
Synthesis and Chemopreventive Potential of 5-FU/Genistein Hybrids on Colorectal Cancer Cells
 and
and
Abstract
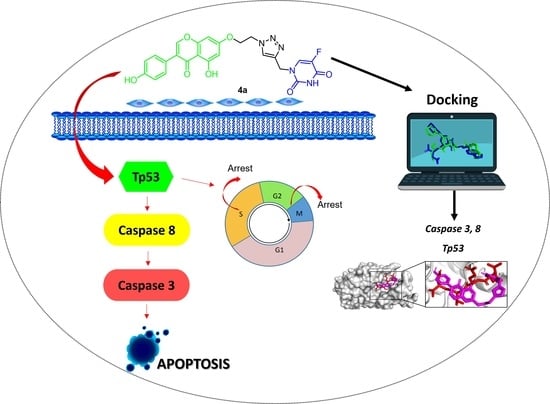
:
1. Introduction
2. Results and Discussion
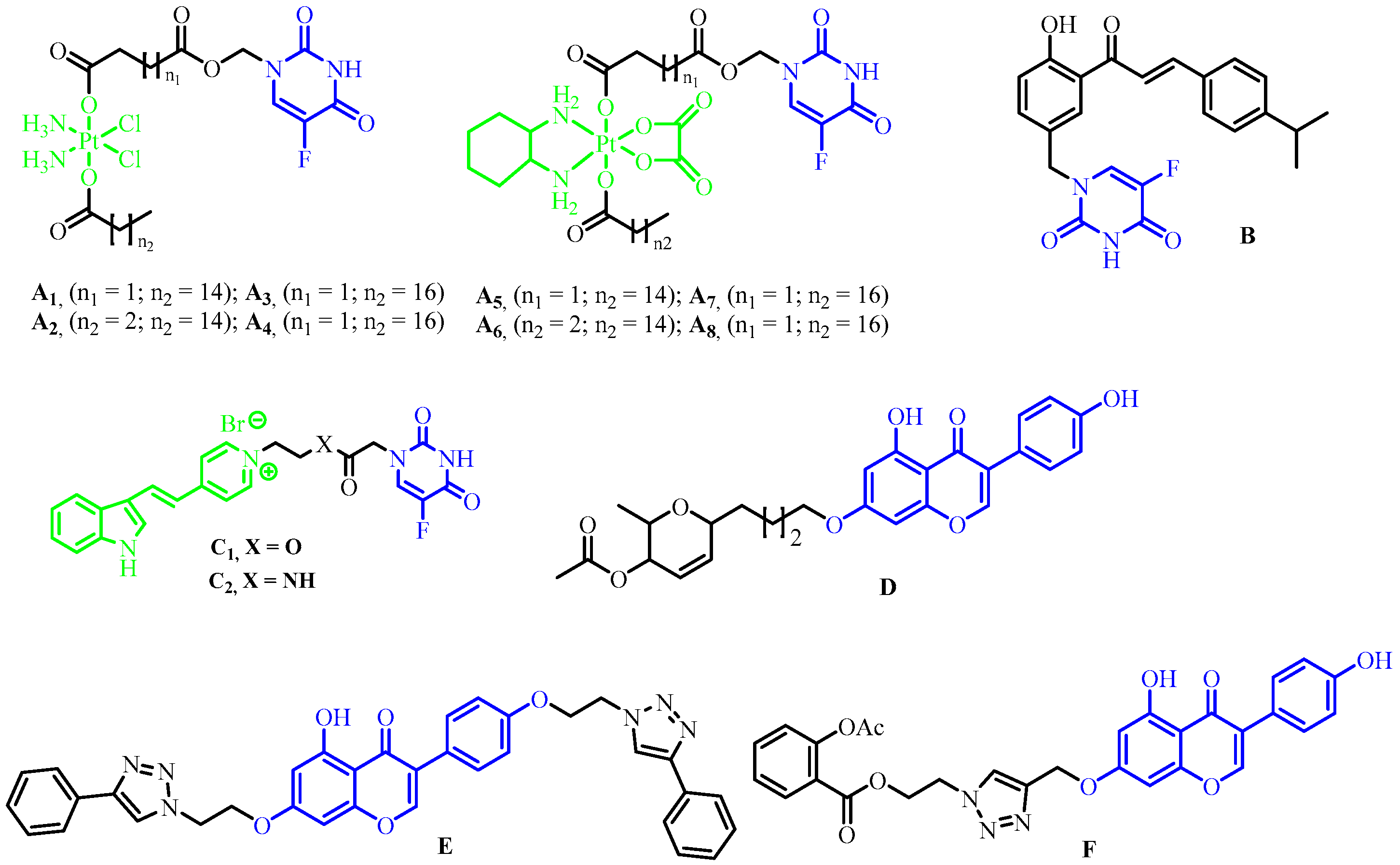
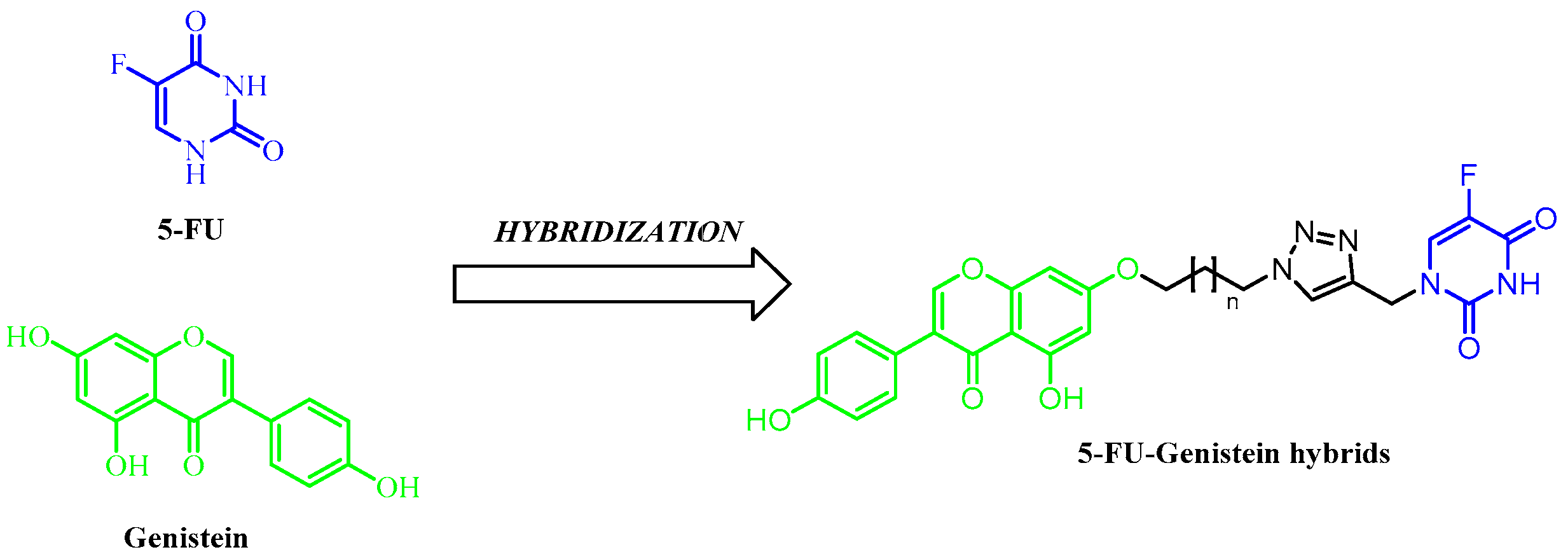
2.1. Chemistry
2.2. Biological Activity
2.2.1. Cytotoxicity of 5-FU/Genistein Hybrids on SW480, SW620, HaCaT and CHO-K1 Cell Lines
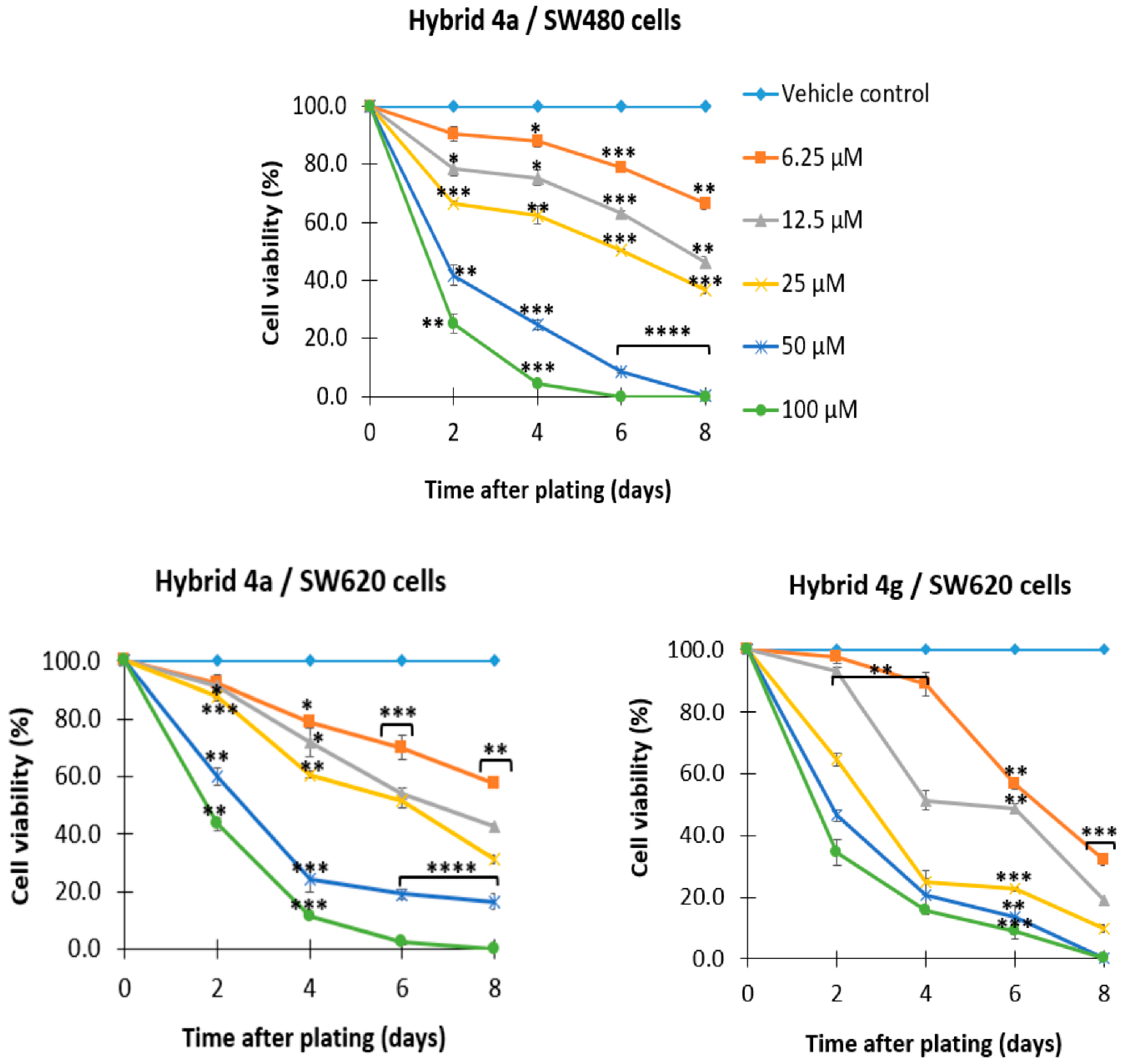
2.2.2. 5-FU/Genistein Hybrids Induce Antiproliferative Effect on SW480 and SW620 Cells
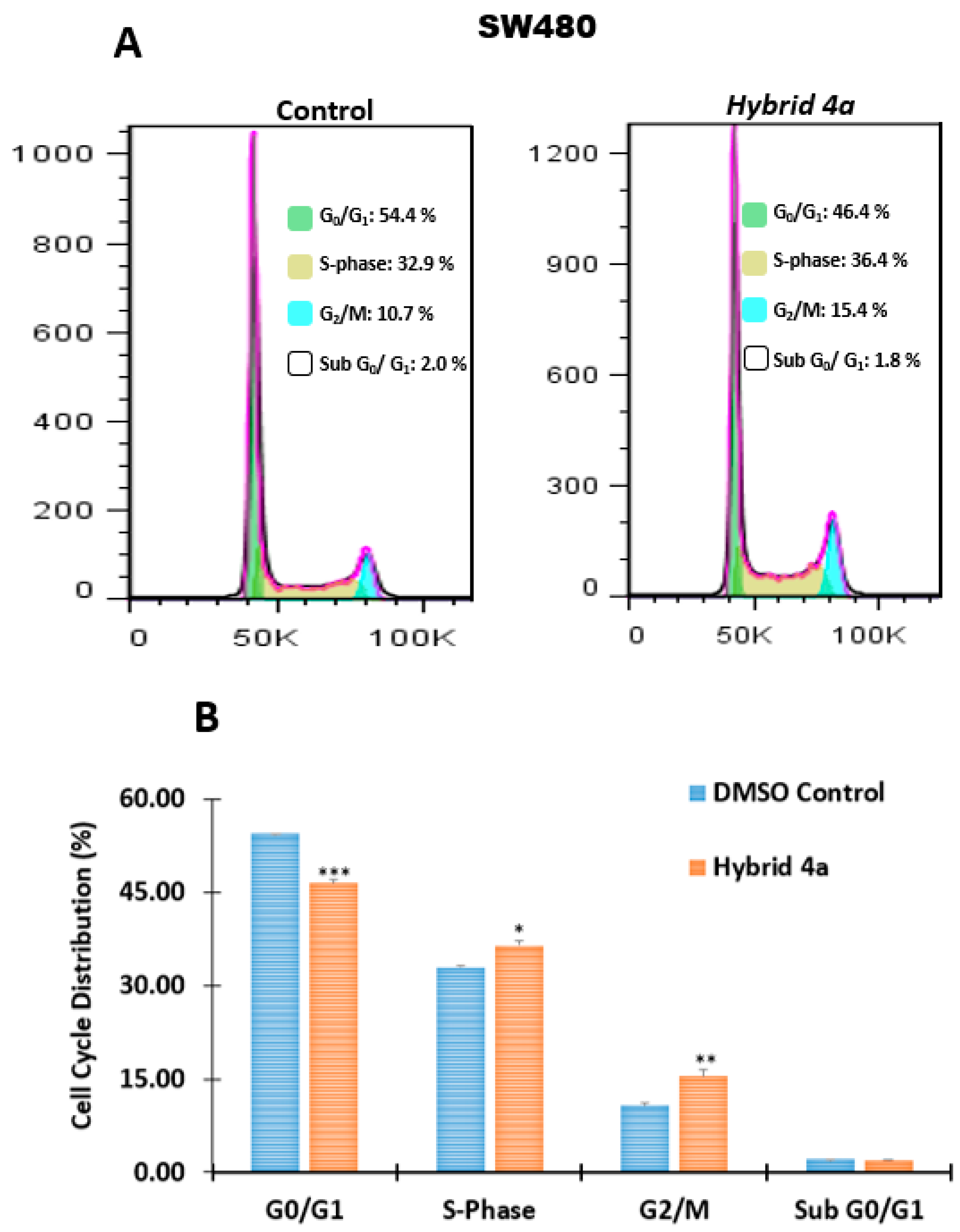
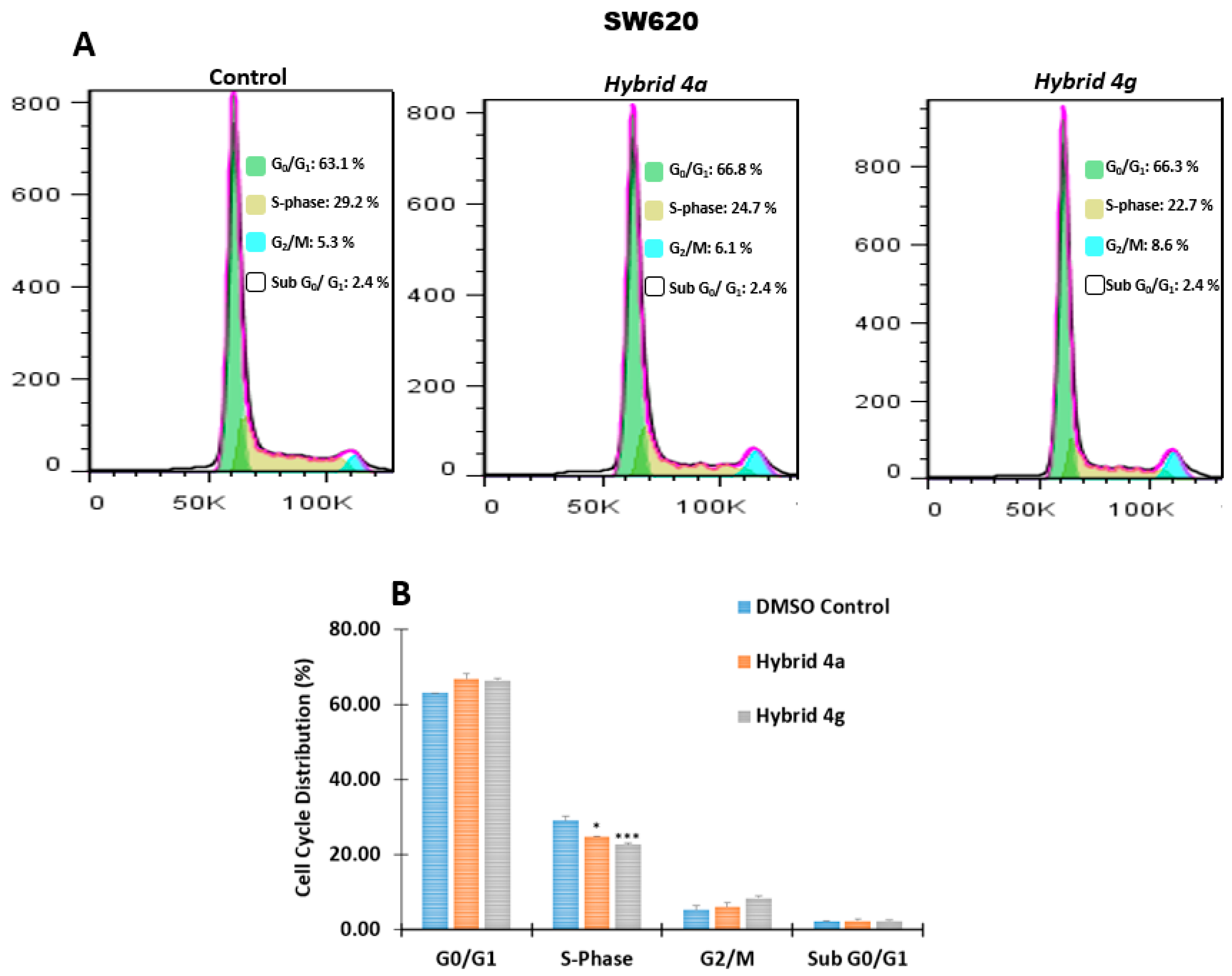
2.2.3. Effect of Hybrids 4a and 4g on Cell Cycle Distribution
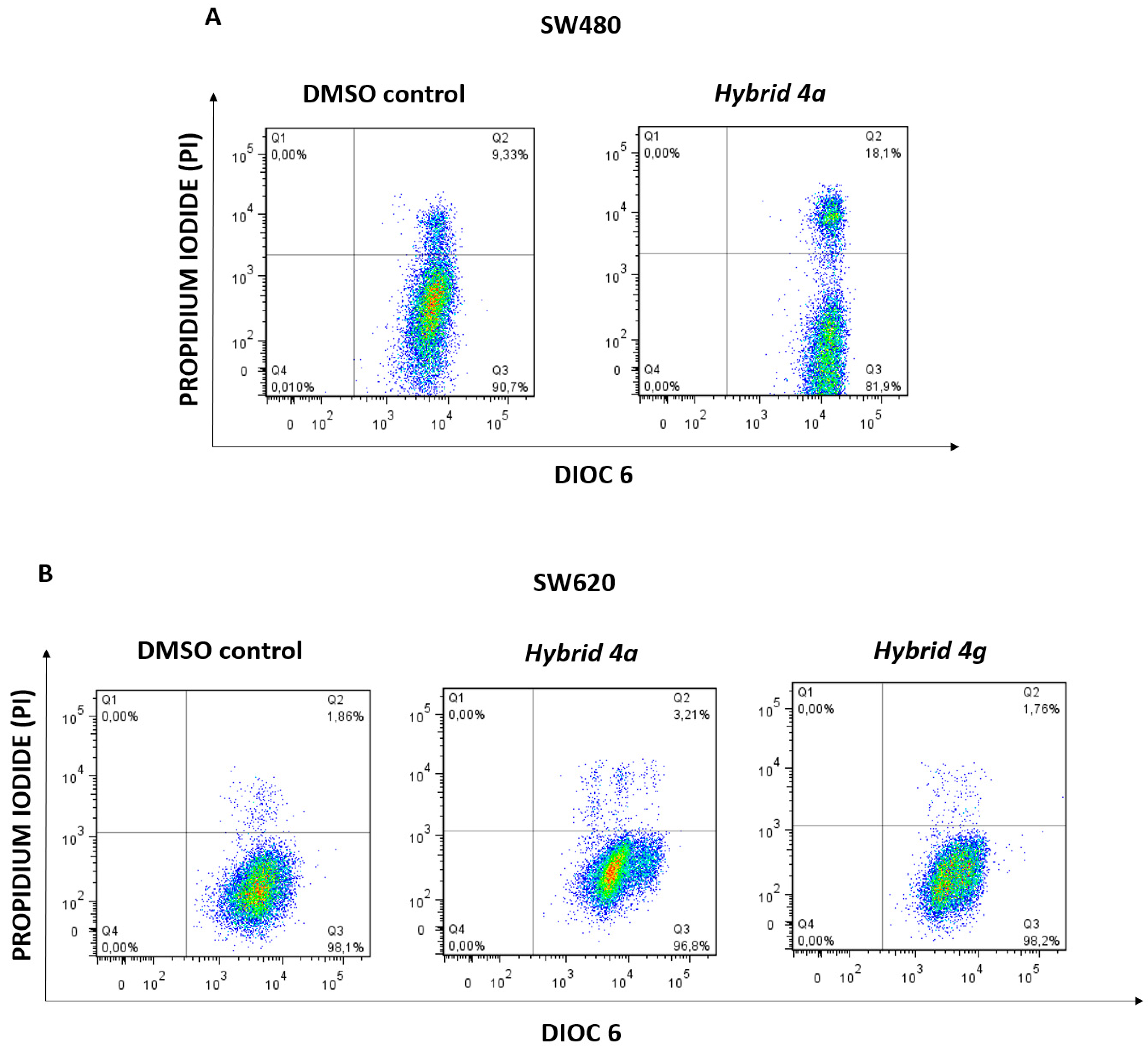
2.2.4. Changes in Mitochondrial Membrane Potential (ΔΨm) and Plasma Membrane Integrity
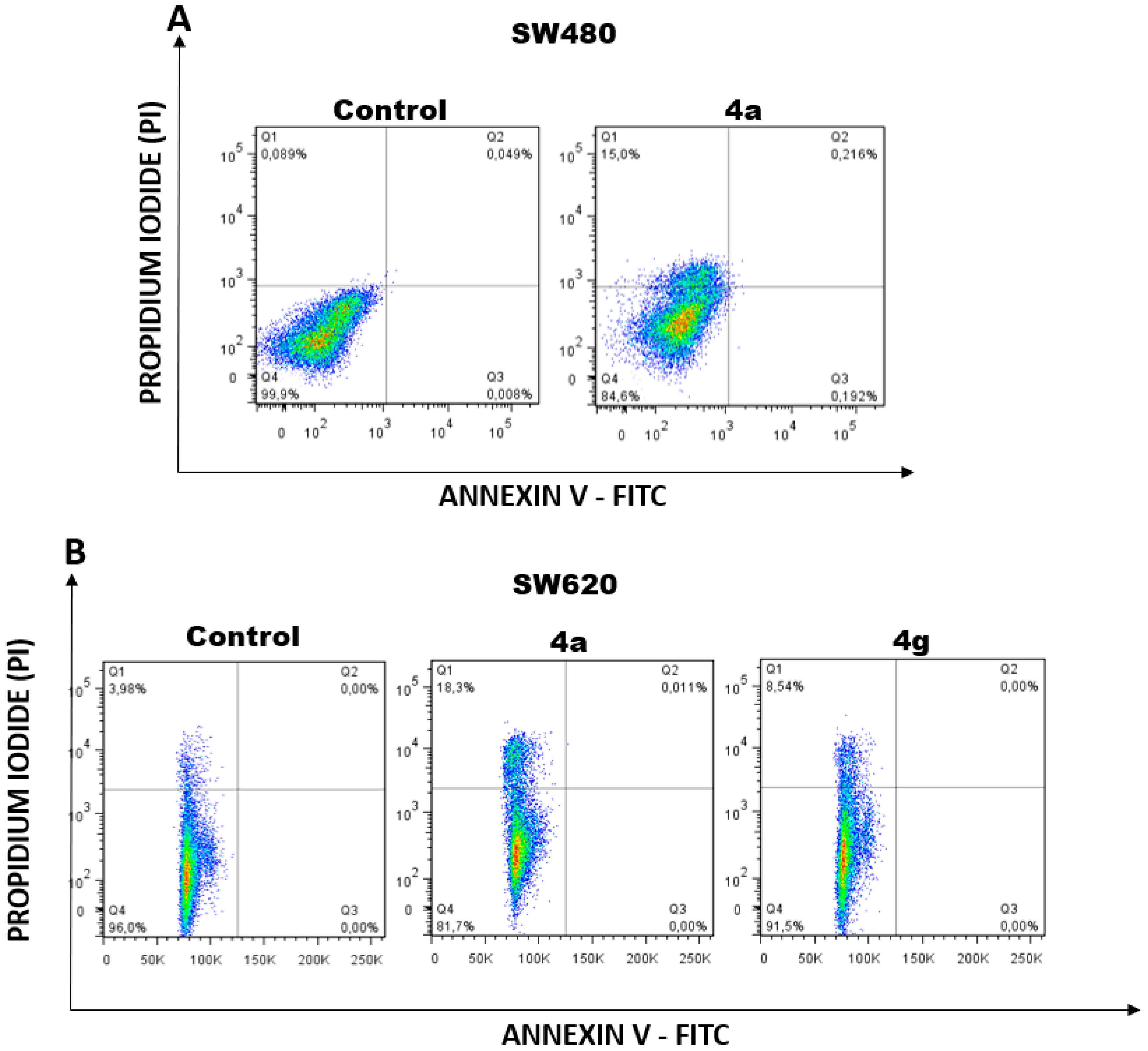
2.2.5. Apoptosis Induction by Hybrids 4a and 4g
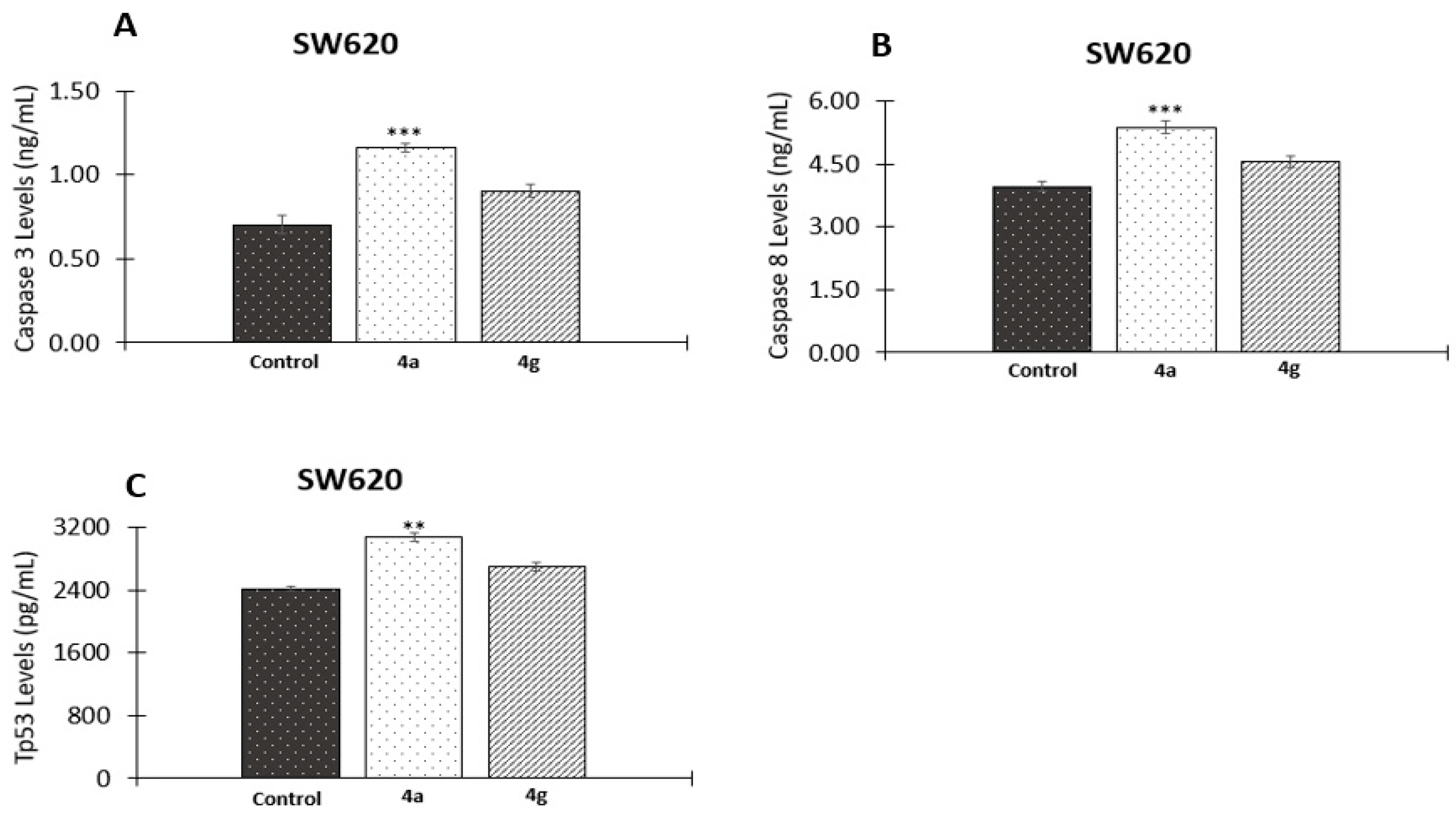
2.2.6. Effect of 5-FU/Genistein Hybrids on the Expression Apoptotic Biomarkers
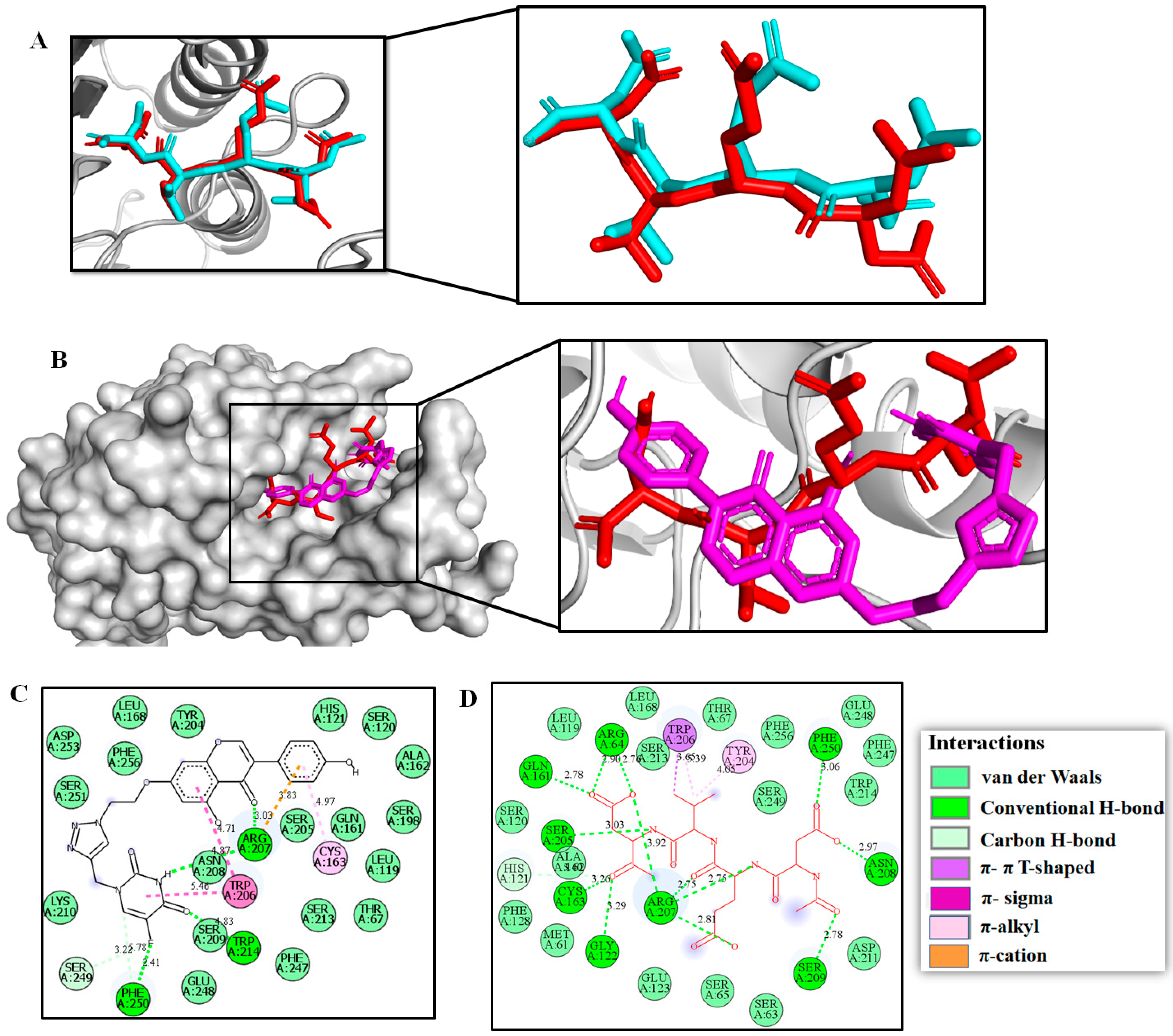
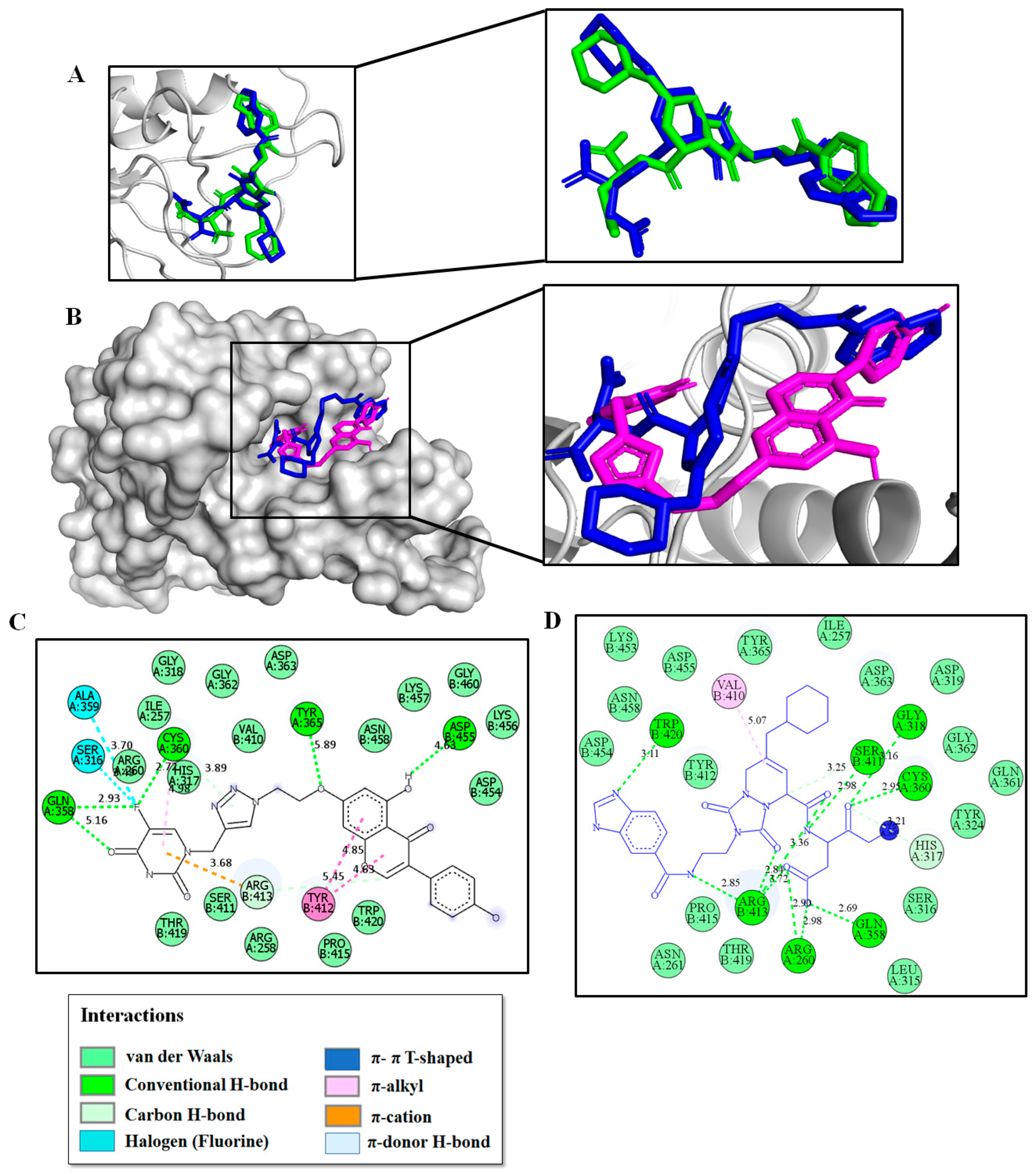
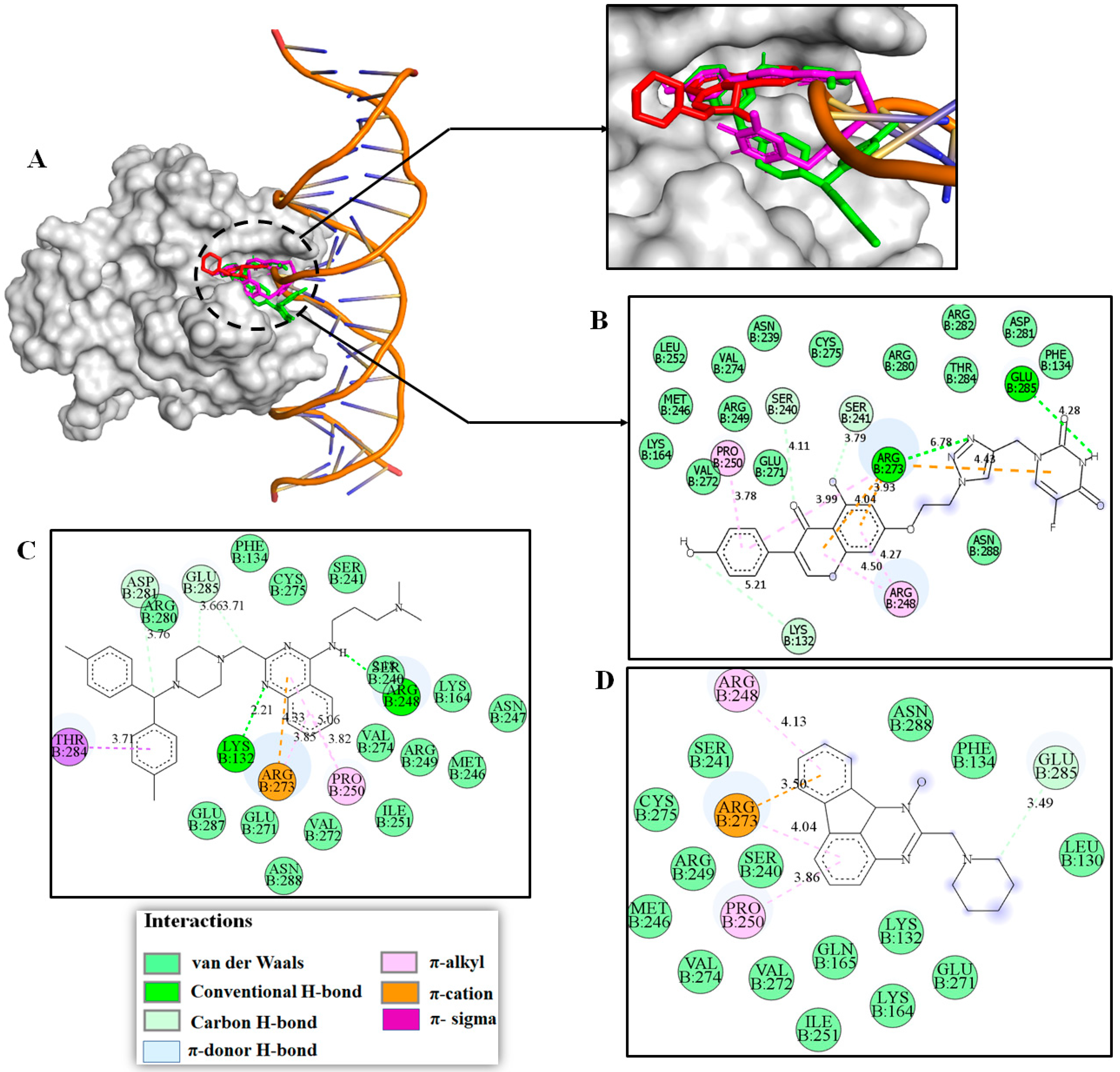
2.3. Molecular Modeling Studies
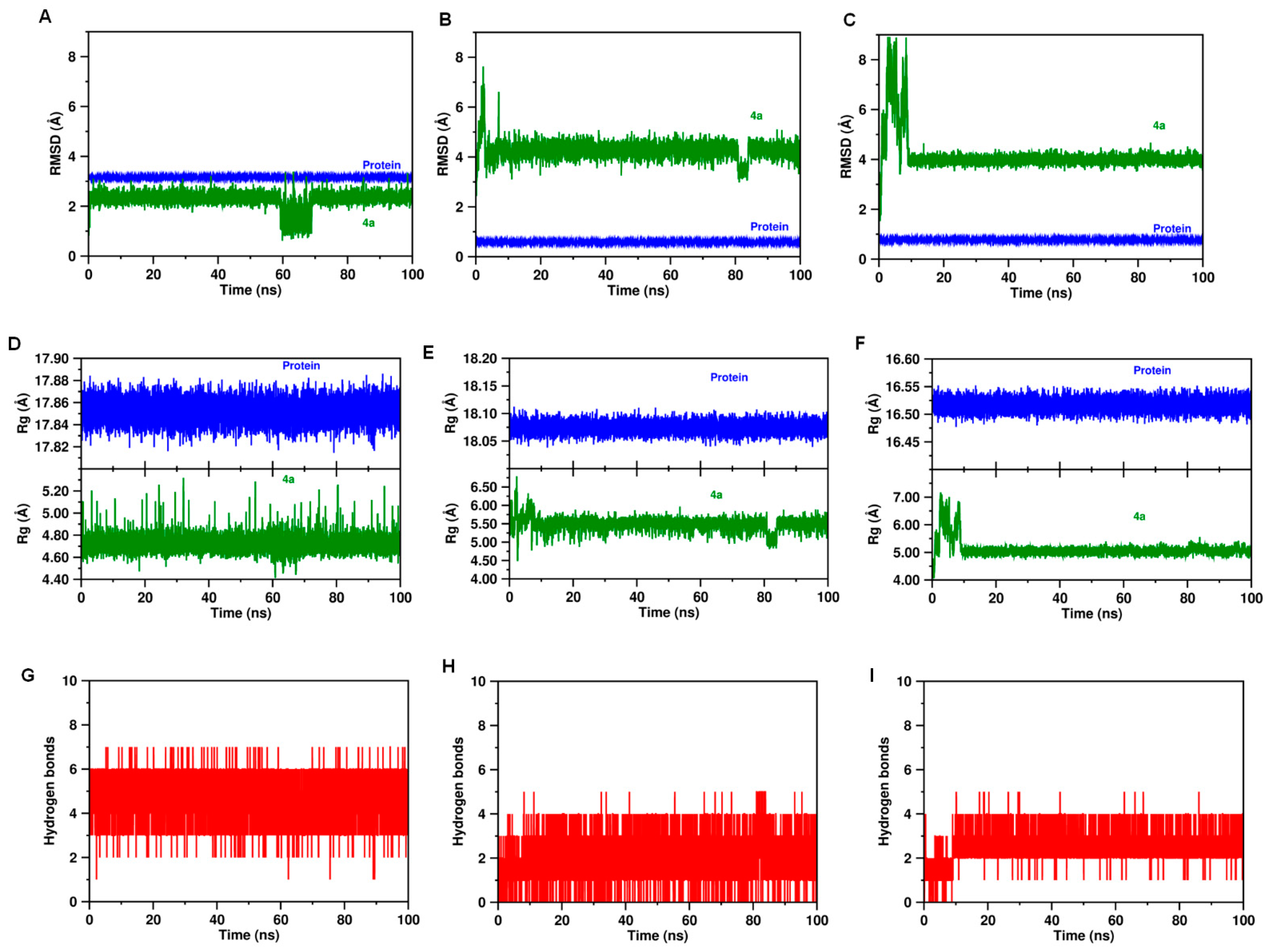
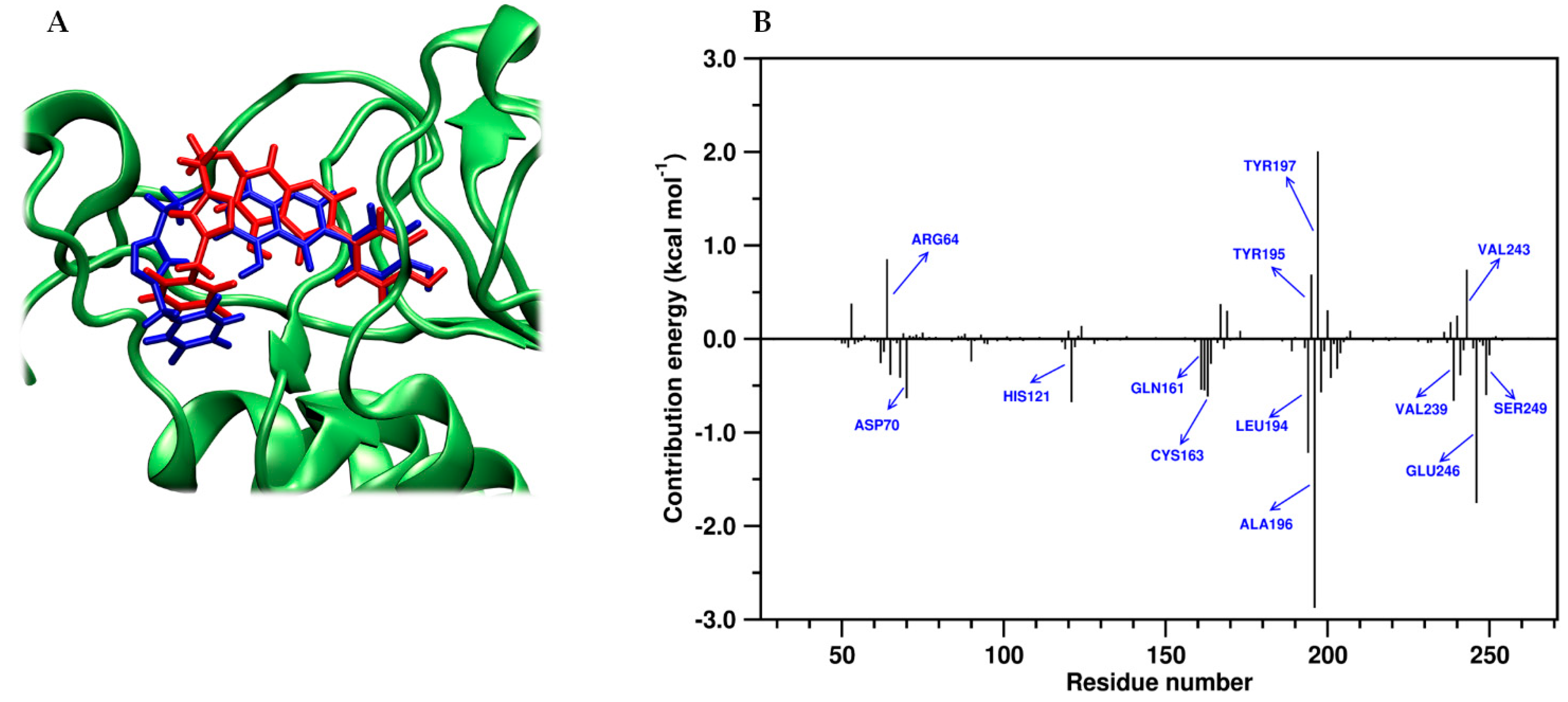
2.4. Molecular Dynamics and Post-MM-PBSA Studies
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General Remarks
3.1.2. General Procedure for the Synthesis of Genistein-Bromoalkyl (1a–h)
3.1.3. General Procedure for the Synthesis of Genistein-Alkylazides (2a–h)
3.1.4. Syntheses of Propargyl-5-FU (3):
3.1.5. General Procedure for the Synthesis of 5-FU-Genistein Hybrids (4a–h)
3.2. In Vitro Biological Assays
3.2.1. Cell Lines and Culture Conditions
3.2.2. Cytotoxic and Antiproliferative Activity
3.2.3. Effect of 5-FU/Genistein Hybrids on Cell Cycle Distribution
3.2.4. Measurement of Mitochondrial Membrane Potential (ΔΨm)
3.2.5. Cell Death Induction by Hybrids 4a and 4g
3.2.6. Determination of Apoptotic Biomarkers
3.2.7. Statistical Analysis
3.3. Computational Methods
3.3.1. Docking Protocol
3.3.2. Molecular Dynamics (MD) Studies
3.3.3. Binding Energy Calculation (MM/PBSA Protocol)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [CrossRef] [PubMed]
- Campos, J.; Domínguez, J.F.; Gallo, M.A.; Espinosa, A. From a classic approach in cancer chemotherapy towards differentiation therapy: Acyclic and cyclic seven-membered 5-fluorouracil O, N-acetals. Curr. Pharm. Des. 2000, 6, 1797. [Google Scholar] [CrossRef] [PubMed]
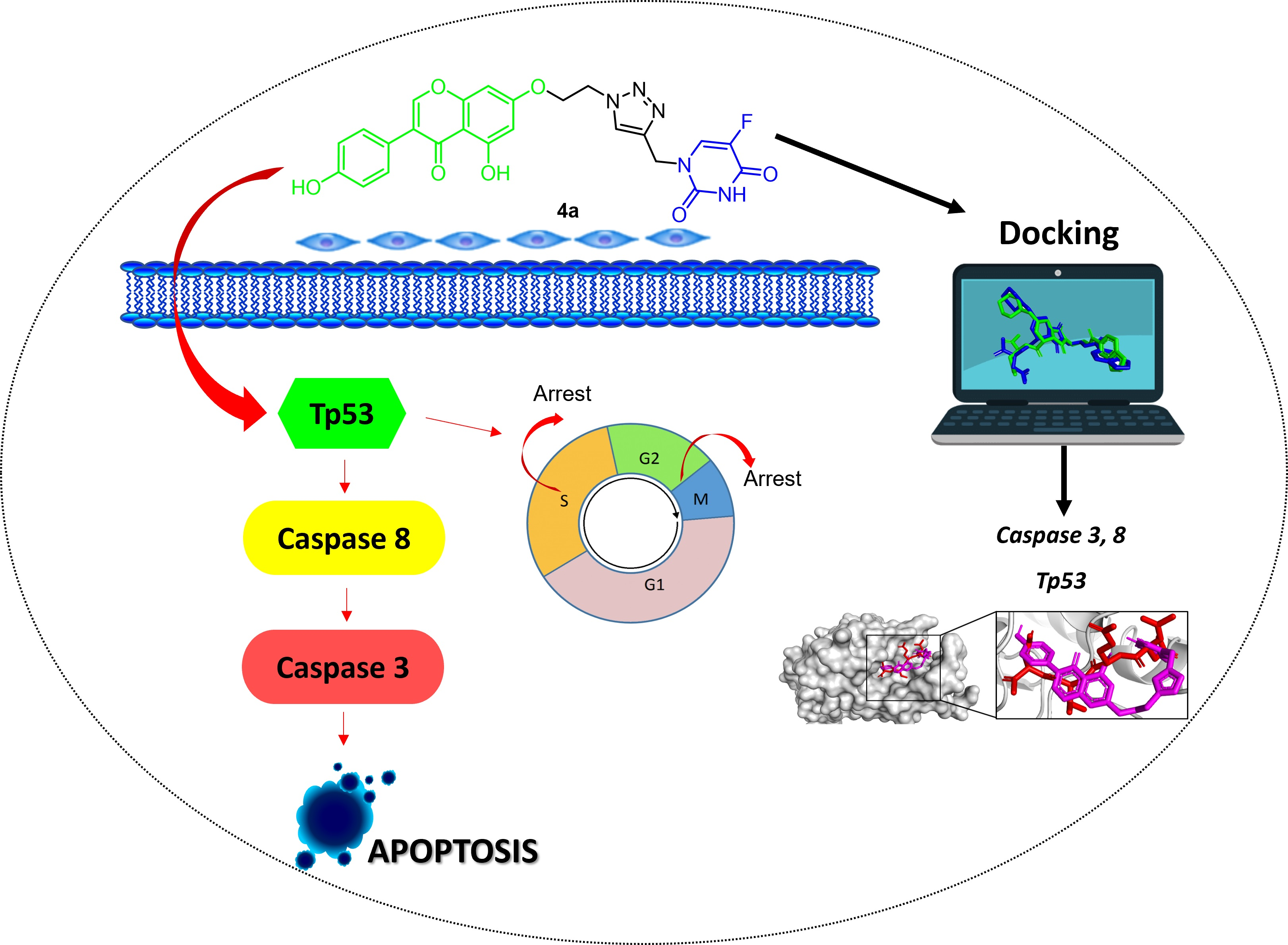
- Cardona-G, W.; Herrera-R, A.; Castrillón-L, W.; Ramírez-Malule, H. Chemistry and Anticancer Activity of Hybrid Molecules and Derivatives Based on 5-Fluorouracil. Curr. Med. Chem. 2021, 51, 5551–5601. [Google Scholar] [CrossRef]
- Dixon, R.A.; Ferreira, D. Genistein. Phytochemistry 2002, 60, 205–211. [Google Scholar] [CrossRef]
- Banerjee, S.; Li, Y.; Wang, Z.; Sarkar, F.H. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269, 226–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Jiang, B.H.; King, S.M.; Chen, Y.C. Inhibition of cell growth and VEGF expression in ovarian cancer cells by flavonoids. Nutr. Cancer 2008, 60, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Amran, D.; de Blas, E.; Aller, P. Regulation of genistein induced differentiation in human acute myeloid leukemia cells (HL60,NB4) Protein kinase modulation and reactive oxygen species generation. Biochem. Pharmacol. 2009, 77, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, C.W.; Jeon, S.Y.; Go, R.E.; Hwang, K.A.; Choi, K.C. Chemopreventive and chemotherapeutic effcts of genistein, a soy isoflvone, upon cancer development and progression in preclinical animal models. Lab. Anim. Res. 2014, 30, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; et al. Genistein and cancer: Current status, challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336–1341. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Pedrosa, M.; Duarte da Cruz, R.M.; de Oliveira Viana, J.; de Moura, R.O.; Ishiki, H.M.; Barbosa Filho, J.M.; Diniz, M.F.; Scotti, M.T.; Scotti, L.; Bezerra Mendonca, F. Hybrid Compounds as Direct Multitarget Ligands: A Review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Song, X.; Liu, R.; Xu, J. Fuplatin: An Efficient and Low-Toxic Dual-Prodrug. J. Med. Chem. 2019, 62, 4543–4554. [Google Scholar] [CrossRef]
- Van Chinh, L.; Ngoc, T.; Thi, N.; Huu, L.; Tien, V. New Chalcones Containing 5-Fluorouracil Exhibiting in vitro Anti-Cancer Activity. Lett. Org. Chem. 2015, 12, 251–261. [Google Scholar] [CrossRef]
- Wang, J.; Fan, X.; Yang, L.; He, H.; Huang, R.; Jiang, F.-L.; Liu, Y. Conjugated 5-Fluorouracil with Mitochondria- Targeting Lipophilic Cation: Design, Synthesis and Biological Evaluation. MedChemComm 2016, 7, 2016–2019. [Google Scholar] [CrossRef]
- Szeja, W.; Grynkiewicz, G.; Bieg, T.; Swierk, P.; Byczek, A.; Papaj, K.; Kitel, R.; Rusin, A. Synthesis and cytotoxicity of 2,3-enopyranosyl C-linked conjugates of genistein. Molecules 2014, 19, 7072–7093. [Google Scholar] [CrossRef] [Green Version]
- Marik, R.; Allu, M.; Anchoori, R.; Stearns, V.; Umbricht, C.B.; Khan, S. Potent genistein derivatives as inhibitors of estrogen receptor alpha-positive breast cancer. Cancer Biol. Ther. 2011, 11, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Gómez-R, L.; Moreno-Q, G.; Herrera-R, A.; Castrillón-L, W.; Yepes, A.F.; Cardona, G.W. New Hybrid Scaffolds Based on ASA/Genistein: Synthesis, Cytotoxic Effect, Molecular Docking, Drug-likeness and in silico ADME/tox Modeling. J. App. Pharm. Sci. 2022, 12, 15–30. [Google Scholar]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Lin, L.P.; Wu, F.H.; Liang, J.Y. The first examples of ilexgenin A hybrids as a new class of multi-potent, anti-platelet agents. Chin. Chem. Lett. 2013, 24, 723–726. [Google Scholar] [CrossRef]
- García, E.; Coa, J.C.; Otero, E.; Carda, M.; Vélez, I.D.; Robledo, S.M.; Cardona, W.I. Synthesis and antiprotozoal activity of furanchalcone–quinoline, furanchalcone–chromone and furanchalcone–imidazole hybrids. Med. Chem. Res. 2018, 27, 497–511. [Google Scholar] [CrossRef]
- Ding, Y.; Li, S.; Ge, W.; Liu, Z.; Zhang, X.; Wang, M.; Chen, T.; Chen, Y.; Zhang, Q. Design and synthesis of parthenolide and 5-fluorouracil conjugates as potential anticancer agents against drug resistant hepatocellular carcinoma. Eur. J. Med. Chem. 2019, 183, 111706. [Google Scholar]
- Pessel, F.; Billault, I.; Scherrmann, M.C. Total synthesis of triazole-linked C-glycosyl flavonoids in alternative solvents and environmental assessment in terms of reaction, workup and purification. Green Chem. 2016, 18, 5558–5568. [Google Scholar] [CrossRef]
- Xu, Y.; Dang, R.; Guan, J.; Xu, Z.; Zhao, S.; Hu, Y. Isatin-(thio)semicarbazide/oxime-1H-1,2,3-triazole-coumarin Hybrids: Design, Synthesis, and in vitro Anti-mycobacterial Evaluation. J. Heterocycl. Chem. 2018, 55, 1069–1073. [Google Scholar] [CrossRef]
- Halay, E.; Ay, E.; Şalva, E.; Ay, K.; Karayıldırım, T. Syntheses of 1,2,3-triazole-bridged pyranose sugars with purine and pyrimidine nucleobases and evaluation of their anticancer potential. Nucleosides Nucleotides Nucleic Acids 2017, 36, 598–619. [Google Scholar]
- Shinde, V.; Mhaske, P.C.; Singh, A.; Sarkar, D.; Mahulikar, P. Synthesis and biological evaluation of new 4-(4-(1-benzyl-1 H-1,2,3- triazol-4-yl)pheny l)-2-phenyl thiazole derivatives. J. Heterocycl. Chem. 2019, 56, 3093–3101. [Google Scholar] [CrossRef]
- Ndreshkjana, B.; Çapci, A.; Klein, V.; Chanvorachote, P.; Muenzner, J.K.; Huebner, K.; Steinmann, S.; Erlenbach-wuensch, K.; Geppert, C.I.; Agaimy, A.; et al. Combination of 5- Fluorouracil and Thymoquinone Targets Stem Cell Gene Signature in Colorectal Cancer Cells. Cell Death Dis. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Kulangara Kandi, S.; Vélez Gerena, C.E.; Zayas, B.; Malhotra, S.V.; Rawat, D.S. C5-curcuminoid-4-aminoquinoline based molecular hybrids: Design, synthesis and mechanistic investigation of anticancer activity. New J. Chem. 2015, 39, 224–234. [Google Scholar] [CrossRef]
- Zhang, Q.; Bao, J.; Yang, J. Genistein-triggered anticancer activity against liver cancer cell line HepG2 involves ROS generation, mitochondrial apoptosis, G2/M cell cycle arrest and inhibition of cell migration. Arch. Med. Sci. 2019, 15, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Polkowski, K.; Popiołkiewicz, J.; Krzeczyński, P.; Ramza, J.; Pucko, W.; Zegrocka-Stendel, O.; Boryski, J.; Skierski, J.S.; Mazurek, A.P.; Grynkiewicz, G. Cytostatic and cytotoxic activity of synthetic genistein glycosides against human cancer cell lines. Cancer Lett. 2004, 203, 59–69. [Google Scholar] [CrossRef]
- Antosiak, A.; Milowska, K.; Maczynska, K.; Rozalska, S.; Gabryelak, T. Cytotoxic activity of genistein-8-C-glucoside form Lupinus luteus L. and genistein against human SK-OV-3 ovarian carcinoma cell line. Med. Chem. Res. 2017, 26, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, G.; Yao, L.; Ruan, K.; Song, G.; Mao, Y.; Bao, S. Genistein induces G2/M cell cycle arrest and apoptosis of human ovarian cancer cells via activation of DNA damage checkpoint pathways. Cell Biol. Int. 2009, 33, 1237–1244. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (∆ψm) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Herrera-R, A.; Naranjo, T.W.; Maldonado, M.E.; Moreno-Q, G.; Yepes, A.; Cardona, G.W. Styrylcoumarin 7-SC2 induces apoptosis in SW480 human colon adenocarcinoma cells and inhibits azoxymethane-induced aberrant crypt foci formation in BALB/c mice. Med. Chem. Res. 2020, 29, 377–395. [Google Scholar] [CrossRef]
- García-Gutiérrez, N.; Maldonado-Celis, M.E.; Rojas-López, M.; Loarca-Piña, G.F.; Campos-Vega, R. The fermented non-digestible fraction of spent coffee grounds induces apoptosis in human colon cancer cells (SW480). J. Funct. Foods 2017, 30, 237–246. [Google Scholar] [CrossRef]
- Yoon, H.; Moon, S.C.; Kim, N.D.; Park, B.S.; Jeong, M.H.; Yoo, Y.H. Genistein induces apoptosis of RPE-J cells by opening mitochondrial PTP. Biochem. Biophys Res. Com. 2000, 276, 151–156. [Google Scholar] [CrossRef]
- Baxa, D.M.; Luo, X.; Yoshimura, F.K. Genistein induces apoptosis in T lymphoma cells via mitochondrial damage. Nutr. Cancer 2005, 51, 93–101. [Google Scholar] [CrossRef]
- Suzuki, R.; Kang, Y.; Li, X.; Roife, D.; Zhang, R.; Fleming, J.B. Genistein potentiates the antitumor effect of 5-Fluorouracil by inducing apoptosis and autophagy in human pancreatic cancer cells. Anticancer. Res. 2014, 34, 4685–4692. [Google Scholar]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Wang, Q.; Wu, H.; Cogswell, J.; Lu, L.; Jhanwar-Uniyal, M.; Dai, W. Reactive Oxygen Species-induced Phosphorylation of p53 on Serine 20 Is Mediated in Part by Polo-like Kinase. J. Biol. Chem. 2001, 276, 36194–36199. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, G.; Wiman, K.G. Reactivation of mutant p53: Molecular mechanisms and therapeutic potential. Oncogene 2007, 26, 2243–2254. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.T.; Ha, J.; Park, O.J. Combination of 5-fluorouracil and genistein induces apoptosis synergistically in chemo-resistant cancer cells through the modulation of AMPK and COX-2 signaling pathways. Biochem. Biophys Res. Commun. 2005, 332, 433–440. [Google Scholar] [CrossRef]
- Wang, Z.; Watt, W.; Brooks, N.A.; Harris, M.S.; Urban, J.; Boatman, D.; McMillan, M.; Kahn, M.; Heinrikson, R.L.; Finzel, B.C.; et al. Kinetic and structural characterization of caspase-3 and caspase-8 inhibition by a novel class of irreversible inhibitors. Biochim. Biophys. Acta 2010, 1804, 1817–1831. [Google Scholar] [CrossRef]
- Li, Q.; Karim, R.M.; Cheng, M.; Das, M.; Chen, L.; Zhang, C.; Lawrence, H.R.; Daughdrill, G.W.; Schonbrunn, E.; Ji, H.; et al. Inhibition of p53 DNA binding by a small molecule protects mice from radiation toxicity. Oncogene 2020, 39, 5187–5200. [Google Scholar] [CrossRef]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Maciag, J.J.; Mackenzie, S.H.; Tucker, M.B.; Schipper, J.L.; Swartz, P.; Clark, A.C. Tunable allosteric library of caspase-3 identifies coupling between conserved water molecules and conformational selection. Proc. Natl. Acad. Sci. USA 2016, 113, E6080–E6088. [Google Scholar] [CrossRef] [Green Version]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Moo-Puc, R.; Robledo, D.; Freile-Pelegrín, Y. In vitro cytotoxic and antiproliferative activities of marine macroalgae from Yucatán, Mexico. Cienc. Mar. 2009, 35, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Ramirez, A.; Yepes-Pérez, A.F.; Quintero-Saumeth, J.; Moreno-Quintero, G.; Naranjo, T.W.; Cardona-Galeano, W. Colorectal Cancer Chemoprevention by S-Allyl Cysteine–Caffeic Acid Hybrids: In vitro Biological Activity and and In Silico Studies. Sci. Pharm. 2022, 90, 40. [Google Scholar] [CrossRef]
- Hernández, C.; Moreno, G.; Herrera-R, A.; Cardona-G, W. New Hybrids Based on Curcumin and Resveratrol: Synthesis, Cytotoxicity and Antiproliferative Activity against Colorectal Cancer Cells. Molecules 2021, 26, 2661. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Herrera-R, A.; Moreno, G.; Araque, P.; Vásquez, I.; Naranjo, E.; Alzate, F.; Cardona-G, W. In vitro Chemopreventive Potential of a Chromone from Bomarea setacea (ALSTROEMERIACEAE) against Colorectal Cancer. Iran. J. Pharm. Res. 2021, 20, 254–267. [Google Scholar]
- Moreno-Q, G.; Herrera-R, A.; Yepes, A.F.; Naranjo, T.W.; Cardona-G, W. Proapoptotic Effect and Molecular Docking Analysis of Curcumin–Resveratrol Hybrids in Colorectal Cancer Chemoprevention. Molecules 2022, 27, 3486. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 3455–3461. [Google Scholar] [CrossRef] [Green Version]
- Yepes, A.F.; Quintero-Saumeth, J.; Cardona-Galeano, W. Biologically Active Quinoline-Hydrazone Conjugates as Potential Trypanosoma cruzi DHFR-TS Inhibitors: Docking, Molecular Dynamics, MM/PBSA and Drug-Likeness Studies. ChemistrySelect 2021, 6, 2928–2938. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 48 h | ||||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | IC50 µM | Selectivity Index (SI) | ||||||
| SW480 | SW620 | HaCaT | CHO-K1 | HaCaT/SW480 | CHO-K1/SW480 | HaCaT/SW620 | CHO-K1/620 | |
| 4a | 62.73 ± 7.26 | 50.58 ± 1.33 | 64.64 ± 1.07 | 71.10 ± 10.51 | 1.03 | 1.13 | 1.28 | 1.41 |
| 4b | >100 | >100 | >100 | >100 | NA | NA | NA | NA |
| 4c | >100 | >100 | >100 | >100 | NA | NA | NA | NA |
| 4d | >100 | >100 | >100 | >100 | NA | NA | NA | NA |
| 4e | >100 | >100 | >100 | >100 | NA | NA | NA | NA |
| 4f | 97.48 ± 5.04 | 47.41 ± 2.19 | 50.02 ± 7.17 | >100 | 0.51 | >1.02 | 1.05 | >2.11 |
| 4g | >100 | 36.84 ± 0.71 | >100 | >100 | NA | NA | >2.71 | >2.71 |
| 4h | >100 | 83.64 ± 8.05 | >100 | >100 | NA | NA | >1.20 | >1.20 |
| Mixture (5-FU Genistein) * | 32.71 ± 3.01 | 37.76 ± 6.19 | 5.89 ± 1.37 | 15.45 ± 1.71 | 0.18 | 0.47 | 0.16 | 0.42 |
| Genistein | 48.13 ± 3.03 | 75.84 ± 5.83 | 45.91 ± 1.97 | 48.44 ± 4.13 | 0.95 | 1.01 | 0.63 | 0.64 |
| 5-FU | 174.30 ± 19.10 | 180.90 ± 18.80 | 118.67 ± 2.84 | 173.20 ± 14.61 | 0.68 | 0.99 | 0.96 | 0.96 |
| Target Protein | Ligands (Docking Score, kcal/mol) | ||||
|---|---|---|---|---|---|
| 4a | Ac-DEVD-CMK a | MMX-9 b | NSC194598 c | SCH529074 d | |
| Caspase-3 | −9.7 −8.7 −7.9 | −8.2 | |||
| Caspase-8 | −8.9 | ||||
| Mutant p53 | −7.3 | −7.0 | |||
| Energy Contribution | Value (kcal.mol−1) |
|---|---|
| ΔGvdw a | −41.29 ±0.09 |
| ΔGElectr b | −8.11 ± 0.03 |
| ΔGPolar c | 30.70 ± 0.06 |
| ΔGSASA d | −4.15 ± 001 |
| ΔGbind e | −22.84 ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Quintero, G.; Castrillón-Lopez, W.; Herrera-Ramirez, A.; Yepes-Pérez, A.F.; Quintero-Saumeth, J.; Cardona-Galeano, W. Synthesis and Chemopreventive Potential of 5-FU/Genistein Hybrids on Colorectal Cancer Cells. Pharmaceuticals 2022, 15, 1299. https://doi.org/10.3390/ph15101299
Moreno-Quintero G, Castrillón-Lopez W, Herrera-Ramirez A, Yepes-Pérez AF, Quintero-Saumeth J, Cardona-Galeano W. Synthesis and Chemopreventive Potential of 5-FU/Genistein Hybrids on Colorectal Cancer Cells. Pharmaceuticals. 2022; 15(10):1299. https://doi.org/10.3390/ph15101299
Chicago/Turabian StyleMoreno-Quintero, Gustavo, Wilson Castrillón-Lopez, Angie Herrera-Ramirez, Andrés F. Yepes-Pérez, Jorge Quintero-Saumeth, and Wilson Cardona-Galeano. 2022. "Synthesis and Chemopreventive Potential of 5-FU/Genistein Hybrids on Colorectal Cancer Cells" Pharmaceuticals 15, no. 10: 1299. https://doi.org/10.3390/ph15101299
APA StyleMoreno-Quintero, G., Castrillón-Lopez, W., Herrera-Ramirez, A., Yepes-Pérez, A. F., Quintero-Saumeth, J., & Cardona-Galeano, W. (2022). Synthesis and Chemopreventive Potential of 5-FU/Genistein Hybrids on Colorectal Cancer Cells. Pharmaceuticals, 15(10), 1299. https://doi.org/10.3390/ph15101299