagpal, R.; Hemalatha, R.; Verma, V.; Kumar, A.; Singh, S.; Marotta, F.; Jain, S.; Yadav, H. Targeted cancer therapies: The future of cancer treatment. Acta Biomed 2012, 83, 220–233. [Google Scholar] [PubMed]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Tomasello, C.; Baldessari, C.; Napolitano, M.; Orsi, G.; Grizzi, G.; Bertolini, F.; Barbieri, F.; Cascinu, S. Resistance to EGFR inhibitors in non-small cell lung cancer: Clinical management and future perspectives. Crit. Rev. Oncol./Hematol. 2018, 123, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Liu, F.; Li, L.; Ding, C.; Chen, K.; Sun, Q.; Shen, Z.; Tan, Y.; Tan, C.; Jiang, Y. A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell Death Dis. 2015, 6, e1686. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Burness, M.L.; Grushko, T.A.; Olopade, O.I. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: Promising clinical target or only a marker? Cancer J. 2010, 16, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Zahra, F.T.; Sajib, M.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Chung, E.; Yamashita, H.; Incio, J.; Duyverman, A.M.; Song, Y.; Farrar, C.T.; Huang, Y.; Ager, E.; Kamoun, W.; et al. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc. Natl. Acad. Sci. USA 2012, 109, E3119–E3127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pike, A. Pyridones in drug discovery: Recent advances. Bioorganic Med. Chem. Lett. 2021, 38, 127849. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent advances in the synthetic and medicinal perspective of quinolones: A review. Bioorganic Chem. 2019, 1, 103291. [Google Scholar] [CrossRef] [PubMed]
- El-Said, M.S.; El-Gazzar, M.G.; Al-Dosari, M.S.; Ghorab, M.M. Synthesis, anticancer activity and radiosensitizing evaluation of some new 2-pyridone derivatives. Arzneimittelforschung 2012, 62, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Mirković, J.M.; Mijin, D.Ž.; Petrović, S.D. Properties and synthesis of milrinone. Hem. Ind. 2013, 67, 17–25. [Google Scholar] [CrossRef]
- Forrestall, K.L.; Burley, D.E.; Cash, M.K.; Pottie, I.R.; Darvesh, S. 2-Pyridone natural products as inhibitors of SARS-CoV-2 main protease. Chem. Biol. Interact. 2021, 335, 109348. [Google Scholar] [CrossRef] [PubMed]
- Mahía, A.; Peña-Díaz, S.; Navarro, S.; Galano-Frutos, J.J.; Pallarés, I.; Pujols, J.; Díaz-de-Villegas, M.D.; Gálvez, J.A.; Ventura, S.; Sancho, J. Design, synthesis and structure-activity evaluation of novel 2-pyridone-based inhibitors of α-synuclein aggregation with potentially improved BBB permeability. Bioorganic Chem. 2021, 117, 105472. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Wan, E.; Foo, S.; Nathan, M.R.; Welti, J.C.; Frentzas, S.; Vermeulen, P.B.; Preece, N.; Springer, C.J.; Powles, T.; et al. Preclinical evidence that trametinib enhances the response to antiangiogenic tyrosine kinase inhibitors in renal cell carcinoma. Mol. Cancer Ther. 2016, 15, 172–183. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N.; et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ, γ, is clinically active in advanced hematologic malignancies. Blood J. Am. Soc. Hematol. 2018, 131, 877–887. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell 2019, 35, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.F.; Shao, M.H.; Gao, P.T.; Ma, J.; Li, H.J.; Li, G.L.; Han, B.H.; Yuan, C.G. The important roles of RET, VEGFR2 and the RAF/MEK/ERK pathway in cancer treatment with sorafenib. Acta Pharmacol. Sin. 2012, 33, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Holmes, F.A.; Ejlertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckwitz, G.; Chia, S.K.; Mansi, J.; Barrios, C.H.; et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1688–1700. [Google Scholar] [CrossRef]
- Flefel, E.M.; Abbas, H.-A.S.; Abdel Mageid, R.E.; Zaghary, W.A. Synthesis and cytotoxic effect of some novel 1, 2-dihydropyridin-3-carbonitrile and nicotinonitrile derivatives. Molecules 2016, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Qin, C.; Sin, J.E.; Yang, X.; Tao, L.; Zeng, X.; Zhang, P.; Gao, C.M.; Jiang, Y.Y.; Zhang, C.; et al. Discovery of novel dual VEGFR2 and Src inhibitors using a multistep virtual screening approach. Future Med. Chem. 2017, 9, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, T.; Tatibana, M. Metabolic fate of pyrimidines and purines in dietary nucleic acids ingested by mice. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1978, 521, 55–66. [Google Scholar] [CrossRef]
- Elsayed, N.M.; Abou El Ella, D.A.; Serya, R.A.; Tolba, M.F.; Shalaby, R.; Abouzid, K.A. Design, synthesis and biological evaluation of indazole–pyrimidine based derivatives as anticancer agents with anti-angiogenic and antiproliferative activities. MedChemComm 2016, 7, 881–899. [Google Scholar] [CrossRef]
- Bhattacharya, S.K.; Cox, E.D.; Kath, J.C.; Mathiowetz, A.M.; Morris, J.; Moyer, J.D.; Pustilnik, L.R.; Rafidi, K.; Richter, D.T.; Su, C.; et al. Achieving selectivity between highly homologous tyrosine kinases: A novel selective erbB2 inhibitor. Biochem. Biophys. Res. Commun. 2003, 307, 267–273. [Google Scholar] [CrossRef]
- Barbacci, E.G.; Pustilnik, L.R.; Rossi, A.M.; Emerson, E.; Miller, P.E.; Boscoe, B.P.; Cox, E.D.; Iwata, K.K.; Jani, J.P.; Provoncha, K.; et al. The biological and biochemical effects of CP-654577, a selective erbB2 kinase inhibitor, on human breast cancer cells. Cancer Res. 2003, 63, 4450–4459. [Google Scholar]
- Modi, S.J.; Modh, D.H.; Kulkarni, V.M. Insights into the structural features of anticancer 1,6-naphthyridines and pyridopyrimidines as VEGFR-2 inhibitors: 3D-QSAR study. J. Appl. Pharm. Sci. 2020, 10, 1–22. [Google Scholar]
- Tamura, Y.; Omori, N.; Kouyama, N.; Nishiura, Y.; Hayashi, K.; Watanabe, K.; Tanaka, Y.; Chiba, T.; Yukioka, H.; Sato, H.; et al. Design, synthesis and identification of novel benzimidazole derivatives as highly potent NPY Y5 receptor antagonists with attractive in vitro ADME profiles. Bioorganic Med. Chem. Lett. 2012, 22, 5498–5502. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, W.; Masuda, N.; Miyamoto, S.; Shiina, Y.; Kikuchi, S.; Mihara, T.; Moriguchi, H.; Fushiki, H.; Murakami, Y.; Amano, Y.; et al. Synthesis, SAR study, and biological evaluation of novel quinoline derivatives as phosphodiesterase 10A inhibitors with reduced CYP3A4 inhibition. Bioorganic Med. Chem. 2015, 23, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Murray, L.J.; Ndubaku, C.O.; O’Brien, T.; Blackwood, E.; Wang, W.; Aliagas, I.; Gazzard, L.; Crawford, J.J.; Drobnick, J.; et al. Chemically diverse group I p21-activated kinase (PAK) inhibitors impart acute cardiovascular toxicity with a narrow therapeutic window. J. Med. Chem. 2016, 59, 5520–5541. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.E.; Cubbon, R.M.; Cummings, R.T.; Wicker, L.S.; Frankshun, R.; Cunningham, B.R.; Cameron, P.M.; Meinke, P.T.; Liverton, N.; Weng, Y.; et al. Photochemical preparation of a pyridone containing tetracycle: A Jak protein kinase inhibitor. Bioorganic Med. Chem. Lett. 2002, 12, 1219–1223. [Google Scholar] [CrossRef]
- Siu, T.; Kozina, E.S.; Jung, J.; Rosenstein, C.; Mathur, A.; Altman, M.D.; Chan, G.; Xu, L.; Bachman, E.; Mo, J.R.; et al. The discovery of tricyclic pyridone JAK2 inhibitors. Part 1: Hit to lead. Bioorganic Med. Chem. Lett. 2010, 20, 7421–7425. [Google Scholar] [CrossRef]
- Bach, J.; Eastwood, P.; González, J.; Gómez, E.; Alonso, J.A.; Fonquerna, S.; Lozoya, E.; Orellana, A.; Maldonado, M.; Calaf, E.; et al. Identification of 2-imidazopyridine and 2-aminopyridone purinones as potent pan-janus kinase (JAK) inhibitors for the inhaled treatment of respiratory diseases. J. Med. Chem. 2019, 62, 9045–9060. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef]
- Teno, N.; Miyake, T.; Ehara, T.; Irie, O.; Sakaki, J.; Ohmori, O.; Gunji, H.; Matsuura, N.; Masuya, K.; Hitomi, Y.; et al. Novel scaffold for cathepsin K inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 6096–6100. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Modern synthetic methods for fluorine-substituted target molecules. Angew. Chem. Int. Ed. 2005, 44, 214–231. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. Former. Pestic. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Y.A.; Ismail, M.M.; El-Sehrawi, H.M.; Noaman, E.; Bayomi, A.H.; Shawer, T.Z. Novel Pirfenidone Analogues: Synthesis of Pyridin-2-ones for the Treatment of Pulmonary Fibrosis. Arch. Pharm. Int. J. Pharm. Med. Chem. 2006, 339, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Mekheimer, R.A.; Al-Sheikh, M.A.; Medrasi, H.Y.; Alsofyani, N.H. A novel synthesis of highly functionalized pyridines by a one-pot, three-component tandem reaction of aldehydes, malononitrile and N-alkyl-2-cyanoacetamides under microwave irradiation. Molecules 2018, 23, 619. [Google Scholar] [CrossRef] [PubMed]
- Khatri, T.T.; Shah, V.H. One pot synthesis of novel cyanopyridones as an intermediate of bioactive pyrido [2,3-d] pyrimidines. J. Korean Chem. Soc. 2014, 58, 366–376. [Google Scholar] [CrossRef]
- Ammar, Y.A.; El-Sharief, A.S.; Mohamed, Y.A.; Salem, M.A.; Al-Sehemi, A.G.; El-Gaby, M.S. Cyanoacetanilides intermediates in heterocyclic synthesis. Part 1: A facile synthesis of polysubstituted and condensed pyridones. J. Chin. Chem. Soc. Taipei 2004, 51, 975–982. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Eremeev, A.V.; Piskunova, I.P.; El’kinson, R.S. Synthesis of 2-amino-1-azirines and their reactions with carboxylic acids. Chem. Heterocycl. Compd. 1985, 21, 998–1002. [Google Scholar] [CrossRef]
- Schellhase, M.; Boehm, R.; Pech, R. Thieno compounds. Part 4: 3-aryl-3,4-dihydro-4-oxobenzo (b) thieno (2, 3-d) pyrimidines. Chem. Inf. 1984, 15, 19–21. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Taghour, S.M.; Elkady, H.; Eldehna, W.M.; El-Deeb, N.M.; Kenawy, A.M.; Elkaeed, E.B.; Alsfouk, A.A.; Alesawy, M.S.; Metwaly, A.M.; Eissa, I.H. Design and synthesis of thiazolidine-2,4-diones hybrids with 1,2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: In-vitro anticancer evaluation and in-silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1903–1917. [Google Scholar] [CrossRef]
- Yousef, G.R.; Ibrahim, A.; Khalifa, M.M.; Eldehna, W.M.; Gobaara, I.M.M.; Mehany, A.B.M.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.H. Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: Virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Doghish, A.S.; El-Sayyad, G.S.; Sallam, A.A.; Khalil, W.F.; El Rouby, W.M. Graphene oxide and its nanocomposites with EDTA or chitosan induce apoptosis in MCF-7 human breast cancer. RSC Adv. 2021, 11, 29052–29064. [Google Scholar] [CrossRef] [PubMed]
- Kassab, A.E.; Gedawy, E.M.; Hamed, M.I.; Doghish, A.S.; Hassan, R.A. Design, synthesis, anticancer evaluation, and molecular modelling studies of novel tolmetin derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 922–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Scholz, C.; Knorr, S.; Hamacher, K.; Schmidt, B. DOCKTITE—A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Modeling 2015, 55, 398–406. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Ibrahim, T.M.; Al-Rashood, S.T.; Alharbi, A.; Eskandrani, R.O.; Eldehna, W.M. In silico identification of novel SARS-CoV-2 2′-O-methyltransferase (nsp16) inhibitors: Structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. J. Enzym. Inhib. Med. Chem. 2021, 36, 727–736. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Eldehna, W.M.; Al-Rashood, S.T.; Alharbi, A.; Eskandrani, R.O.; Alkahtani, H.M.; Elkaeed, E.B.; Abou-Seri, S.M. Multi-stage structure-based virtual screening approach towards identification of potential SARS-CoV-2 NSP13 helicase inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 563–572. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Comp. | R | IC50 (µM) | |
|---|---|---|---|
| MCF-7 | HepG2 | ||
| 5a | H | 1.77 ± 0.10 | 2.71 ± 0.15 |
| 5b | 4-OH | 24.68 ± 1.33 | 11.37 ± 0.61 |
| 5c | 4-NO2 | 20.23 ± 1.09 | 4.01 ± 0.22 |
| 5d | 4-Cl | 13.70 ± 0.74 | 22.52 ± 1.22 |
| 5e | 2,4-(Cl)2 | 1.39 ± 0.08 | 10.70 ± 0.58 |
| 5f | 3,4,5-(OCH3)3 | 54.22 ± 2.93 | 40.60 ± 2.19 |
| 6a | 4-OH | 55.33 ± 2.99 | 7.27 ± 0.39 |
| 6b | 4-Cl | 13.99 ± 0.76 | 2.68 ± 0.14 |
| 7a | H | 10.68 ± 0.58 | 16.30 ± 0.88 |
| 7b | 4-Cl | 6.22 ± 0.34 | 19.58 ± 1.06 |
| 7c | 2,4-(Cl)2 | 9.28 ± 0.50 | 5.04 ± 0.27 |
| 7d | 4-OCOCH3 | 43.43 ± 2.35 | 52.51 ± 2.84 |
| 7e | 3,4,5-(OCH3)3 | 27.78 ± 1.50 | 71.93 ± 3.88 |
| Taxol | - | 8.48 ± 0.46 | 14.60 ± 0.79 |
| Comp. | IC50 (µM) a | |
|---|---|---|
| VEGFR-2 | HER-2 | |
| 5a | 0.217 ± 0.020 | 0.168 ± 0.009 |
| 5e | 0.124 ± 0.011 | 0.077 ± 0.003 |
| Lapatinib | 0.182 ± 0.010 | 0.131 ± 0.012 |
| Comp. | MCF-7 | |
|---|---|---|
| VEGFR-2 (pg/mL) | HER-2 (pg/mL) | |
| 5a | 884.30 ± 29.3 | 299.50 ± 4.7 |
| 5e | 510.00 ± 20.2 | 148.60 ± 5.5 |
| Taxol | 660.10 ± 31.4 | 199.10 ± 8.6 |
| Control | 1573.00 ± 36.9 | 559.00 ± 33.9 |
| Complex | ΔEbinding (kJ/mol) | ΔEelectrostatic (kJ/mol) | ΔEvan der Waal (kJ/mol) | ΔEpolar solvation (kJ/mol) | SASA (kJ/mol) |
|---|---|---|---|---|---|
| 5e–HER-2 | −290.9 ± 2.8 | −110.1±1.9 | −251.3 ± 4.2 | 101.8 ± 1.5 | −31.5 ± 0.5 |
| TAK285–HER-2 | −293.8 ± 2.2 | −130.1±1.8 | −234.1 ± 3.3 | 99.8 ± 0.9 | −29.4 ± 0.4 |
| 5e–VEGFR-2 | −284.1 ± 2.6 | −109.7±1.7 | −248.6 ± 4.3 | 104.9 ± 1.4 | −30.7 ± 0.2 |
| Sorafenib–VEGFR-2 | −313.2 ± 3.5 | −140.1±2.1 | −260.1 ± 5.9 | 112.7 ± 1.6 | −25.7 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Warhi, T.; Sallam, A.-A.M.; Hemeda, L.R.; El Hassab, M.A.; Aljaeed, N.; Alotaibi, O.J.; Doghish, A.S.; Noshy, M.; Eldehna, W.M.; Ibrahim, M.H. Identification of Novel Cyanopyridones and Pyrido[2,3-d]Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2022, 15, 1262. https://doi.org/10.3390/ph15101262
Al-Warhi T, Sallam A-AM, Hemeda LR, El Hassab MA, Aljaeed N, Alotaibi OJ, Doghish AS, Noshy M, Eldehna WM, Ibrahim MH. Identification of Novel Cyanopyridones and Pyrido[2,3-d]Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals. 2022; 15(10):1262. https://doi.org/10.3390/ph15101262
Chicago/Turabian StyleAl-Warhi, Tarfah, Al-Aliaa M. Sallam, Loah R. Hemeda, Mahmoud A. El Hassab, Nada Aljaeed, Ohoud J. Alotaibi, Ahmed S. Doghish, Mina Noshy, Wagdy M. Eldehna, and Mona H. Ibrahim. 2022. "Identification of Novel Cyanopyridones and Pyrido[2,3-d]Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies" Pharmaceuticals 15, no. 10: 1262. https://doi.org/10.3390/ph15101262
APA StyleAl-Warhi, T., Sallam, A.-A. M., Hemeda, L. R., El Hassab, M. A., Aljaeed, N., Alotaibi, O. J., Doghish, A. S., Noshy, M., Eldehna, W. M., & Ibrahim, M. H. (2022). Identification of Novel Cyanopyridones and Pyrido[2,3-d]Pyrimidines as Anticancer Agents with Dual VEGFR-2/HER-2 Inhibitory Action: Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals, 15(10), 1262. https://doi.org/10.3390/ph15101262