4. Materials and Methods
Chemistry, General. Unless otherwise mentioned, THF was dried with sodium/benzophenone and was freshly distilled before use. Thin layer chromatography (TLC): silica gel 60 F
254 plates (Merck). Flash chromatography (FC): silica gel 60, 40–63 µm (Macherey-Nagel). Reversed phase thin layer chromatography (RP-TLC): silica gel 60 RP-18 F
254S plates (Merck). Automatic flash column chromatography: Isolera One (Biotage); brackets include eluent, cartridge-type. Melting point (
m,.p.): melting point apparatus SMP 3 (Stuart Scientific), uncorrected.
1H NMR (400 MHz),
1H NMR (600 MHz), and
13C NMR (151 MHz): Agilent DD2 400 and 600 MHz spectrometers; chemical shifts (δ) are reported in ppm against the reference substance tetramethylsilane and calculated using the solvent residual peak of the undeuterated solvent. IR: IR Prestige-21 (Shimadzu). HRMS: MicrOTOF-QII (Bruker). HPLC method to determine the purity of compounds: equipment 1: pump: L-7100, degasser: L-7614, autosampler: L-7200, UV detector: L-7400, interface: D-7000, data transfer: D-line, data acquisition: HSMS software (all from LaChrom, Merck Hitachi); equipment 2: pump: LPG-3400SD, degasser: DG-1210, autosampler: ACC-3000T, UV detector: VWD-3400RS, interface: Dionex UltiMate 3000, data acquisition: Chromeleon 7 (Thermo Fisher Scientific); column: LiChrospher 60 RP-select B (5 μm), LiChroCART 250–4 mm cartridge; flow rate: 1.0 mL/min; injection volume: 5.0 μL; detection at λ = 210 nm; solvents: A: demineralized water with 0.05% (
v/
v) trifluoroacetic acid, B: acetonitrile with 0.05% (
v/
v) trifluoroacetic acid; gradient elution (% A): 0–4 min: 90%; 4–29 min: gradient from 90 to 0%; 29–31 min: 0%; 31–31.5 min: gradient from 0 to 90%; 31.5–40 min: 90% [
14,
17].
General procedure A. Synthesis of β-ketonitriles. If not explicitly described otherwise, under N2, NaH (0.98 eq, 60% mineral oil dispersion) was added to a stirred solution of the respective ester (1.00 eq.) and dry CH3CN (0.98 eq.) in dry THF at rt. The resulting suspension was refluxed for 16 h, allowed to cool to rt, filtrated and washed with Et2O (3×). The product was dried in vacuo to yield the sodium enolate of β-ketonitrile, which was used without further purification in the next step.
General procedure B. Synthesis pyrazol-5-amines. If not explicitly described otherwise, the sodium enolate of the β-ketonitrile (1.00 eq.) was dissolved in aqueous HCl (1M) and stirred at rt for 5 min. The pH was adjusted to 4–5 with aqueous NaOH (2M), and the aqueous phase was extracted with EtOAc (6×). The combined organic layers were dried (Na2SO4), filtered and quickly concentrated in vacuo as decomposition of the β-ketonitrile was observed. To the orange oily residue (if the keto-form of β-ketonitrile was used, 1.00 eq of the keto-form was taken from this step on) was added EtOH and NH2NH2·H2O (2.00–3.00 eq.) and the solution was refluxed for 16 h. After cooling down, the reaction mixture was concentrated in vacuo and the residue was either crystallized or extracted and purified by flash column chromatography.
General procedure C. Reductive amination with pyrazol-5-amines. If not explicitly described otherwise, molecular sieve (3Å), the respective aldehyde (1.50 eq.) and AcOH (1.00 eq.) were added to a solution of the respective aminopyrazole (1.00 eq.) in dry EtOH. The reaction mixture was stirred at rt for 24 h. At 0 °C, NaBH4 (6.00 eq.) was added, and the reaction mixture was stirred at rt for 16 h and then quenched with H2O. The suspension was filtered over Celite® and washed with CH3OH (1×), EtOAc (2×), aqueous HCl (4M) and H2O (1×). The organic layer was washed with aqueous HCl (4M, 3×) and the combined aqueous layers were neutralized using aqueous KOH (8M). The aqueous layer was extracted with EtOAc (5×), the combined organic layers were dried (Na2SO4), filtered, concentrated in vacuo, and the residue was purified by flash column chromatography.
General procedure D. Einhorn acylation using acid chlorides. If not explicitly described otherwise, respective aminotriazole or aminopyrazole (1.00 eq.) was suspended in dry pyridine/dry THF mixture at 0 °C. A solution of respective acid chloride (1.00–1.40 eq.) in dry THF (1 mL) was added to this suspension dropwise via a syringe pump (1 mL/h) and afterwards, the suspension was stirred for 1 h at rt. The reaction mixture was quenched with H2O. If a precipitate was formed, it was filtered off, washed with H2O (3×) and dried in vacuo. Otherwise, the aqueous layer was extracted with EtOAc (3×), the combined organic layers were dried (N2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography.
General procedure E. Under N2, an azole exhibiting the 2-iodobenzoyl moiety (1.00 eq.), CuI (0.20 eq.), Cs2CO3 (2.00 eq.) and 1,10-phenanthroline (0.20 eq.) were dissolved in dry DMF. The reaction mixture was stirred at 80 °C until TLC analysis indicated the complete consumption of the azole bearing 2-iodobenzoyl moiety. After cooling, the suspension was diluted with H2O (30 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography.
General procedure F. If not explicitly described otherwise, the respective acylated 1,2,4-triazol-5-amine (1.00 eq.), CuI (0.20 eq.), Cs2CO3 (2.00 eq.) and 1,10-phenanthroline (0.20 eq.) were dissolved in dry DMF (2 mL) at room temperature. The reaction mixture was flushed with N2 and was subjected to microwave irradiation (150 °C, 1 h, max 300 W). After the heated mixture was cooled to room temperature, the solvent was evaporated under reduced pressure. The crude product was purified by flash column chromatography (CH2Cl2/CH3OH = 97/3 or 100/0 → 90/10).
sodium 2-cyano-1-(pyridin-3-yl)ethen-1-olate (6a). According to general procedure A, NaH (2.59 g, 64.8 mmol), ester 5a (9.01 mL, 66.2 mmol) and dry CH3CN (3.39 mL, 64.8 mmol) were reacted in dry THF (110 mL) yielding sodium enolate 6a as a beige solid (9.84 g, 58.5 mmol, 88%). M.p.: >300 °C. TLC: Rf = 0.37 (CH2Cl2/CH3OH = 95/5). 1HNMR (600 MHz, DMSO-d6): δ (ppm) = 4.03 (s, 1H, CH), 7.27 (ddd, J = 7.8/4.7/0.9 Hz, 1H, 5-Hpyridyl), 7.93 (dt, J = 7.9/2.0 Hz, 1H, 4-Hpyridyl), 8.43 (dd, J = 4.7/1.7 Hz, 1H, 6-Hpyridyl), 8.78 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl). The signals of the major isomer are given. 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 52.7 (1C, CH), 122.8 (1C, C-5pyridyl), 126.8 (1C, CN), 133.0 (1C, C-4pyridyl), 137.6 (1C, C-3pyidyl), 147.3 (1C, C-2pyridyl), 148.8 (1C, C-6pyridyl), 177.4 (1C, CONa). The signals of the major isomer are given. IR (neat): ṽ [cm−1] = 2167, 1593, 1581, 1527, 1442, 1408, 1091, 1026, 1010, 821, 717, 694. HRMS (APCI): m/z = 147.0553 calculated for [C8H6N2O+H]+, found: 147.0553.
sodium 2-cyano-1-phenylethen-1-olate (6b). According to general procedure A, NaH (261 mg, 6.53 mmol), ester 5b (952 μL, 6.66 mmol) and dry CH3CN (341 μL, 6.53 mmol) were reacted in dry THF (11 mL) yielding sodium enolate 6b as a colorless solid (837 mg, 5.01 mmol, 75%). M.p.: 266–268 °C (decomp.). TLC: Rf = 0.37 (CH/EtOAc = 60/40). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 3.96 (s, 1H, CH), 7.22–7.25 (m, 3H, 2/4/6-Hphenyl), 7.58–7.62 (m, 2H, 3/5-Hphenyl). The signals of the major isomer are given. 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 51.5 (1C, CH), 125.6 (2C, C-3/5phenyl), 127.3 (2C, C-2/6phenyl), 127.6 (1C, CN), 127.9 (1C, C-4phenyl), 143.1 (1C, C-1phenyl), 179.9 (1C, CONa). The signals of the major isomer are given. IR (neat): ṽ [cm−1] = 3271, 3213, 2164, 1550, 1508, 1481, 1442, 1404, 1226, 1002, 879, 694. HRMS (APCI): m/z = 146.0600 calculated for [C9H7NO+H]+, found: 146.0611.
sodium 2-cyano-1-cyclohexylethen-1-olate (6c). According to general procedure A, NaH (2.76 g, 68.9 mmol), ester 5c (10.0 g, 70.3 mmol) and dry CH3CN (3.6 mL, 68.9 mmol) were reacted in dry THF (120 mL) yielding sodium enolate 6c as a colorless solid (8.82 g, 72%). %). M.p.: >300 °C. TLC: Rf = 0.29 (EtOAc/CH = 20/80). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 1.81–1.70 (m, 1H, Hcyclohexyl); 1.68–1.50 (m, 4H, Hcyclohexyl); 1.36–1.00 (m, 6H, Hcyclohexyl), the signal of Henolate cannot be seen on the spectrum. 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.9 (1C, C-ONa); 128.2 (1C, C≡N); 46.0 (1C, Ccyclohexyl); 30.3 (2C, Ccyclohexyl); 26.2 (1C, Cyclohexyl); 25.9 (2C, Ccyclohexyl), the signal of Cenolate cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 2926, 2851, 2166, 1560, 1537, 1501, 1443, 1414, 1356, 1281, 945, 773, 712. HRMS (APCI): m/z = 152.1074, calculated for C9H14NO+ [M + H]+ 152.1070.
3-(pyridin-3-yl)-1H-pyrazol-5-amine (7a). According to general procedure B, β-ketonitrile sodium enolate 6a (8.00 g, 47.2 mmol, 1.00 eq.) was dissolved in aqueous HCl (120 mL, 1M), followed by work-up as described above. The orange oily residue of the keto-form of β-ketonitrile 6a and NH2NH2·H2O (6.94 mL, 143 mmol, 3.00 eq.) were reacted in EtOH (87 mL). Then, the obtained residue was crystallized using EtOH and cyclohexane. The precipitate was collected by filtration, washed with cold EtOH (2×), and dried in vacuo yielding 4.68 g of 7a, that was used without further purification. The filtrate was concentrated in vacuo and purified by flash column chromatography (CH2Cl2/CH3OH = 1/0 → 90/10) yielding 1.00 g of 7a (combined 5.68 g, 35.5 mmol, 75%) as a beige solid. M.p.: 145 °C. TLC: Rf = 0.30 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.96 (bs, 2H, NH2), 5.82 (bs, 1H, 4-Hpyrazolyl), 7.38 (dd, J = 7.9/4.8 Hz, 1H, 5-Hpyridyl), 8.01 (dt, J = 8.0/2.0 Hz, 1H, 4-Hpyridyl), 8.45 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl), 8.88 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl), 11.81 (bs, 1H, NH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 123.7 (1C, C-5pyridyl), 131.8 (1C, C-4pyridyl), 146.0 (1C, C-2pyridyl), 148.1 (1C, C-6pyridyl). The signals for C-3pyridyl and C-3/4/5pyrazolyl are not seen in the spectrum. IR (neat): ṽ [cm−1] = 3390, 3290, 3194, 3116, 3047, 2858, 1631, 1612, 1573, 1516, 1469, 1192, 995, 956, 810, 771, 736, 702, 671. HRMS (APCI): m/z = 161.0822 calculated for [M + H]+, found: 161.0841. HPLC: tR = 3.3 min, purity: 99.8%.
3-phenyl-1H-pyrazol-5-amine (7b). According to general procedure B, β-ketonitrile keto-form 6b (500 mg, 3.44 mmol) and NH2NH2·H2O (345 µL, 6.89 mmol, 2.00 eq.) were reacted in EtOH (6.20 mL). The reaction mixture was quenched with H2O, the aqueous phase was extracted with EtOAc (3×), the combined organic layers were dried (Na2SO4), filtrated and concentrated in vacuo. The residue was purified by flash column chromatography (CH2Cl2/CH3OH = 1/0 → 91/9) yielded 7b as a yellow solid (515 mg, 3.24 mmol, 94%). M.p.: 128 °C. TLC: Rf = 0.14 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.70 (bs, 2H, NH2), 5.74 (bs, 1H, 4-Hpyrazolyl), 7.19–7.29 (m, 1H, 4-Hphenyl), 7.32–7.40 (m, 2H, 3/5-Hphenyl), 7.61–7.69 (m, 2H, 2/6-Hphenyl), 11.68 (bs, 1H, NH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 124.7 (2C, C-2/6phenyl), 127.2 (1C, C-4phenyl), 128.6 (2C, C-3/5phenyl). The signals for C-1phenyl and C-3/4/5pyrazolyl are not seen in the spectrum. IR (neat): ṽ [cm−1] = 3398, 3294, 3197, 1620, 1562, 1504, 1465, 1095, 1076, 1056, 995, 956, 918, 759, 694. HRMS (APCI): m/z = 160.0869 calculated for [M + H]+, found: 160.0874. HPLC: tR = 11.1 min, purity: 99.7%.
3-cyclohexyl-1H-pyrazol-5-amine (7c). According to general procedure B, β-ketonitrile sodium enolate 6c (6.92 g, 40 mmol, 1.00 eq.) was dissolved in aqueous HCl (120 mL, 1M), followed by work-up as described above. The residue of the keto-form of β-ketonitrile 6c and NH2NH2·H2O (4.0 mL, 79.9 mmol, 2.00 eq.) were reacted in EtOH (80 mL). The product was purified by flash column chromatography (CH2Cl2/CH3OH = 1/0 → 90/10) yielding 7b as a reddish oil (5.01 g, 76%). TLC: Rf = 0.17 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 11.02 (bs, 1H, NHpyrazolyl); 5.15 (s, 1H, 4-Hpyrazolyl); 4.39 (bs, 2H, NH2); 2.21 (tdt, J = 12.0/8.4/4.2 Hz, 1H, Hcyclohexyl); 1.88–1.75 (m, 2H, Hcyclohexyl); 1.75–1.66 (m, 2H, Hcyclohexyl); 1.66–1.60 (m, 1H, Hcyclohexyl); 1.35–1.23 (m, 4H, Hcyclohexyl); 1.22–1.10 (m, 1H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 153.6 (1C, C-5pyrazolyl); 149.9 (1C, C-3pyrazolyl); 87.9 (1C, C-4pyrazolyl); 35.2 (1C, Ccyclohexyl); 32.3 (2C, Ccyclohexyl); 25.6 (1C, Cyclohexyl); 25.5 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3206, 2922, 2851, 1684, 1574, 1485, 1447, 1373, 1001, 986, 891, 758. HRMS (APCI): m/z = 166.1328, calculated for C9H16N3+ [M + H]+ 166.1339. HPLC: tR = 12.9 min, purity: 96.2%.
N-benzyl-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8a). According to general procedure C, aminopyrazole 7a (100 mg, 624 µmol), benzaldehyde (95.6 µL, 936 µmol) and AcOH (35.7 µL, 624 µmol) were reacted in EtOH (3.0 mL). After reduction with NaBH4 (142 mg, 3.75 mmol) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 90/10) 8a was obtained as a colorless solid (137 mg, 547 µmol, 88%). M.p.: 135–136 °C. TLC: Rf = 0.30 (CH2Cl2/CH3OH = 93/7). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.27 (d, J = 6.4 Hz, 2H, CH2), 5.82 (bs, 2 × 0.50H, 4-Hpyrazolyl, NHCH2), 6.03 (bs, 0.50H, 4-Hpyrazolyl), 6.21 (bs, 0.5H, NHCH2), 7.17–7.25 (m, 1H, 4-Hphenyl), 7.27–7.34 (m, 2H, 3/5-Hphenyl), 7.35–7.45 (m, 3H, 2/6-Hphenyl, 5-Hpyridyl), 8.00 (d, J = 8.1 Hz, 1H, 4-Hpyridyl), 8.39–8.50 (m, 1H, 6-Hpyridyl), 8.84–8.89 (m, 1H, 2-Hpyridyl), 11.88 (bs, 0.50H, NH), 12.14 (bs, 0.50H, NH). The ratio of tautomers is 1:1. 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 48.0 (1C, CH2), 83.5 (0.5C, C-4pyrazolyl), 88.9 (0.5C, C-4pyrazolyl), 123.8 (1C, C-5pyridyl), 126.8 (1C, C-4phenyl), 127.4 (2C, C-2/6phenyl), 128.1 (2C, C-3/5phenyl), 131.8 (1C, C-4pyridyl), 139.1 (1C, C-3pyridyl), 140.0 (0.5C, C-1phenyl), 141.3 (0.5C, C-1phenyl), 146.0 (1C, C-2pyridyl), 148.5 (1C, C-6pyridyl), 157.8 (1C, C-5pyrazolyl). The signals of the major tautomer are given, except of C-4pyrazolyl. The signal for C-3pyrazolyl is not seen in the spectrum. IR (neat): ṽ [cm−1] = 3209, 3132, 3062, 2943, 2839, 2719, 1585, 1573, 1446, 1361, 1219, 1141, 1118, 1029, 956, 810, 736, 702. HRMS (APCI): m/z = 251.1291 calculated for [M + H]+, found: 251.1313. HPLC: tR = 13.2 min, purity: 99.6%.
N-(4-methoxybenzyl)-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8b). According to general procedure C, aminopyrazole 7a (600 mg, 3.75 mmol, 1.00 eq.), 4-methoxybenzaldehyde (683 µL, 5.62 mmol) and AcOH (214 µL, 3.75 mmol) were reacted in EtOH (19.0 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 94/6 → 90/10) 8b was obtained as a beige solid (731 mg, 70%). M.p.: 134 °C. TLC: Rf = 0.33 (CH2Cl2/CH3OH = 94/6). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 11.98 (bs, 1H, NHpyrazolyl); 8.87 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.45 (dd, J = 4.7/1.6 Hz, 1H, 6-Hpyridyl); 8.00 (ddd, J = 7.9/2.3/1.7 Hz, 1H, 4-Hpyridyl); 7.39 (dd, J = 8.0/4.8 Hz, 1H, 5-Hpyridyl); 7.33–7.25 (m, 2H, 2/6-Hmethoxyphenyl); 6.92–6.83 (m, 2H, 3/5-Hmethoxyphenyl); 5.91 (s, 1H, 4-Hpyrazolyl); 4.18 (d, J = 6.3 Hz, 2H, CH2); 3.72 (s, 3H, CH3), the signal of NH cannot be seen on the spectrum. 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 158.0 (1C, C-4methoxyphenyl); 148.1 (1C, C-6pyridyl); 146.0 (1C, C-2pyridyl); 132.3 (1C, C-1methoxyphenyl); 131.8 (1C, C-4pyridyl); 130.5 (1C, C-3pyridyl); 128.6 (2C, C-2/6methoxyphenyl); 123.6 (1C, C-5pyridyl); 113.5 (2C, C-3/5methoxyphenyl); 54.9 (1C, CH3); 47.2 (1C, CH2), the signals of C-3pyridyl, C-3pyrazolyl and C-5pyrazolyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3240, 3213, 3132, 2835, 1603, 1574, 1514, 1462, 1441, 1398, 1302, 1244, 1180, 1118, 1084, 1030, 959, 864, 824, 804, 752. HRMS (APCI): m/z = 281.1380, calculated for C16H17N4O+ [M + H]+ 281.1397. HPLC: tR = 13.4 min, purity: 99.7%.
N-(naphthalen-1-ylmethyl)-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8c). According to general procedure C, aminopyrazole 7a (600 mg, 3.75 mmol, 1.00 eq.), 1-naphthaldehyde (763 µL, 5.62 mmol) and AcOH (214 µL, 3.75 mmol) were reacted in EtOH (19.0 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 100/0 → 90/10) 8c was obtained as a beige solid (772 mg, 69%). M.p.: 78 °C. TLC: Rf = 0.29 (CH2Cl2/CH3OH = 94/6). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 12.05 (bs, 1H, NHpyrazolyl); 8.89 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.46 (dd, J = 4.8/1.6 Hz, 1H, 6-Hpyridyl); 8.18 (d, J = 8.3 Hz, 1H, 9-Hnaphthyl); 8.02 (ddd, J = 8.0/2.3/1.6 Hz, 1H, 4-Hpyridyl); 7.96–7.92 (m, 1H, 6-Hnaphthyl); 7.83 (d, J = 8.2 Hz, 1H, 4-Hnaphthyl); 7.60–7.55 (m, 2H, 7/8-Hnaphthyl); 7.55–7.52 (m, 1H, 2-Hnaphthyl); 7.46 (dd, J = 8.2/7.0 Hz, 1H, 3-Hnaphthyl); 7.40 (ddd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl); 6.00 (s, 1H, 4-Hpyrazolyl); 4.74 (d, J = 5.9 Hz, 2H, CH2), the signal of NH cannot be seen on the spectrum. 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 146.0 (1C, C-2pyridyl); 131.8 (1C, C-4pyridyl); 133.3 (1C, C-10naphthyl); 131.1 (1C, C-5naphthyl); 128.4 (1C, C-6naphthyl); 127.2 (1C, C-4naphthyl); 125.9 (1C, C-7naphthyl); 125.6 (1C, C-2naphthyl); 125.3 (1C, C-3naphthyl); 125.1 (1C, C-8naphthyl); 123.7 (1C, C-5pyridyl); 123.6 (1C, C-9naphthyl); 45.5 (1C, CH2), the signals of C-1naphthyl, C-4naphthyl, C-6pyridyl, C-3pyridyl, C-3pyrazolyl and C-5pyrazolyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 2980, 2970, 2887, 1597, 1566, 1546, 1512, 1503, 1462, 1441, 1382, 1261, 1163, 1086, 1072, 1024, 955, 791, 752, 706. HRMS (APCI): m/z = 301.1477, calculated for C19H17N4+ [M + H]+ 301.1448. HPLC: tR = 15.9 min, purity: 99.3%.
N-(furan-2-ylmethyl)-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8d). According to general procedure C, aminopyrazole 7a (600 mg, 3.75 mmol, 1.00 eq.), furaldehyde (465 µL, 5.62 mmol) and AcOH (214 µL, 3.75 mmol) were reacted in EtOH (19.0 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 94/6) 8d was obtained as a beige solid (687 mg, 76%). M.p.: 170 °C. TLC: Rf = 0.34 (CH2Cl2/CH3OH = 94/6). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 12.06 (bs, 1H, NHpyrazolyl); 8.89 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.46 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 8.02 (ddd, J = 7.9/2.3/1.7 Hz, 1H, 4-Hpyridyl); 7.56 (dd, J = 1.9/0.9 Hz, 1H, 5-Hfuranyl); 7.40 (dd, J = 8.0/4.8 Hz, 1H, 5-Hpyridyl); 6.37 (dd, J = 3.2/1.9 Hz, 1H, 4-Hfuranyl); 6.30 (dd, J = 3.2 Hz, 1H, 3-Hfuranyl); 5.98 (s, 1H, 4-Hpyrazolyl); 5.89 (s, 1H, NH); 4.24 (dd, J = 6.3/0.9 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 153.6 (1C, C-2furanyl); 148.2 (1C, C-6pyridyl); 146.0 (1C, C-2pyridyl); 141.7 (1C, C-5furanyl); 131.8 (1C, C-4pyridyl); 123.7 (1C, C-5pyridyl); 110.3 (1C, C-4furanyl); 106.7 (1C, C-3furanyl); 41.0 (1C, CH2), the signals of C-3pyridyl, C-3pyrazolyl, C-4pyrazolyl and C-5pyrazolyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3194, 1601, 1587, 1573, 1503, 1445, 1352, 1188, 1118, 1080, 1032, 957, 916, 881, 810, 758, 741, 723. HRMS (APCI): m/z = 241.1086, calculated for C13H13N4O+ [M + H]+ 241.1084. HPLC: tR = 11.1 min, purity: 99.1%.
N-[(5-chlorothiophen-2-yl)methyl]-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8e). According to general procedure C, aminopyrazole 7a (100 mg, 624 µmol), 5-chlorothiophene-2-carbaldehyde (99.5 µL, 936 µmol) and AcOH (35.7 µL, 624 µmol) were reacted in EtOH (3.0 mL). After reduction with NaBH4 (142 mg, 3.75 mmol) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 90/10) 8e was obtained as a colorless solid (142 mg, 488 µmol, 78%). M.p.: 158–159 °C. TLC: Rf = 0.32 (CH2Cl2/CH3OH = 93/7). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.37 (d, J = 6.4 Hz, 2H, CH2), 5.96 (bs, 0.7H, NHCH2), 6.04 (bs, 1H, 4-Hpyrazolyl), 6.28 (bs, 0.3H, NHCH2*), 6.84–6.91 (m, 1H, 4-Hthiophenyl), 6.91–6.97 (m, 1H, 3-Hthiophenyl), 7.35–7.45 (m, 1H, 5-Hpyridyl), 8.02 (dt, J = 8.0/2.0 Hz, 1H, 4-Hpyridyl), 8.43–8.52 (m, 1H, 6-Hpyridyl), 8.89 (d, J = 2.4 Hz, 1H, 2-Hpyridyl), 11.97 (bs, 0.3H, NH*), 12.26 (bs, 0.7H, NH). The ratio of tautomers is 7:3, the minor tautomer is marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 43.0 (1C, CH2), 89.2 (1C, C-4pyrazolyl), 123.8 (1C, C-5pyridyl), 124.4 (1C, C-4thiophenyl), 126.1 (1C, C-3thiophenyl), 131.9 (1C, C-4pyridyl), 139.4 (1C, C-3pyridyl), 146.0 (1C, C-2thiophenyl), 146.0 (1C, C-2pyridyl), 148.5 (1C, C-6pyridyl), 157.0 (1C, C-5pyrazolyl). The signals of the major tautomer are given. The signals for C-3pyrazolyl and C-5thiophenyl are seen in the spectrum. IR (neat): ṽ [cm−1] = 3278, 3059, 2816, 1562, 1535, 1489, 1450, 1419, 1354, 1099, 991, 956, 786, 756, 702, 632. HRMS (APCI): m/z = 291.0466 calculated for [M + H]+, found: 291.0463. HPLC: tR = 14.6 min, purity: 99.2%.
N-(cyclohexylmethyl)-3-(pyridin-3-yl)-1H-pyrazol-5-amine (8f). According to general procedure C, aminopyrazole 7a (250 mg, 1.56 mmol) and cyclohexane carbaldehyde (284 µL, 2.34 mmol) were reacted in EtOH (7.50 mL). No AcOH was added. After reduction with NaBH4 (354 mg, 9.36 mmol) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 9/1) 8f was obtained as a colorless solid (294 mg, 1.15 mmol, 73%). M.p.: 124 °C. TLC: Rf = 0.26 (CH2Cl2/CH3OH = 93/7). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 0.86-0.95 (m, 2H, 2/6-Hcyclohexyl), 1.10–1.24 (m, 3H, 3/4/5-Hcyclohexyl), 1.48–1.56 (m, 1H, 1-Hcyclohexyl), 1.59–1.64 (m, 1H, 4-Hcyclohexyl), 1.65–1.71 (m, 2H, 3/5-Hcyclohexyl), 1.74–1.79 (m, 2H, 2/6-Hcyclohexyl), 2.88 (t, J = 6.5 Hz, 1H, CH2), 5.17 (bs, 0.4H, NHCH2*), 5.61 (bs, 0.6H, NHCH2), 5.77 (bs, 0.6H, 4-Hpyrazolyl), 5.97 (bs, 0.4H, 4-Hpyrazolyl*), 7.32–7.46 (m, 1H, 5-Hpyridyl), 8.02 (d, J = 7.9 Hz, 1H, 4-Hpyridyl), 8.40–8.49 (m, 1H, 6-Hpyridyl), 8.84–8.94 (m, 1H, 2-Hpyridyl), 11.66 (bs, 0.6H, NH), 12.06 (bs, 0.4H, NH*). The ratio of tautomers is 6:4, the minor tautomer is marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 25.5 (2C, C-3/5cyclohexyl), 26.2 (1C, C-4cyclohexyl), 30.6 (2C, C-2/6cyclohexyl), 37.3 (1C, C-1cyclohexyl), 50.9 (1C, CH2), 82.7 (0.6C, C-4pyrazolyl), 88.5 (0.4C, C-4pyrazolyl), 123.6 (1C, C-5pyridyl), 131.8 (1C, C-5pyridyl), 146.0 (1C, C-5pyridyl), 147.9 (1C, C-6pyridyl), 151.4 (1C, C-3pyrazolyl), 158.4 (1C, C-5pyrazolyl). The signals of the major tautomer are given, except of C-4pyrazolyl. IR (neat): ṽ [cm−1] = 2920, 2843, 1593, 1442, 1188, 1145, 1118, 1029, 960, 806, 736, 705. HRMS (APCI): m/z = 257.1761 calculated for [M + H]+, found: 257.1746. HPLC: tR = 15.4 min, purity: 98.1%.
N-[(5-chlorothiophen-2-yl)methyl]-3-phenyl-1H-pyrazol-5-amine (8g). According to general procedure C, aminopyrazole 7b (200 mg, 1.26 mmol, 1.00 eq.), 5-chlorothiophene-2-carbaldehyde (200 µL, 1.88 mmol) and AcOH (72 µL, 1.26 mmol) were reacted in EtOH (6.3 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 9/1) 8g was obtained as a beige solid (288 mg, 79%). M.p.: 84–85 °C. TLC: Rf = 0.48 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 12.01 (bs, 1H, NHpyrazolyl); 7.64 (m, 2H, Hphenyl); 7.39 (m, 2H, Hphenyl); 7.29 (m, 1H, Hphenyl); 6.92 (d, J = 3.7 Hz, 1H, 3-Hchlorothiophenyl); 6.89 (d, J = 3.7 Hz, 1H, 4-Hchlorothiophenyl); 5.93 (bs, 1H, NH); 5.90 (bs, 1H, 4-Hpyrazolyl); 4.37 (d, J = 5.5 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 144.3 (1C, C-2chlorothiophenyl); 128.6 (2C, Cphenyl); 127.5 (1C, Cphenyl); 126.1 (1C, C-4chlorothiophenyl); 124.6 (2C, Cphenyl); 124.4 (1C, C-3chlorothiophenyl); 87.4 (1C, C-4pyrazolyl); 43.1 (1C, CH2), the signals of C-5chlorothiophenyl, C-3pyridyl, C-3pyrazolyl and C-5pyrazolyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3671, 2361, 1690, 1599, 1570, 1539, 1489, 1460, 1435, 1184, 1061, 1007, 930, 853, 746. HRMS (APCI): m/z = 290.0469, calculated for C14H13ClN3S+ [M + H]+ 290.0513. HPLC: tR = 18.2 min, purity: 96.7%.
N-benzyl-3-cyclohexyl-1H-pyrazol-5-amine (8h). According to general procedure C, aminopyrazole 7c (500 mg, 3.03 mmol), benzaldehyde (200 µL, 4.54 mmol) and AcOH (460 µL, 3.03 mmol) were reacted in EtOH (15 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 9/1) 8h was obtained as a beige oil (297 mg, 38%). TLC: Rf = 0.20 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 7.36–7.32 (m, 2H, 2/6-Hphenyl); 7.31–7.26 (m, 2H, 3/5-Hphenyl); 7.22–7.16 (m, 1H, 4-Hphenyl); 5.25 (s, 1H, 4-Hpyrazolyl); 4.19 (s, 2H, CH2); 2.44 (dd, J = 8.9/5.5 Hz, 1H, Hcyclohexyl); 1.89–1.79 (m, 2H, Hcyclohexyl); 1.78–1.68 (m, 2H, Hcyclohexyl); 1.67–1.59 (m, 1H, Hcyclohexyl); 1.34–1.22 (m, 4H, Hcyclohexyl); 1.22–1.09 (m, 1H, Hcyclohexyl), the signals of NHpyrazolyl and NH cannot be seen on the spectrum. 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 141.6 (1C, C-1phenyl); 128.5 (2C, C-3/5phenyl); 127.8 (2C, C-2/6phenyl); 126.8 (1C, C-4phenyl); 87.5 (1C, C-4pyrazolyl); 48.1 (2C, CH2), 35.6 (1C, Ccyclohexyl); 32.8 (2C, Ccyclohexyl); 26.1 (1C, Ccyclohexyl); 26.0 (2C, Ccyclohexyl), the signals of C-3pyrazolyl, C-5pyrazolyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3225, 2924, 2851, 2361, 1738, 1628, 1574, 1495, 1449, 1354, 1117, 986, 891, 733. HRMS (APCI): m/z = 256.1832, calculated for C16H22N3+ [M + H]+ 256.1808.
N-[(5-chlorothiophen-2-yl)methyl]-3-cyclohexyl-1H-pyrazol-5-amine (8i). According to general procedure C, aminopyrazole 7c (100 mg, 605 µmol), 5-chlorothiophene-2-carbaldehyde (95 µL, 908 µmol) and AcOH (35 µL, 605 µmol) were reacted in EtOH (3 mL). After reduction with NaBH4 (6.00 eq.) and flash column chromatography (CH2Cl2/CH3OH = 1/0 → 9/1) 8i was obtained as a yellowish oil (77 mg, 43%). TLC: Rf = 0.28 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 6.90 (d, J = 3.7 Hz, 1H, 4-Hchlorothiophenyl); 6.83 (d, J = 3.7 Hz, 1H, 3-Hchlorothiophenyl); 5.59 (s, 1H, NH); 5.25 (s, 1H, 4-Hpyrazolyl); 4.28 (d, J = 6.0 Hz, 2H, CH2); 2.45 (m, 1H, Hcyclohexyl); 2.48–2.41 (m, 1H, Hcyclohexyl); 1.89–1.80 (m, 2H, Hcyclohexyl); 1.771.68 (m, 2H, Hcyclohexyl); 1.68–1.60 (m, 1H, Hcyclohexyl); 1.34–1.25 (m, 4H, Hcyclohexyl); 1.24–1.13 (m, 1H, Hcyclohexyl), the signal of NHpyrazolyl cannot be seen on the spectrum. 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 43.6 (1C, CH2); 35.5 (1C, Ccyclohexyl); 32.8 (2C, Ccyclohexyl); 26.1 (1C, Ccyclohexyl); 26.0 (2C, Ccyclohexyl), the signals of C-3pyrazolyl, C-4pyrazolyl, C-5pyrazolyl, C-2chlorothiophenyl, C-3chlorothiophenyl, C-4chlorothiophenyl, C-5chlorothiophenyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3227, 2924, 2851, 1738, 1574, 1520, 1506, 1449, 1206, 1115, 1059, 989, 891, 789. HRMS (APCI): m/z = 296.0982, calculated for C14H19ClN3S+ [M + H]+ 296.0983.
[5-(benzylamino)-3-(pyridin-3-yl)-1H-pyrazol-1-yl](2-iodophenyl)methanone (9a). According to general procedure D, aminopyrazole 8a (409 mg, 1.63 mmol) was acylated using 2-iodobenzoyl chloride (522 mg, 1.96 mmol) in dry pyridine/THF (18 mL/6 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9a as a yellowish solid (505 mg, 64%). M.p.: 115 °C. TLC: Rf = 0.73 (CH/EtOAc = 1/1). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.81 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.54 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 7.96–7.94 (m, 1H, 3-Hbenzoyl); 7.94–7.92 (m, 1H, 4-Hpyridyl); 7.92–7.89 (m, 1H, NH); 7.59 (dd, J = 7.6/1.7 Hz, 1H, 6-Hbenzoyl); 7.54 (td, J = 7.5/1.1 Hz, 1H, 5-Hbenzoyl); 7.50–7.47 (m, 2H, 2/6-Hphenyl); 7.41–7.39 (m, 1H, 5-Hpyridyl); 7.37 (dd, J = 8.3/7.1 Hz, 2H, 3/5-Hphenyl); 7.31–7.28 (m, 1H, 4-Hphenyl); 7.27 (dt, J = 8.6/1.5 Hz, 1H, 4-Hbenzoyl); 6.10 (s, 1H, 4-Hpyrazolyl); 4.51 (d, J = 6.1 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.5 (1C, C=O); 153.3 (1C, C-5pyrazolyl); 152.3 (1C, C-3pyrazolyl); 150.0 (1C, C-6pyridyl); 147.0 (1C, C-2pyridyl); 140.6 (1C, C-1benzoyl); 138.6 (1C, C-1phenyl); 138.4 (1C, C-3benzoyl); 133.0 (1C, C-4pyridyl); 131.4 (1C, C-4phenyl); 128.7 (1C, C-6benzoyl); 128.4 (2C, C-3/5phenyl); 127.6 (1C, C-5benzoyl); 127.5 (1C, C-3pyridyl); 127.3 (2C, C-2/6phenyl); 127.0 (1C, C-4benzoyl); 123.7 (1C, C-5pyridyl); 93.6 (1C, C-2benzoyl); 83.8 (1C, C-4pyrazolyl); 47.8 (1C, CH2). IR (neat): ṽ [cm−1] = 3385, 1684, 1607, 1574, 1526, 1495, 1449, 1429, 1366, 1020, 937, 924, 727. HRMS (APCI): m/z = 481.0511, calculated for C22H18IN4O+ [M + H]+ 481.0520. HPLC: tR = 19.8 min, purity: 93%.
(2-iodophenyl){5-[(4-methoxybenzyl)amino]-3-(pyridin-3-yl)-1H-pyrazol-1-yl}methanone (9b). According to general procedure D, aminopyrazole 8b (500 mg, 1.78 mmol) was acylated using 2-iodobenzoyl chloride (570 mg, 2.14 mmol) in dry pyridine/THF (10 mL/20 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9b as a yellowish solid (484 mg, 53%). M.p.: 136–137 °C. TLC: Rf = 0.67 (CH/EtOAc = 1/1). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 8.82 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.55 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 7.96–7.92 (m, 1H, 4-Hpyridyl); 7.94–7.92 (m, 1H, 3-Hbenzoyl); 7.80 (t, J = 6.2 Hz, 1H, NH); 7.57 (dd, J = 7.7/1.7 Hz, 1H, 6-Hbenzoyl); 7.53 (td, J = 7.4/1.1 Hz, 1H, 5-Hbenzoyl); 7.43–7.42 (m, 2H, 2/6-Hmethoxyphenyl); 7.39 (dd, J = 4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.29 (ddd, J = 7.9/7.2/2.0 Hz, 1H, 4-Hbenzoyl); 6.94–6.91 (m, 2H, 3/5-Hmethoxyphenyl); 6.12 (s, 1H, 4-Hpyrazolyl); 4.42 (d, J = 6.1 Hz, 2H, CH2); 3.73 (s, 3H, CH3). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 170.5 (1C, C=O); 158.4 (1C, C-4methoxyphenyl); 153.2 (1C, C-5pyrazolyl); 152.3 (1C, C-3pyrazolyl); 150.0 (1C, C-6pyridyl); 147.0 (1C, C-2pyridyl); 140.6 (1C, C-1benzoyl); 138.4 (1C, C-3benzoyl); 133.1 (1C, C-4pyridyl); 131.4 (1C, C-4benzoyl); 130.4 (1C, C-1methoxyphenyl); 128.8 (2C, C-2/6methoxyphenyl); 128.7 (1C, C-6benzoyl); 127.6 (1C, C-5benzoyl); 127.5 (1C, C-3pyridyl), 123.7 (1C, C-5pyridyl), 113.8 (2C, C-3/5methoxyphenyl); 93.6 (1C, C-2benzoyl); 83.8 (1C, C-4pyrazolyl); 55.0 (1C, CH3); 47.3 (1C, CH2). IR (neat): ṽ [cm−1] = 3408, 1684, 1597, 1576, 1512, 1418, 1362, 1240, 1179, 1016, 937, 920, 814, 737, 721, 704. HRMS (APCI): m/z = 511.0617, calculated for C23H20IN4O2+ [M + H]+ 511.0626. HPLC: tR = 19.7 min, purity: 90.3%.
(2-iodophenyl)(5-{[(naphthalen-1-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)methanone (9c). According to general procedure D, aminopyrazole 8c (500 mg, 1.66 mmol) was acylated using 2-iodobenzoyl chloride (532 mg, 2.00 mmol) in dry pyridine/THF (19 mL/9 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9c as a yellowish solid (358 mg, 40%). M.p.: 137 °C. TLC: Rf = 0.35 (CH/EtOAc = 1/1). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.80 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.51 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 8.26–8.20 (m, 1H, 9-Hnaphthyl); 7.99–7.90 (m, 3H, 4-Hpyridyl, 3-Hbenzoyl, 6-Hnaphthyl); 7.88–7.82 (m, 2H, NH, 4-Hnaphthyl); 7.66–7.46 (m, 6H, 6-Hbenzoyl, 5-Hbenzoyl, 2-Hnaphthyl, 3-Hnaphthyl, 7-Hnaphtyl, 8-Hnaphthyl); 7.36 (ddd, J = 8.0/4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.27 (ddd, J = 8.0/7.3/1.8 Hz, 1H, 4-Hbenzoyl); 6.20 (s, 1H, 4-Hpyrazolyl); 4.98 (d, J = 6.0 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.6 (1C, C=O); 153.4 (1C, C-5pyrazolyl); 152.4 (1C, C-3pyrazolyl); 150.0 (1C, C-6pyridyl); 147.1 (1C, C-2pyridyl); 140.6 (1C, C-1benzoyl); 138.5 (1C, C-3benzoyl); 133.4 (1C, C-10naphthyl); 133.0 (1C, C-4pyridyl); 131.4 (1C, C-4benzoyl); 131.0 (1C, C-5naphthyl); 128.7 (1C, C-6benzoyl); 128.4 (1C, C-6naphthyl); 127.6 (1C, C-5benzoyl); 127.5 (1C, C-3pyridyl); 126.2 (1C, C-7naphthyl); 125.8 (1C, C-2naphthyl); 125.4 (1C, C-3naphthyl); 124.7 (1C, C-8naphthyl); 123.7 (1C, C-5pyridyl); 123.6 (1C, C-9naphthyl); 93.5 (1C, C-2benzoyl); 83.8 (1C, C-4pyrazolyl); 46.1 (1C, CH2), the signals of C-1naphthyl and C-4naphthyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 1692, 1599, 1568, 1489, 1435, 1368, 1342, 1184, 1063, 1005, 930, 853, 748, 704. HRMS (APCI): m/z = 531.0648, calculated for C26H20IN4O+ [M + H]+ 531.0676. HPLC: tR = 21.2 min, purity: 94.2%.
{5-[(furan-2-ylmethyl)amino]-3-(pyridin-3-yl)-1H-pyrazol-1-yl}(2-iodophenyl)methanone (9d). According to general procedure D, aminopyrazole 8d (500 mg, 2.08 mmol) was acylated using 2-iodobenzoyl chloride (665 mg, 2.5 mmol) in dry pyridine/THF (22 mL/13 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9d as a yellowish solid (792 mg, 81%). M.p.: 103–104 °C. TLC: Rf = 0.68 (CH/EtOAc = 1/1). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.85 (dd, J = 2.2/0.8 Hz, 1H, 2-Hpyridyl); 8.56 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 7.98–7.96 (m, 1H, 4-Hpyridyl); 7.95 (dd, J = 8.1/1.1 Hz, 1H, 3-Hbenzoyl); 7.71 (t, J = 6.2 Hz, 1H, NH); 7.64 (dd, J = 1.9/0.9 Hz, 1H, 5-Hfuranyl); 7.57 (dd, J = 7.6/1.8 Hz, 1H, 6-Hbenzoyl); 7.53 (td, J = 7.5/1.1 Hz, 1H, 5-Hbenzoyl); 7.42 (ddd, J = 8.0/4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.29 (ddd, J = 8.0/7.4/1.8 Hz, 1H, 4-Hbenzoyl); 6.52–6.49 (m, 1H, 3-Hfuranyl); 6.43 (dd, J = 3.2/1.8 Hz, 1H, 4-Hfuranyl); 6.24 (s, 1H, 4-Hpyrazolyl); 4.49 (d, J = 6.0 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.5 (1C, C=O); 152.9 (1C, C-5pyrazolyl); 152.3 (1C, C-3pyrazolyl); 151.5 (1C, C-1furanyl); 150.0 (1C, C-6pyridyl); 147.0 (1C, C-2pyridyl); 142.5 (1C, C-4furanyl); 140.5 (1C, C-1benzoyl); 138.4 (1C, C-3benzoyl); 133.0 (1C, C-4pyridyl); 131.5 (1C, C-4benzoyl); 128.7 (1C, C-6benzoyl); 127.6 (1C, C-5benzoyl); 127.5 (1C, C-3pyridyl); 123.8 (1C, C-5pyridyl); 120.4 (1C, C-2furanyl); 107.9 (1C, C-3furanyl); 93.5 (1C, C-2benzoyl); 84.0 (1C, C-4pyrazolyl); 41.1 (1C, CH2). IR (neat): ṽ [cm−1] = 3381, 1692, 1603, 1576, 1522, 1501, 1373, 1238, 1196, 1153, 939, 901, 810, 735. HRMS (APCI): m/z = 471.0287, calculated for C20H16IN4O2+ [M + H]+ 471.0313. HPLC: tR = 18.7 min, purity: 95.6%.
(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)(2-iodophenyl)methanone (9e). According to general procedure D, aminopyrazole 8e (142 mg, 488 µmol) was acylated using 2-iodobenzoyl chloride (156 mg, 586 µmol) in dry pyridine/THF (4.50 mL/2.25 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9e as a yellowish solid (146 mg, 280 µmol, 57%). M.p.: 105–106 °C. TLC: Rf = 0.18 (CH/EtOAc = 70/30). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.60 (d, J = 6.5 Hz, 2H, CH2), 6.24 (s, 1H, 4-Hpyrazolyl), 6.99 (d, J = 3.7 Hz, 1H, 4-Hthiophenyl), 7.10–7.12 (m, 1H, 3-Hthiophenyl), 7.29 (td, J = 7.6/1.8 Hz, 1H, 4-Hiodobenzoyl), 7.41 (ddd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl), 7.53 (td, J = 7.5/1.1 Hz, 1H, 5-Hiodobenzoyl), 7.58 (dd, J = 7.6/1.7 Hz, 1H, 6-Hiodobenzoyl), 7.93–8.01 (m, 3H, NH, 4-Hpyridyl, 3-Hiodobenzoyl), 8.56 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl), 8.84 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 43.3 (1C, CH2), 84.2 (1C, C-4pyrazolyl), 93.6 (1C, C-2iodobenzoyl), 123.8 (1C, C-5pyridyl), 126.2 (1C, C-3thiophenyl), 126.3 (1C, C-4thiophenyl), 127.0 (1C, C-5thiophenyl), 127.5 (1C, C-3pyridyl), 127.7 (1C, C-5iodobenzoyl), 128.8 (1C, C-6iodobenzoyl), 131.5 (1C, C-4iodobenzoyl), 133.1 (1C, C-4pyridyl), 138.4 (1C, C-3iodobenzoyl), 140.6 (1C, C-1iodobenzoyl), 141.5 (1C, C-2thiophenyl), 147.1 (1C, C-2pyridyl), 150.1 (1C, C-6pyridyl), 152.3 (1C, C-3pyrazolyl), 152.6 (1C, C-5pyrazolyl), 170.4 (1C, CON). IR (neat): ṽ [cm−1] = 3379, 1685, 1589, 1516, 1419, 1350, 1230, 1018, 1002, 937, 918, 794, 740. HRMS (APCI): m/z = 520.9694 calculated for [M + H]+, found: 520.9716. HPLC: tR = 20.5 min, purity: 95.8%.
{5-[(cyclohexylmethyl)amino]-3-(pyridin-3-yl)-1H-pyrazol-1-yl}(2-iodophenyl)methanone (9f). According to general procedure D, aminopyrazole 8f (130 mg, 507 µmol) was acylated using 2-iodobenzoyl chloride (162 mg, 609 µmol) in dry pyridine/THF (6 mL/3 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9e as a beige solid (92 mg, 37%). M.p.: 123–124 °C. TLC: Rf = 0.48 (CH/EtOAc = 1/1). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.87 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.56 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 7.99 (dt, J = 8.0/2.0 Hz, 1H, 3-Hbenzoyl); 7.94 (dd, J = 8.0/1.0 Hz, 1H, 4-Hpyridyl); 7.57 (dd, J = 7.7/2.0 Hz, 1H, 6-Hbenzoyl); 7.53 (td, J = 7.4/1.0 Hz, 1H, 5-Hbenzoyl); 7.41 (ddd, J = 8.0/4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.32–7.29 (m, 1H, 4-Hbenzoyl); 7.29–7.26 (m, 1H, NH); 6.17 (s, 1H, 4-Hpyrazolyl); 3.14 (t, J = 6.5 Hz, 2H, CH2); 1.86–1.60 (m, 6H, Hcyclohexyl); 1.32–1.13 (m, 3H, Hcyclohexyl); 1.09–0.95 (m, 2H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.7 (1C, C=O); 153.8 (1C, C-5pyrazolyl); 152.5 (1C, C-3pyrazolyl); 150.0 (1C, C-6pyridyl); 147.1 (1C, C-2pyridyl); 140.7 (1C, C-1benzoyl); 138.2 (1C, C-4pyridyl); 133.1 (1C, C-3benzoyl); 131.4 (1C, C-4benzoyl); 128.7 (1C, C-6benzoyl); 127.7 (1C, C-5benzoyl); 123.7 (1C, C-5pyridyl); 93.5 (1C, C-2benzoyl); 83.2 (1C, C-4pyrazolyl); 50.8 (1C, CH2); 36.5 (1C, Ccyclohexyl); 30.2 (2C, Ccyclohexyl); 26.0 (1C, Ccyclohexyl); 25.3 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3217, 3152, 2920, 2837, 2361, 1738, 1686, 1601, 1576, 1524, 1489, 1366, 1229, 1209, 1182, 1016, 853, 746, 725. HRMS (APCI): m/z = 487.0986, calculated for C22H24IN4O+ [M + H]+ 487.0989. HPLC: tR = 22.0 min, purity: 99.3%.
(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-phenyl-1H-pyrazol-1-yl)(2-iodophenyl)methanone (9g). According to general procedure D, aminopyrazole 8g (160 mg, 552 µmol) was acylated using 2-iodobenzoyl chloride (177 mg, 663 µmol) in dry pyridine/THF (6 mL/3 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9g as a colorless solid (121 mg, 42%). M.p.: 137–138 °C. TLC: Rf = 0.47 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 7.94 (d, J = 7.9 Hz, 1H, 3-Hbenzoyl); 7.88 (t, J = 6.3 Hz, 1H, NH); 7.65–7.59 (m, 2H, Hphenyl); 7.85–7.49 (m, 2H, 5/6-Hbenzoyl); 7.41–7.33 (m, 3H, Hphenyl); 7.28 (td, J = 7.4/2.0 Hz, 1H, 4-Hbenzoyl); 7.13–7.06 (m, 1H, 3-Hchlorothiophenyl); 6.99 (d, J = 3.8/0.8 Hz, 1H, 4-Hchlorothiophenyl); 6.11 (s, 1H, 4-Hpyrazolyl); 4.60 (d, J = 6.3 Hz, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.4 (1C, C=O); 154.6 (1C, C-5pyrazolyl); 152.4 (1C, C-3pyrazolyl); 141.6 (1C, C-2chlorothiophenyl); 140.8 (1C, C-1benzoyl); 138.4 (1C, C-3benzoyl); 131.7 (1C, C-phenyl); 131.4 (1C, C-4benzoyl); 129.2 (1C, Cphenyl); 128.7 (1C, C-6benzoyl); 128.6 (2C, Cphenyl); 127.6 (1C, C-5benzoyl); 126.9 (1C, C-5chlorothiophenyl); 126.2 (1C, C-4chlorothiophenyl); 126.0 (1C, C-3chlorothiophenyl); 125.9 (2C, C-phenyl); 93.5 (1C, C-2benzoyl); 84.3 (1C, C-4pyrazolyl); 43.3 (1C, CH2). IR (neat): ṽ [cm−1] = 3389, 1682, 1597, 1578, 1520, 1503, 1449, 1395, 1234, 1165, 1059, 1001, 935, 918, 800, 733. HRMS (APCI): m/z = 519.9739, calculated for C21H16ClIN3OS+ [M + H]+ 519.9742. HPLC: tR = 24.9 min, purity: 96.7%.
[5-(benzylamino)-3-cyclohexyl-1H-pyrazol-1-yl](2-iodophenyl)methanone (9h). According to general procedure D, aminopyrazole 8h (100 mg, 392 µmol) was acylated using 2-iodobenzoyl chloride (125 mg, 470 µmol) in dry pyridine/THF (4 mL/2 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9g as a colorless oil (103 mg, 54%). TLC: Rf = 0.60 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 7.91–7.86 (m, 1H, 3-Hbenzoyl); 7.59 (t, J = 6.2 Hz, 1H, NH); 7.51–7.45 (m, 2H, 5/6-Hbenzoyl); 7.44–7.39 (m, 2H, 2/6-Hphenyl); 7.39–7.33 (m, 2H, 3/5-Hphenyl); 7.29–7.25 (m, 1H, 4-Hphenyl); 7.23 (ddd, J = 7.9/7.0/2.1 Hz, 1H, 4-Hbenzoyl); 5.33 (s, 1H, 4-Hpyrazolyl); 4.39 (d, J = 6.0 Hz, 2H, CH2); 2.32–2.22 (m, 1H, Hcyclohexyl), 1.73–1.62 (m, 4H, Hcyclohexyl), 1.58 (d, J = 12.7 Hz, 1H, Hcyclohexyl), 1.29–1.17 (m, 4H, Hcyclohexyl), 1.18–1.07 (m, 0H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.9 (1C, C=O); 163.3 (1C, C-5pyrazolyl); 152.9 (1C, C-3pyrazolyl); 141.7 (1C, C-1benzoyl); 139.3 (1C, C-1phenyl); 138.8 (1C, C-3benzoyl); 131.6 (1C, C-4benzol); 128.9 (3C, C-3/5phenyl, C-6benzoyl); 128.0 (1C, C-5benzoyl); 127.8 (2C, C-2/6phenyl); 127.5 (1C, C-4phenyl); 94.0 (1C, C-2benzoyl); 84.5 (1C, C-4pyrazolyl); 48.3 (1C, CH2); 37.6 (1C, Ccyclohexyl); 32.1 (2C, Ccyclohexyl); 26.8 (1C, Ccyclohexyl); 25.8 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3387, 2924, 2849, 1682, 1593, 1524, 1466, 1431, 1373, 1290, 1229, 1202, 1165, 1119, 1016, 974, 926, 891, 856, 824, 735. HRMS (APCI): m/z = 486.1004, calculated for C23H25IN3O+ [M + H]+ 486.1037. HPLC: tR = 25.5 min, purity: 97.0%.
(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-cyclohexyl-1H-pyrazol-1-yl)(2-iodophenyl)methanone (9i). According to general procedure D, aminopyrazole 8i (72 mg, 243 µmol) was acylated using 2-iodobenzoyl chloride (78 mg, 292 µmol) in dry pyridine/THF (3 mL/1.5 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 9i as a colorless oil (52 mg, 41%). TLC: Rf = 0.56 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 7.88 (d, J = 7.8/1.0 Hz, 1H, 3-Hbenzoyl); 7.66 (t, J = 6.3 Hz, 1H, NH); 7.51–7.43 (m, 2H, 5/6-Hbenzoyl); 7.23 (ddd, J = 8.0/7.2/2.0 Hz, 1H, 4-Hbenzoyl); 7.03–6.99 (m, 1H, 3-Hchlorothiophenyl); 6.98 (d, J = 3.7 Hz, 1H, 4-Hchlorothiophenyl); 5.47 (s, 1H, 4-Hpyrazolyl); 4.50 (d, J = 6.1 Hz, 2H, CH2); 2.33–2.23 (m, 1H, Hcyclohexyl), 1.78–1.63 (m, 4H, Hcyclohexyl), 1.62–1.54 (m, 1H, Hcyclohexyl), 1.32–1.20 (m, 4H, Hcyclohexyl), 1.20–1.09 (m, 1H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 170.7 (1C, C=O); 163.2 (1C, C-5pyrazolyl); 152.2 (1C, C-3pyrazolyl); 142.3 (1C, C-2chlorothiophenyl); 141.6 (1C, C-1benzoyl); 138.8 (1C, C-3benzoyl); 131.6 (1C, C-4benzoyl); 129.0 (1C, C-6benzoyl); 128.1 (1C, C-5benzoyl); 127.3 (1C, C-5chlorothiophenyl); 126.8 (1C, C-4chlorothiophenyl); 126.3 (1C, C-3chlorothiophenyl); 94.0 (1C, C-2benzoyl); 85.0 (1C, C-4pyrazolyl); 43.8 (1C, CH2); 37.6 (1C, Ccyclohexyl); 32.1 (2C, Ccyclohexyl); 26.0 (1C, Ccyclohexyl); 25.8 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3379, 2924, 2849, 1682, 1591, 1524, 1449, 1371, 1348, 1117, 1061, 974, 920, 856, 793, 741. HRMS (APCI): m/z = 526.0207, calculated for C21H22ClIN3OS+ [M + H]+ 526.0211. HPLC: tR = 26.1 min, purity: 96.0%.
4-benzyl-2-(pyridin-3-yl)pyrazolo[5,1b]quinazolin-9(4H)-one (10a). According to general procedure E, 9a (63.2 mg, 131 µmol), CuI (5.0 mg, 26.3 µmol, 0.20 eq.), Cs2CO3 (85.7 mg, 263 µmol, 2.00 eq.) and 1,10-phenanthroline (4.7 mg, 26.3 µmol, 0.20 eq.) were dissolved in dry DMF (2 mL) and stirred at 80 °C for 3 h. Flash column chromatography (CH2Cl2/CH3OH = 96/4) yielded 10a as a colorless solid (37.9 mg, 82%). M.p.: 266 °C. TLC: Rf = 0.44 (CH2Cl2/CH3OH = 96/4). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 9.22 (bs, 1H, 2-Hpyridyl); 8.68 (bs, 1H, 6-Hpyridyl); 8.40–8.37 (m, 1H, 4-Hpyridyl); 8.37–8.32 (m, 1H, 8-Hquinazolinyl); 7.80 (ddd, J = 8.7/7.1/1.7 Hz, 1H, 6-Hquinazolinyl); 7.59–7.54 (m, 1H, 5-Hpyridyl); 7.52 (d, J = 8.7 Hz, 1H, 5-Hquinazolinyl); 7.39–7.35 (m, 1H, 7-Hquinazolinyl); 7.34 (dd, J = 0.9 Hz, 2H, 3/5-Hphenyl); 7.34–7.33 (m, 2H, 2/6-Hphenyl); 7.28 (dd, J = 8.6/3.3 Hz, 1H, 4-Hphenyl); 7.14 (s, 1H, 3-Hquinazolinyl); 5.64 (s, 2H, CH2). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 155.1 (1C, C-9quinazolinyl, C=O); 152.4 (1C, C-2quinazolinyl); 150.2 (1C, C-6pyridyl); 147.3 (1C, C-2pyridyl); 146.1 (1C, C-3aquinazolinyl); 139.7 (1C, C-4aquinazolinyl); 135.3 (1C, C-6quinazolinyl); 135.1 (1C, C-1phenyl); 133.4 (1C, C-4pyridyl); 128.9 (2C, C-3/5phenyl); 128.4 (1C, C-8quinazolinyl); 126.4 (2C, C-2/6phenyl); 124.2 (1C, C-5pyridyl); 122.0 (1C, C-7quinazolinyl); 114.3 (1C, C-5quinazolinyl); 113.4 (1C, C-8aquinazolinyl); 86.3 (1C, C-3quinazolinyl); 50.7 (1C, CH2), the signal of C-3pyridyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3096, 1695, 1601, 1566, 1491, 1435, 1369, 1180, 1026, 951, 934, 854, 797, 745, 706. HRMS (APCI): m/z = 353.1408, calculated for C22H17N4O+ [M + H]+ 353.1397. HPLC: tR = 16.6 min, purity: 94.3%.
4-(4-methoxybenzyl)-2-(pyridin-3-yl)pyrazolo[5,1-b]quinazolin-9(4H)-one (10b). According to general procedure E, 9b (150 mg, 294 µmol), CuI (11.2 mg, 58.8 µmol, 0.20 eq.), Cs2CO3 (192 mg, 588 µmol, 2.00 eq.) and 1,10-phenanthroline (10.6 mg, 58.8 µmol, 0.20 eq.) were dissolved in dry DMF (6 mL) and stirred at 80 °C for 90 min. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10b as a colorless solid (39 mg, 35%). M.p.: 245 °C. TLC: Rf = 0.43 (CH2Cl2/CH3OH = 95/5). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 9.24 (bs, 1H, 2-Hpyridyl); 8.69 (bs, 1H, 6-Hpyridyl); 8.38 (d, J = 8.0 Hz, 1H, 4-Hpyridyl); 8.35 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 7.81 (ddd, J = 8.7/7.1/1.7 Hz, 1H, 6-Hquinazolinyl); 7.63–7.51 (m, 2H, 5-Hpyridyl, 5-Hquinazolinyl); 7.36 (ddd, J = 8.0/7.1/0.8 Hz, 1H, 7-Hquinazolinyl); 7.31–7.25 (m, 2H, 2/6-Hmethoxyphenyl); 7.16 (s, 1H, 3-Hquinazolinyl); 6.92–6.85 (m, 2H, 3/5-Hmethoxyphenyl); 5.56 (s, 2H, CH2); 3.69 (s, 3H, CH3). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 158.7 (1C, C-4methoxyphenyl); 155.1 (1C, C-9quinazolinyl, C=O); 152.4 (1C, C-2quinazolinyl); 150.2 (1C, C-6pyridyl); 147.3 (1C, C-2pyridyl); 146.0 (1C, C-3aquinazolinyl); 139.6 (1C, C-4aquinazolinyl); 135.2 (1C, C-6quinazolinyl); 133.4 (1C, C-4pyridyl); 128.3 (1C, C-8quinazolinyl); 127.9 (2C, C-2/6methoxyphenyl); 127.0 (1C, C-1methoxyphenyl); 121.8 (1C, C-7quinazolinyl); 114.4 (1C, C-5quinazolinyl); 114.2 (2C, C-3/5methoxyphenyl); 113.3 (1C, C-8aquinazolinyl); 86.2 (1C, C-3quinazolinyl); 55.0 (3C, CH3); 50.1 (1C, CH2), the signals of C-3pyridyl and C-5pyridyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3098, 3013, 2361, 1714, 1695, 1512, 1491, 1433, 1371, 1346, 1290, 1250, 1182, 1022, 928, 856, 743, 706. HRMS (APCI): m/z = 383.1502, calculated for C23H19N4O2+ [M + H]+ 383.1503. HPLC: tR = 16.8 min, purity: 99.5%.
4-[(naphthalen-1-yl)methyl]-2-(pyridin-3-yl)pyrazolo[5,1-b]quinazolin-9(4H)-one (10c). According to general procedure E, 9c (150 mg, 283 µmol), CuI (10.8 mg, 56.6 µmol, 0.20 eq.), Cs2CO3 (184 mg, 566 µmol, 2.00 eq.) and 1,10-phenanthroline (10.2 mg, 56.6 µmol, 0.20 eq.) were dissolved in dry DMF (6 mL) and stirred at 80 °C for 90 min. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10c as a beige solid (63 mg, 55%). M.p.: 252 °C. TLC: Rf = 0.41 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.15 (d, J = 2.1 Hz, 1H, 2-Hpyridyl); 8.60 (dd, J = 4.9/1.6 Hz, 1H, 6-Hpyridyl); 8.41 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 8.37–8.31 (m, 2H, 9-Hnaphthyl, 4-Hpyridyl); 8.07–8.01 (m, 1H, Hnaphthyl); 7.86 (d, J = 8.3 Hz, 1H, Hnaphthyl); 7.78–7.70 (m, 2H, 5/6-Hquinazolinyl); 7.67 (ddd, J = 8.1/6.9/1.1 Hz, 1H, Hnaphthyl); 7.53–7.47 (m, 1H, 5-Hpyridyl); 7.37 (ddd, J = 8.0/7.1/0.8 Hz, 1H, 7-Hquinazolinyl); 7.33–7.25 (m, 2H, Hnaphthyl); 7.06 (s, 1H, 3-Hquinazolinyl); 6.92 (m, 1H, Hnaphthyl); 6.12 (s, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 155.2 (1C, C-9quinazolinyl, C=O); 152.4 (1C, C-2quinazolinyl); 150.2 (1C, C-6pyridyl); 147.4 (1C, C-2pyridyl); 146.0 (1C, C-3aquinazolinyl); 139.9 (1C, C-4aquinazolinyl); 135.2 (1C, C-6quinazolinyl); 133.4 (1C, C-4pyridyl); 130.2 (1C, Cnaphthyl); 130.2 (1C, Cnaphthyl); 129.5 (1C, Cnaphthyl); 128.6 (1C, Cnaphthyl); 128.4 (1C, C-8quinazolinyl); 127.7 (1C, C-3pyridyl); 126.4 (1C, C-5quinazolinyl); 126.2 (1C, Cnaphthyl); 125.5 (1C, Cnaphthyl); 123.9 (1C, C-5pyridyl); 123.3 (1C, C-9naphthyl); 122.1 (1C, C-7quinazolinyl); 121.7 (1C, Cnaphthyl); 114.2 (1C, Cnaphthyl); 113.5 (1C, C-8aquinazolinyl); 86.2 (1C, C-3quinazolinyl); 49.2 (1C, CH2), the signals of C-1naphthyl and C-4naphthyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 1707, 1601, 1566, 1493, 1435, 1412, 1296, 1184, 1024, 932, 787, 766, 706. HRMS (APCI): m/z = 403.1511, calculated for C26H19N4O+ [M + H]+ 403.1553. HPLC: tR = 18.7 min, purity: 99.2%.
4-[(furan-2-yl)methyl]-2-(pyridin-3-yl)pyrazolo[5,1-b]quinazolin-9(4H)-one (10d). According to general procedure E, 9d (100 mg, 213 µmol), CuI (8.1 mg, 42.5 µmol, 0.20 eq.), Cs2CO3 (139 mg, 425 µmol, 2.00 eq.) and 1,10-phenanthroline (7.7 mg, 42.5 µmol, 0.20 eq.) were dissolved in dry DMF (4 mL) and stirred at 80 °C for 45 min. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10d as a beige solid (31 mg, 43%). M.p.: 272 °C. TLC: Rf = 0.52 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.26 (bs, 1H, 2-Hpyridyl); 8.70 (bs, 1H, 6-Hpyridyl); 8.40 (dt, J = 7.9/1.6 Hz, 1H, 8-Hquinazolinyl); 8.33 (dt, J = 8.1/1.1 Hz, 1H, 4-Hpyridyl); 7.94–7.87 (m, 2H, 5/6-Hquinazolinyl); 7.62 (dd, J = 1.8/0.8 Hz, 1H, 5-Hfuranyl); 7.58 (dd, J = 8.0/4.4 Hz, 1H, 7-Hquinazolinyl); 7.39 (ddd, J = 8.0/4.7/3.3 Hz, 1H, 5-Hpyridyl); 7.24 (s, 1H, 3-Hquinazolinyl); 6.75 (dd, J = 3.3/0.8 Hz, 1H, 3-Hfuranyl); 6.43 (dd, J = 3.3/1.8 Hz, 1H, 4-Hfuranyl); 5.62 (s, 2H, CH2). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 155.0 (1C, C-9quinazolinyl, C=O); 152.3 (1C, C-2quinazolinyl); 150.2 (1C, C-6pyridyl); 148.4 (1C, C-2furanyl); 147.4 (1C, C-2pyridyl); 145.3 (1C, C-3aquinazolinyl); 143.5 (1C, C-5furanyl); 139.5 (1C, C-4aquinazolinyl); 135.1 (1C, C-6quinazolinyl); 133.5 (1C, C-4pyridyl); 128.3 (1C, C-8quinazolinyl); 122.1 (1C, C-5pyridyl); 114.3 (1C, C-5quinazolinyl); 113.2 (1C, C-8aquinazolinyl); 110.6 (1C, C-4furanyl); 109.9 (1C, C-3furanyl); 86.6 (1C, C-3quinazolinyl); 44.0 (1C, CH2), the signals of C-3pyridyl and C-7quinazolinyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 2970, 2363, 1738, 1692, 1599, 1568, 1489, 1435, 1368, 1344, 1230, 1180, 949, 851, 748. HRMS (APCI): m/z = 343.1152, calculated for C20H15N4O2+ [M + H]+ 343.1190. HPLC: tR = 15.5 min, purity: 99.5%.
4-[(5-chlorothiophen-2-yl)methyl]-2-(pyridin-3-yl)pyrazolo[5,1-b]quinazolin-9(4H)-one (10e). According to general procedure E, 9e (50.0 mg, 96.0 µmol), CuI (3.7 mg, 19.2 µmol, 0.20 eq.), Cs2CO3 (62.6 mg, 192 µmol, 2.00 eq.) and 1,10-phenanthroline (3.5 mg, 19.2 µmol, 0.20 eq.) were dissolved in dry DMF (2 mL) and stirred at 80 °C for 30 min. Flash column chromatography (CH2Cl2/CH3OH = 96/4) yielded 10e as a colorless solid (34 mg, 90%). M.p.: 248–249 °C. TLC: Rf = 0.27 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 5.75 (s, 2H, CH2), 7.00 (d, J = 3.8 Hz, 1H, 4-Hthiophenyl), 7.24 (d, J = 3.9 Hz, 1H, 3-Hthiophenyl), 7.26 (s, 1H, 3-Hquinazolinyl), 7.40 (ddd, J = 8.0/6.9/0.9 Hz, 1H, 7-Hquinazolinyl), 7.57 (ddd, J = 7.9/4.7/0.9 Hz, 1H, 5-Hpyridyl), 7.82 (d, J = 8.5 Hz, 1H, 5-Hquinazolinyl), 7.90 (ddd, J = 8.6/7.0/1.7 Hz, 1H, 6-Hquinazolinyl), 8.35 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl), 8.38 (dt, J = 7.9/1.9 Hz, 1H, 4-Hpyridyl), 8.67 (dd, J = 4.9/1.6 Hz, 1H, 6-Hpyridyl), 9.21 (d, J = 2.4 Hz, 1H, 2-Hpyridyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 46.1 (1C, CH2), 86.5 (1C, C-3quinazolinyl), 113.5 (1C, C-8aquinazolinyl), 114.2 (1C, C-5quinazolinyl), 122.4 (1C, C-7quinazolinyl), 124.1 (1C, C-5pyridyl), 126.5 (1C, C-4thiophenyl), 127.7 (1C, C-3thiophenyl), 127.8 (1C, C-3pyridyl), 127.9 (1C, C-5thiophenyl), 128.6 (1C, C-8quinazolinyl), 133.5 (1C, C-4pyridyl), 135.3 (1C, C-6quinazolinyl), 136.4 (1C, C-2thiophenyl), 139.1 (1C, C-4aquinazolinyl), 145.2 (1C, C-3aquinazolinyl), 147.4 (1C, C-2pyridyl), 150.4 (1C, C-6pyridyl), 152.5 (1C, C-2quinazolinyl), 155.0 (1C, CON). IR (neat): ṽ [cm−1] = 1689, 1600, 1570, 1489, 1477, 1454, 1435, 1411, 1342, 1292, 1184, 1002, 817, 744, 702. HRMS (APCI): m/z = 393.0571 calculated for [M + H]+, found: 393.0550. HPLC: tR = 17.8 min, purity: 97.9%.
4-(cyclohexylmethyl)-2-(pyridin-3-yl)pyrazolo[5,1-b]quinazolin-9(4H)-one (10f). According to general procedure E, 9f (65.6 mg, 135 µmol), CuI (5.1 mg, 27.0 µmol, 0.20 eq.), Cs2CO3 (87.9 mg, 270 µmol, 2.00 eq.) and 1,10-phenanthroline (4.9 mg, 27.0 µmol, 0.20 eq.) were dissolved in dry DMF (3 mL) and stirred at 80 °C for 2 h. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10f as a yellowish solid (13.5 mg, 28%). M.p.: 235–236 °C. TLC: Rf = 0.38 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.32 (bs, 1H, 2-Hpyridyl); 8.73 (bs, 1H, 6-Hpyridyl); 8.42 (d, J = 7.9 Hz, 1H, 4-Hpyridyl); 8.33 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 7.88 (ddd, J = 8.7/7.0/1.7 Hz, 2H, 6-Hquinazolinyl); 7.73 (d, J = 8.6 Hz, 1H, 5-Hquinazolinyl); 7.63–7.53 (m, 1H, 5-Hpyridyl); 7.37 (ddd, J = 7.9/7.0/0.8 Hz, 1H, 7-Hquinazolinyl); 7.13 (s, 1H, 3-Hquinazolinyl); 4.21 (d, J = 7.4 Hz, 2H, CH2); 2.08–1.95 (m, 1H, Hcyclohexyl); 1.74–1.56 (m, 4H, Hcyclohexyl); 1.27–1.08 (m, 6H, Hcyclohexyl). IR (neat): ṽ [cm−1] = 3109, 2924, 2853, 2361, 1738, 1692, 1564, 1489, 1433, 1184, 1024, 874, 818, 746, 706. HRMS (APCI): m/z = 359.1963, calculated for C22H23N4O+ [M + H]+ 359.1866. HPLC: tR = 18.6 min, purity: 98.6%.
4-[(5-chlorothiophen-2-yl)methyl]-2-phenylpyrazolo[5,1-b]quinazolin-9(4H)-one (10g). According to general procedure E, 9g (100 mg, 192 µmol), CuI (7.3 mg, 38.5 µmol, 0.20 eq.), Cs2CO3 (125 mg, 385 µmol, 2.00 eq.) and 1,10-phenanthroline (6.9 mg, 38.5 µmol, 0.20 eq.) were dissolved in dry DMF (4 mL) and stirred at 80 °C for 2 h. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10f as a beige solid (63.3 mg, 84%). M.p.: 251–252 °C. TLC: Rf = 0.48 (CH2Cl2/CH3OH = 98/2). 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 8.34 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 8.06–8.00 (m, 2H, Hphenyl); 7.88 (ddd, J = 8.6/7.0/1.6 Hz, 1H, 6-Hquinazolinyl); 7.80 (d, J = 8.6 Hz, 1H, 5-Hquinazolinyl); 7.57–7.51 (m, 2H, Hphenyl); 7.49–7.43 (m, 1H, Hphenyl); 7.39 (ddd, J = 7.9/7.0/0.9 Hz, 1H, 7-Hquinazolinyl); 7.23 (d, J = 3.9 Hz, 1H, 3-Hchlorothiophenyl); 7.15 (s, 1H, 3-Hquinazolinyl); 7.00 (d, J = 3.8 Hz, 1H, 4-Hchlorothiophenyl); 5.75 (s, 2H, CH2). 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 154.9 (1C, C-9quinazolinyl, C=O); 144.9 C-3aquinazolinyl); 139.1 (1C, C-4aquinazolinyl); 136.5 (1C, C-2chlorothiophenyl); 135.1 (1C, C-6quinazolinyl); 131.8 (1C, Cphenyl); 129.4 (1C, Cphenyl); 128.8 (2C, Cphenyl); 128.5 (1C, C-8quinazolinyl); 127.8 (1C, C-5chlorothiophenyl); 127.8 (1C, C-3pyridyl); 127.6 (1C, C-3chlorothiophenyl); 126.4 (1C, C-4chlorothiophenyl); 126.3 (2C, Cphenyl); 122.1 (1C, C-7quinazolinyl); 114.0 (1C, C-5quinazolinyl); 113.4 (1C, C-8aquinazolinyl); 86.3 (1C, C-3quinazolinyl); 45.9 (1C, CH2), the signal of C-2quinazolinyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3389, 1688, 1599, 1491, 1454, 1425, 1371, 1346, 1294, 1186, 1059, 1003, 974, 932, 854, 814, 739. HRMS (APCI): m/z = 392.0617, calculated for C21H15ClN3OS+ [M + H]+ 392.0619. HPLC: tR = 22.2 min, purity: 97.4%.
4-benzyl-2-cyclohexylpyrazolo[5,1-b]quinazolin-9(4H)-one (10h). According to general procedure E, 9h (80 mg, 165 µmol), CuI (6.3 mg, 33.0 µmol, 0.20 eq.), Cs2CO3 (107 mg, 330 µmol, 2.00 eq.) and 1,10-phenanthroline (5.9 mg, 33.0 µmol, 0.20 eq.) were dissolved in dry DMF (3 mL) and stirred at 80 °C for 6.5 h. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10h as a beige solid (34.4 mg, 58%). M.p.: 180–181 °C. TLC: Rf = 0.57 (CH2Cl2/CH3OH = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 8.30 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 7.75 (ddd, J = 8.7/7.1/1.7 Hz, 1H, 6-Hquinazolinyl); 7.47 (d, J = 8.6 Hz, 1H, 5-Hquinazolinyl); 7.35–7.22 (m, 6H, 2/6-Hphenyl, 3/5-Hphenyl, 4-Hphenyl, 7-Hquinazolinyl); 6.32 (s, 1H, 3-Hquinazolinyl); 5.55 (s, 2H, CH2), 2.75–2.64 (m, 1H, Hcyclohexyl); 2.02–1.92 (m, 2H. Hcyclohexyl); 1.83–1.72 (m, 2H, Hcyclohexyl); 1.71–1.62 (m, 1H, Hcyclohexyl); 1.51–1.31 (m, 4H, Hcyclohexyl), 1.28–1.18 (m, 1H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 163.4 (1C, C-2quinazolinyl); 155.5 (1C, C-9quinazolinyl, C=O); 145.5 (1C, C-3aquinazolinyl); 140.2 (1C, C-4aquinazolinyl); 136.0 (C-1phenyl); 135.3 (1C, C-6quinazolinyl); 129.3 (2C, Cphenyl); 128.8 (1C, C-8quinazolinyl); 127.9 (2C, Cphenyl); 126.8 (1C, Cphenyl); 122.1 (1C, C-7quinazolinyl); 114.5 (1C, C-5quinazolinyl); 113.6 (1C, C-8aquinazolinyl); 86.8 (1C, C-3quinazolinyl); 50.9 (1C, CH2); 38.1 (1C, Ccyclohexyl); 32.5 (2C, Ccyclohexyl); 26.1 (2C, Ccyclohexyl); 26.1 (1C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 2924, 2851, 2361, 1738, 1697, 1593, 1570, 1487, 1356, 1287, 1229, 1202, 1175, 1042, 1024, 939, 860, 748, 708. HRMS (APCI): m/z = 358.1892, calculated for C23H24N3O+ [M + H]+ 358.1914. HPLC: tR = 22.2 min, purity: 98.1%.
4-[(5-chlorothiophen-2-yl)methyl]-2-cyclohexylpyrazolo[5,1-b]quinazolin-9(4H)-one (10i). According to general procedure E, 9i (40 mg, 76.1 µmol), CuI (2.9 mg, 15.2 µmol, 0.20 eq.), Cs2CO3 (49.6 mg, 152 µmol, 2.00 eq.) and 1,10-phenanthroline (2.7 mg, 15.2 µmol, 0.20 eq.) were dissolved in dry DMF (2 mL) and stirred at 80 °C for 45 min. Flash column chromatography (CH2Cl2/CH3OH = 97/3) yielded 10i as a colorless solid (21.8 mg, 72%). M.p.: 210 °C. TLC: Rf = 0.56 (CH2Cl2/CH3OH = 95/5). 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 8.29 (dd, J = 8.0/1.6 Hz, 1H, 8-Hquinazolinyl); 7.84 (ddd, J = 8.7/6.9/1.7 Hz, 1H, 6-Hquinazolinyl); 7.76 (d, J = 8.6 Hz, 1H, 5-Hquinazolinyl); 7.34 (t, J = 7.2 Hz, 1H, 7-Hquinazolinyl); 7.16 (d, J = 3.8 Hz, 1H, 3-Hchlorothiophenyl); 6.99 (d, J = 3.8 Hz, 1H, 4-Hchlorothiophenyl); 6.47 (s, 1H, 3-Hquinazolinyl); 5.66 (s, 2H, CH2); 2.77–2.67 (m, 1H, Hcyclohexyl); 2.04–1.92 (m, 2H, Hcyclohexyl); 1.83–1.75 (m, 2H, Hcyclohexyl); 1.73–1.65 (m, 1H, Hcyclohexyl); 1.57–1.30 (m, 4H, Hcyclohexyl); 1.32–1.17 (m, 1H, Hcyclohexyl). 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 163.4 (1C, C-2quinazolinyl); 155.3 (1C, C-9quinazolinyl, C=O); 144.5 C-3aquinazolinyl); 139.5 (1C, C-4aquinazolinyl); 137.2 (1C, C-2chlorothiophenyl); 135.4 (1C, C-6quinazolinyl); 129.0 (1C, C-8quinazolinyl); 128.2 (1C, C-5chlorothiophenyl); 128.0 (1C, C-3chlorothiophenyl); 126.9 (1C, C-4chlorothiophenyl); 122.4 (1C, C-7quinazolinyl); 114.4 (1C, C-5quinazolinyl); 113.8 (1C, C-8aquinazolinyl); 87.1 (1C, C-3quinazolinyl); 46.3 (1C, CH2). IR (neat): ṽ [cm−1] = 2932, 2851, 1697, 1595, 1574, 1487, 1470, 1443, 1389, 1287, 1198, 1061, 974, 856, 746, 712. HRMS (APCI): m/z = 398.1099, calculated for C21H21ClN3OS+ [M + H]+ 398.1088. HPLC: tR = 23.2 min, purity: 97.8%.
2-iodo-N-[3-(pyridin-3-yl)-1H-pyrazol-5-yl]benzamide (14). According to general procedure D, aminopyrazole 7a (925 mg, 1.40 mmol) was acylated using 2-iodobenzoyl chloride (449 mg, 1.69 mmol, 1.20 eq.) in dry pyridine/THF (16 mL/8 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 14 as a colorless solid (344 mg, 63%). M.p.: 205–206 °C. TLC: Rf = 0.09 (CH/EtOAc = 20/80). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 13.08 (bs, 1H, NHpyrazolyl); 10.98 (bs, 1H, NH); 9.00 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.55 (d, J = 4.7 Hz, 1H, 6-Hpyridyl); 8.15 (ddd, J = 8.0/2.3/1.6 Hz, 1H, 4-Hpyridyl); 7.92 (d, J = 7.9 Hz, 1H, 3-Hbenzoyl); 7.54–7.39 (m, 3H, 5/6-Hbenzoyl, 5-Hpyridyl); 7.22 (t, J = 7.5 Hz, 1H, 4-Hbenzoyl), the signal of 4-Hpyrazolyl cannot be seen on the spectrum. 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 167.2 (1C, C=O); 149.4 (1C, C-6pyridyl); 146.7 (1C, C-2pyridyl); 132.8 (1C, C-4pyridyl); 131.5 (1C, C-4benzoyl); 128.7 (1C, C-6benzoyl); 128.4 (1C, C-5benzoyl); 124.4 (1C, C-5pyridyl); 94.1 (1C, C-2benzyol), the signals of C-3pyrazolyl, and C-4pyrazolyl, C-5pyrazolyl, C-3pyridyl and C-1benzoyl cannot be seen on the spectrum. IR (neat): ṽ [cm−1] = 3389, 1684, 1597, 1578, 1522, 1396, 1306, 1016, 961, 895, 799, 735. HRMS (APCI): m/z = 391.0042, calculated for C15H12IN4O+ [M + H]+ 391.0050. HPLC: tR = 13.3 min, purity: 96.2%.
2-(pyridin-3-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (15). According to general procedure E, 14 (150 mg, 384 µmol), CuI (14.6 mg, 76.9 µmol, 0.20 eq.), Cs2CO3 (251 mg, 769 µmol, 2.00 eq.) and 1,10-phenanthroline (13.9 mg, 76.9 µmol, 0.20 eq.) were dissolved in dry DMF (8 mL) and stirred at 80 °C for 1 h. Flash column chromatography (CH2Cl2/CH3OH = 1/0 → 95/5) yielded 15 as a colorless solid (21.3 mg, 21%). M.p.: >300 °C. TLC: Rf = 0.15 (CH2Cl2/CH3OH = 95/5). 1H-NMR (400 MHz, DMSO-d6): δ (ppm) = 12.38 (s, 1H, NH); 9.18 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.60 (dd, J = 4.7/1.7 Hz, 1H, 6-Hpyridyl); 8.34 (dt, J = 7.9/2.0 Hz, 1H, 4-Hpyridyl); 8.22–8.13 (m, 2H, Hquinazolinyl); 7.92 (ddd, J = 8.4/7.3/1.5 Hz, 1H, Hquinazolinyl); 7.56–7.47 (m, 2H, 5-Hpyridyl, Hquinazolinyl); 6.51 (s, 1H, 3-Hquinazolinyl). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 158.9 (1C, C-5quinazolinyl, C=O); 150.4 (1C, C-2quinazolinyl); 150.0 (1C, C-6pyridyl); 147.4 (1C, C-2pyridyl); 140.4 (1C, C-3aquinazolinyl); 137.6 (1C, C-quinazolinyl); 135.6 (1C, C-quinazolinyl); 133.6 (1C, C-4pyridyl); 128.7 (1C, Cquinazolinyl); 128.6 (1C, C-3pyridyl); 126.0 (1C, Cquinazolinyl); 124.4 (1C, C-5pyridyl); 116.9 (1C, Cquinazolinyl); 114.9 (1C, Cquinazolinyl); 86.6 (1C, C-3quinazolinyl). IR (neat): ṽ [cm−1] = 3115, 1659, 1574, 1479, 1454, 1425, 1375, 1321, 1188, 1134, 916, 791, 760, 704. HRMS (APCI): m/z = 263.0948, calculated for C15H11N4O+ [M + H]+ 263.0927. HPLC: tR = 12.9 min, purity: 99.8%.
(5-amino-3-(pyridin-3-yl)-1H-1,2,4-triazol-1-yl)(2-iodophenyl)methanone (16). According to general procedure D, aminotriazole 11 (500 mg, 3.10 mmol) was acylated using 2-iodobenzoyl chloride (1.24 g, 4.65 mmol) in dry pyridine/THF (5 mL/5 mL). After the washing steps, pure product 16 was obtained as a colorless solid (844 mg, 70%). M.p.: 220 °C. TLC: Rf = 0.87 (EtOAc/MeOH= 9/1). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.32 (td, J = 7.7, 1.7 Hz, 1H, 4-Hphenyl), 7.46 (ddd, J = 7.9, 4.8, 0.9 Hz 1H, 5-Hpyridyl), 7.56 (td, J = 7.5, 1.1 Hz, 1H, 5-Hphenyl), 7.64 (dd, J = 7.6, 1.7 Hzl, 1H, 6-Hphenyl), 7.97 (dd, J = 8.0, 1.1 Hz, 1H, 3-Hphenyl), 8.02 (s, 2H, NH2), 8.10 (dt, J = 7.9, 2.0 Hz, 1H, 4-Hpyridyl), 8.63 (dd, J = 4.8, 1.7 Hz, 1H, 6-Hpyridyl), 8.97 (d, J = 2.2 Hz, 1H, 2-Hpyridyl). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 93.3 (1C, C-2phenyl), 123.9 (1C, C-5pyridyl), 125.8 (1C, C-3pyridyl), 127.8 (1C, C-5phenyl), 129.0 (1C, C-6phenyl), 132.0 (1C, C-4phenyl), 133.7 (1C, C-4pyridyl), 138.7 (1C, C-3phenyl), 139.7 (1C, C-1phenyl), 147.4 (1C, C-2pyridyl), 151.0 (1C, C-6pyridyl), 158.2 (1C, C-5triazolyl), 158.4 (1C, C-3triazolyl), 168.6 (1C, CO). IR (neat): ṽ [cm−1] = 3464, 2978, 1697, 1647, 1582, 1524, 1485, 1412, 1358, 1327, 1188, 1138, 1076, 1018, 961, 921, 795, 748, 706, 682, 640. HRMS (APCI): m/z = 391.9957, calculated for C14H11N5O+ [M + H]+ 392.0003.
2-iodo-N-(3-(pyridin-3-yl)-1H-1,2,4-triazol-5-yl)benzamide (16′). Aminotriazole 16 (250 mg, 639.1 μmol) was heated to 260 °C (neat) for 15 min. After cooling to room temperature, the crude product was purified by flash column chromatography (CH2Cl2/CH3OH = 100/0→ 90/10) to yield 16′ as a colorless solid (199 mg, 509.2 μmol, 80%). M.p.: 299–300 °C. TLC: Rf = 0.32 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.27 (t, J = 7.2 Hz, 1H, 4-Hbenzoyl), 7.52 (t, J = 7.2 Hz, 2H, 5-Hbenzoyl/5-Hpyridyl), 7.56 (d, J = 7.0 Hz, 1H, 6-Hbenzoyl), 7.96 (d, J = 7.9 Hz, 1H, 3-Hbenzoyl), 8.29 (d, J = 7.9 Hz, 1H, 4-Hpyridyl), 8.62 (d, J = 3.7 Hz, 1H, 6-Hpyridyl), 9.15 (s, 1H, 2-Hpyridyl), 12.27 (s, 1H, NHtriazolyl), 13.95 (s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 93.5 (1C, C-2benzoyl), 123.9 (1C, C-5pyridyl), 127.0 (1C, C-3pyridyl), 128.0 (1C, C-5benzoyl), 128.5 (1C, C-6benzoyl), 131.8 (1C, C-4benzoyl), 132.9 (1C, C-4pyridyl), 139.2 (1C, C-3benzoyl), 140.5 (1C, C-1benzoyl), 146.7 (1C, C-2pyridyl), 149.2 (1C, C-5triazolyl), 149.9 (1C, C-6pyridyl), 156.0 (1C, C-3triazolyl), 167.7 (1C, C=O). IR (neat): ṽ [cm−1] = 3254, 3057, 1672, 1597, 1425, 1300, 1140, 1084, 1053, 1037, 1016, 889, 795, 750, 667, 638, 629. HRMS (APCI): m/z = 392.0003 calculated for [M + H]+, found: 391.9994. HPLC: tR = 12.8 min, purity: 97.0%.
(5-amino-3-(pyridin-4-yl)-1H-1,2,4-triazol-1-yl)(2-iodophenyl)methanone (17). According to general procedure D, aminotriazole 12 (730 mg, 4.53 mmol) was acylated using 2-iodobenzoyl chloride (1.81 g, 6.80 mmol) in dry pyridine/THF (5 mL/5 mL). After the washing steps, pure product 17 was obtained as a colorless solid (1.56 g, 86%). M.p.: 191–193 °C. TLC: Rf = 0.80 (EtOAc/MeOH = 9/1). 1H NMR (400 MHz, DMSO-d6) δ (in ppm) = 7.33 (t, J = 7.6 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.64 (d, J = 6.7 Hz, 1H), 7.70 (d, J = 5.7 Hz, 2H), 7.97 (d, J = 7.9 Hz, 1H), 8.03 (br s, 2H, NH2), 8.65 (d, J = 5.5 Hz, 2H). 13C-NMR (101 MHz, DMSO-d6): δ (ppm) = 168.8 (NCOmethanone), 158.5 (5-Ctriazolyl), 158.4 (3-Ctriazolyl), 150.3 (2-Cpyridyl and 6-Cpyridyl), 139.6 (1-Cphenyl), 138.7 (3-Cphenyl), 137.2 (4-Cpyridyl), 132.0 (4-Cphenyl), 129.0 (6-Cphenyl), 127.9 (5-Cphenyl), 120.5 (3-Cpyridyl and 5-Cpyridyl), 93.3 (2-Cphenyl). IR (neat): ṽ [cm−1] = 3310, 3163, 1709, 1643, 1524, 1493, 1462, 1420, 1358, 1323, 1138, 1038, 968, 922, 741, 675. HRMS (APCI): m/z = 391.9946, calculated for C14H11N5O+ [M + H]+ 392.0003.
2-iodo-N-(3-(pyridin-4-yl)-1H-1,2,4-triazol-5-yl)benzamide (17′). Aminotriazole 17 (207 mg, 529.2 μmol) was heated to 260 °C (neat) for 15 min. After cooling to room temperature, the crude product was purified by flash column chromatography (CH2Cl2/CH3OH = 100/0→ 90/10) to yield 17′ as a colorless solid (158 mg, 403.9 μmol, 76%). M.p.: >300 °C. TLC: Rf = 0.31 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.27 (t, J = 10.9, 4.3 Hz, 1H, 4-Hbenzoyl), 7.52 (t, J = 7.4 Hz, 1H, 5-Hbenzoyl), 7.64 (d, J = 7.1 Hz, 1H, 6-Hbenzoyl), 7.88 (d, J = 5.9 Hz, 2H, 3-Hpyridyl/5-Hpyridyl), 7.96 (d, J = 7.8 Hz, 1H, 3-Hbenzoyl), 8.68 (d, J = 4.5 Hz, 1H, 2-Hpyridyl/6-Hpyridyl), 12.29 (s, 1H, NHtriazolyl), 14.08 (s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 93.5 (1C, C-2benzoyl), 119.8 (1C, C-3pyridyl/C-5pyridyl), 128.0 (1C, C-5benzoyl), 128.6 (1C, C-6benzoyl), 131.8 (1C, C-4benzoyl), 138.2 (1C, C-4pyridyl), 139.2 (1C, C-3benzoyl), 140.4 (1C, C-1benzoyl), 149.4 (1C, C-5triazolyl), 150.3 (C-2pyridyl/C-6pyridyl), 156.3 (1C, C-3triazolyl), 167.8 (1C, C=O). IR (neat): ṽ [cm−1] = 3347, 1659, 1422, 1306, 1138, 1090, 1053, 1016, 1003, 978, 953, 878, 837, 795, 746, 727, 704, 669, 635. HRMS (APCI): m/z = 392.0003 calculated for [M + H]+, found: 391.9996. HPLC: tR = 12.8 min, purity: 98.6%.
(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-(pyrazin-2-yl)-1H-1,2,4-triazol-1-yl)(2-iodophenyl)methanone (18). According to general procedure D, aminotriazole 13 (150 mg, 512 µmol) was acylated using 2-iodobenzoyl chloride (163 mg, 615 µmol, 1.20 eq.) in dry pyridine/THF (4.80 mL/2.40 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 18 as a yellowish solid (157 mg, 300 µmol, 59%). M.p.: 159–160 °C. TLC: Rf = 0.10 (CH/EtOAc = 70/30). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.80 (d, J = 6.2 Hz, 2H, CH2), 6.99 (d, J = 3.8 Hz, 1H, 4-Hthiophenyl), 7.04–7.06 (m, 1H, 3-Hpyrazinyl), 7.34 (td, J = 7.7/1.6 Hz, 1H, 4-Hiodobenzoyl), 7.57 (td, J = 7.6/1.1 Hz, 1H, 5-Hiodobenzoyl), 7.66 (dd, J = 7.7/1.6 Hz, 1H, 6-Hiodobenzoyl), 7.98 (dd, J = 7.9/1.1 Hz, 1H, 3-Hiodobenzoyl), 8.71 (dd, J = 2.5/1.5 Hz, 1H, 6-Hpyrazinyl), 8.74 (d, J = 2.5 Hz, 1H, 5-Hpyrazinyl), 8.79 (t, J = 6.3 Hz, 1H, NH), 9.16 (d, J = 1.5 Hz, 1H, 3-Hpyrazinyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 41.8 (1C, CH2), 93.3 (1C, C-2iodobenzoyl), 126.2 (1C, C-4thiophenyl), 126.7 (1C, C-3thiophenyl), 127.3 (1C, C-5thiophenyl), 127.9 (1C, C-5iodobenzoyl), 129.0 (1C, C-6iodobenzoyl), 132.1 (1C, C-4iodobenzoyl), 138.7 (1C, C-3iodobenzoyl), 139.4 (1C, C-1iodobenzoyl), 140.5 (1C, C-2thiophenyl), 143.5 (1C, C-3pyrazinyl), 144.1 (1C, C-2pyrazinyl), 144.8 (1C, C-6pyrazinyl), 145.9 (1C, C-5pyrazinyl), 157.8 (1C, C-3/5triazolyl), 157.8 (1C, C-3/5triazolyl), 168.8 (1C, CON). IR (neat): ṽ [cm−1] = 3371, 2924, 1697, 1535, 1373, 1338, 1219, 1157, 1049, 921, 771, 748. HRMS (APCI): m/z = 522.9599 calculated for [M + H]+, found: 522.9613. HPLC: tR = 22.3 min, purity: 93.8%.
4-[(5-chlorothiophen-2-yl)methyl]-2-(pyrazin-2-yl)-[1,2,4]triazolo[5,1-b]quinazolin-9(4H)-one (19). According to general procedure E, 18 (73.0 mg, 140 µmol, 1.00 eq.), CuI (65.3 mg, 27.9 µmol, 0.20 eq.), Cs2CO3 (91.0 mg, 279 µmol, 2.00 eq.) and 1,10-phenanthroline (5.0 mg, 27.9 µmol, 0.20 eq.) were dissolved in dry DMF (2.8 mL) and stirred at 80 °C for 30 min. Flash column chromatography (CH2Cl2/CH3OH = 1/0 → 95/5) yielded 19 as a colorless solid (44 mg, 111 µmol, 80%). M.p.: 246–247 °C (decomp.). TLC: Rf = 0.55 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 5.94 (s, 2H, CH2), 7.00 (d, J = 3.9 Hz, 1H, 4-Hthiophenyl), 7.25–7.27 (m, 1H, 3-Hthiophenyl), 7.51 (ddd, J = 8.0/4.6/3.5 Hz, 1H, 7-Hquinazolinyl), 7.94–7.98 (m, 2H, 5/6-Hquinazolinyl), 8.38 (dt, J = 8.0/1.1 Hz, 1H, 8-Hquinazolinyl), 8.83 (d, J = 2.4 Hz, 1H, 5-Hpyrazinyl), 8.87 (dd, J = 2.5/1.5 Hz, 1H, 6-Hpyrazinyl), 9.45 (d, J = 1.5 Hz, 1H, 3-Hpyrazinyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 44.7 (1C, CH2), 115.1 (1C, C-5/8aquinazolinyl), 115.2 (1C, C-5/8aquinazolinyl), 123.7 (1C, C-7quinazolinyl), 126.4 (1C, C-4thiophenyl), 127.9 (1C, C-3thiophenyl), 128.3 (1C, C-5thiophenyl), 128.5 (1C, C-8quinazolinyl), 135.7 (1C, C-6quinazolinyl), 136.1 (1C, C-2thiophenyl), 138.7 (1C, C-4aquinazolinyl), 143.7 (1C, C-3pyrazinyl), 144.3 (1C, C-2pyrazinyl), 145.0 (1C, C-6pyrazinyl), 146.1 (1C, C-5pyrazinyl), 52.7 (1C, C-3aquinazolinyl), 155.0 (1C, CON), 159.2 (1C, C-2quinazolinyl). IR (neat): ṽ [cm−1] = 3082, 1705, 1604, 1562, 1489, 1442, 1334, 1157, 1014, 933, 860, 794, 759, 717. HRMS (APCI): m/z = 395.0476 calculated for [M + H]+, found: 395.0462. HPLC: tR = 19.7 min, purity: 95.1%.
2-(pyridin-3-yl)-[1,2,4]triazolo[5,1-b]quinazolin-9(4H)-one (20). According to general procedure E, 20 was synthesized using 16 (50 mg, 127.8 μmol, 1.00 eq.), CuI (5 mg, 25.6 μmol, 0.20 eq.), Cs2CO3 (83 mg, 255.6 μmol, 2.00 eq.) and 1,10-phenanthroline (5 mg, 25.6 μmol, 0.20 eq.) in dry DMF (2 mL). The mixture was stirred at 80 °C for 1 h. After the first purification (CH2Cl2/CH3OH = 100/0 → 90/10), product 20 was obtained as a mixture with compound 21 (20 + 21, 27 mg, 101.2 μmol, 79%). According to HPLC measurement 20:21 ratio was 1:1. Synthesis in microwave conditions (procedure F) allowed increasing the yield of the isomers’ mixture up to 92% (20 + 21, 31 mg, 127.8 μmol) with the ratio 7:3. Further purification (CH2Cl2/CH3OH = 97/3) gave pure product 20 as a colorless solid (21 mg, 78.6 μmol, 62%). M.p.: >300 °C. TLC: Rf = 0.33 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.41 (ddd, J = 8.1, 7.2, 1.0 Hz, 1H, 7-Hquinazolinyl), 7.52 (d, J = 8.0 Hz, 1H, 5-Hquinazolinyl), 7.60 (dd, J = 7.8, 4.8 Hz, 1H, 5-Hpyridyl), 7.86 (ddd, J = 8.5, 7.1, 1.5 Hz, 1H, 6-Hquinazolinyl), 8.25 (dd, J = 8.1, 1.2 Hz, 1H, 8-Hquinazolinyl), 8.48 (dt, J = 7.9, 1.9 Hz, 1H, 4-Hpyridyl), 8.73 (d, J = 3.0 Hz, 1H, 6-Hpyridyl), 9.31 (s, 1H, 2-Hpyridyl), 13.42 (s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 113.5 (1C, C-8aquinazolinyl), 116.9 (1C, C-5quinazolinyl), 123.0 (1C, C-7quinazolinyl), 124.1 (1C, C-5pyridyl), 126.0 (1C, C-3pyridyl), 127.5 (1C, C-8quinazolinyl), 134.2 (1C, C-4pyridyl), 135.2 (1C, C-6quinazolinyl), 139.3 (1C, C-4aquinazolinyl), 147.7 (1C, C-2pyridyl), 151.3 (1C, C-6pyridyl), 151.6 (1C, C-3aquinazolinyl), 155.5 (1C, C-9quinazolinyl, C=O), 159.7 (1C, C-2quinazolinyl). IR (neat): ṽ [cm−1] = 3059, 2666, 1651, 1566, 1524, 1481, 1420, 1319, 1204, 1138, 1038, 945, 813, 787, 748, 702, 683, 633. HRMS (APCI): m/z = 264.0880 calculated for [M + H]+, found: 264.0889. HPLC: tR = 11.8 min, purity: 100.0%.
2-(pyridin-3-yl)-[1,2,4]triazolo[1,5-a]quinazolin-5(4H)-one (21). According to general procedure E, 21 was synthesized using 16′ (191 mg, 487.0 μmol, 1.00 eq.), CuI (19 mg, 97.4 μmol, 0.20 eq.), Cs2CO3 (317 mg, 974.0 μmol, 2.00 eq.) and 1,10-phenanthroline (18 mg, 97.4 μmol, 0.20 eq.) in dry DMF (3 mL). The mixture was stirred at 80 °C for 1 h. After purification, product 21 was obtained as a colorless solid (86 mg, 327.4 μmol, 67%); M.p.: >300 °C. TLC: Rf = 0.36 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.52–7.66 (m, 2H, 4-Hpyridyl/7-Hquinazolinyl), 7.97 (t, J = 8.3 Hz, 1H, 8-Hquinazolinyl), 8.10 (d, J = 8.1 Hz, 1H, 9-Hquinazolinyl), 8.21 (d, J = 7.8 Hz, 1H, 6-Hquinazolinyl), 8.41 (d, J = 7.9 Hz, 1H, 4-Hpyridyl), 8.67–8.72 (m, 1H, 6-Hpyridyl), 9.26 (s, 1H, 2-Hpyridyl), 13.18 (s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 114.7 (1C, C-9quinazolinyl), 117.1 (1C, C-5aquinazolinyl), 124.1 (1C, C-5pyridyl), 126.1 (1C, C-3pyridyl), 126.3 (1C, C-7quinazolinyl), 128.4 (1C, C-6quinazolinyl), 133.6 (1C, C-4pyridyl), 135.4 (1C, C-8quinazolinyl), 135.6 (1C, C-9aquinazolinyl), 147.2 (1C, C-2pyridyl), 149.1 (1C, C-3aquinazolinyl), 150.8 (1C, C-6pyridyl), 158.5 (1C, C-2quinazolinyl), 159.7 (1C, C-5quinazolinyl, C=O). IR (neat): ṽ [cm−1] = 2708, 1690, 1609, 1582, 1524, 1481, 1416, 1393, 1319, 1123, 1038, 818, 752, 706, 633. HRMS (APCI): m/z = 264.0880 calculated for [M + H]+, found: 264.0898. HPLC: tR = 12.2 min, purity: 100.0%.
2-(pyridin-4-yl)-[1,2,4]triazolo[5,1-b]quinazolin-9(4H)-one (22). According to general procedure E, 22 was synthesized using 17 (50 mg, 127.8 μmol, 1.00 eq.), CuI (5 mg, 25.6 μmol, 0.20 eq.), Cs2CO3 (83 mg, 255.6 μmol, 2.00 eq.) and 1,10-phenanthroline (5 mg, 25.6 μmol, 0.20 eq.) in dry DMF (2 mL). The mixture was stirred at 80 °C for 1 h. After the first purification (CH2Cl2/CH3OH = 100/0 → 90/10), product 22 was obtained as a mixture with compound 23 (22 + 23, 23 mg, 86.6 μmol, 68%). According to HPLC measurement 22:23 ratio was 3:7. Synthesis in microwave conditions (procedure F) allowed increasing the yield of the isomers’ mixture up to 95% (22 + 23, 32 mg, 121.9 μmol), but the ratio remained the same. Further purification (CH2Cl2/CH3OH/DMEA = 95/4/1) gave pure product 22 as a colorless solid (9 mg, 35.7 μmol, 28%); M.p.: >300 °C. TLC: Rf = 0.29 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.40 (t, J = 7.4 Hz, 1H, 7-Hquinazolinyl), 7.53 (d, J = 8.2 Hz, 1H, 5-Hquinazolinyl), 7.85 (t, J = 7.3 Hz, 1H, 6-Hquinazolinyl), 8.07 (s, 2H, 3-Hpyridyl/5-Hpyridyl), 8.25 (d, J = 7.8 Hz, 1H, 8-Hquinazolinyl), 8.79 (s, 2H, 2-Hpyridyl/6-Hpyridyl), 13.38 (br s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 113.5 (1C, C-8aquinazolinyl), 117.2 (1C, C-5quinazolinyl), 120.8 (2C, C-3pyridyl/C-5pyridyl), 122.9 (1C, C-7quinazolinyl), 127.5 (1C, C-8quinazolinyl), 135.2 (1C, C-6quinazolinyl), 137.4 (1C, C-4pyridyl), 139.8 (1C, C-4aquinazolinyl), 150.6 (2C, C-2pyridyl/C-6pyridyl), 151.9 (1C, C-2quinazolinyl), 155.6 (1C, C-3aquinazolinyl), 159.8 (1C, C-9quinazolinyl, C=O). IR (neat): ṽ [cm−1] = 2978, 2924, 2662, 1697, 1643, 1608, 1570, 1481, 1427, 1389, 1207, 1161, 1065, 1003, 953, 837, 791, 748, 714, 679. HRMS (APCI): m/z = 264.0880 calculated for [M + H]+, found: 264.0862. HPLC: tR = 11.5 min, purity: 100.0%.
2-(pyridin-4-yl)-[1,2,4]triazolo[1,5-a]quinazolin-5(4H)-one (23). According to general procedure E, 23 was synthesized using 17′ (99 mg, 253.1 μmol, 1.00 eq.), CuI (10 mg, 50.6 μmol, 0.20 eq.), Cs2CO3 (165 mg, 506.2 μmol, 2.00 eq.) and 1,10-phenanthroline (9 mg, 50.6 μmol, 0.20 eq.) in dry DMF (2 mL). The mixture was stirred at 80 °C for 1 h. After purification, product 23 was obtained as a colorless solid (28 mg, 107.5 μmol, 42%); M.p.: >300 °C. TLC: Rf = 0.31 (DCM/MeOH = 95/5). 1H NMR (600 MHz, DMSO-d6) δ (in ppm) = 7.58–7.63 (m, 1H, 7-Hquinazolinyl), 7.97 (dd, J = 8.3, 1.2 Hz, 1H, 8-Hquinazolinyl), 8.00 (dd, J = 4.5, 1.6 Hz, 2H, 3-Hpyridyl/5-Hpyridyl), 8.10 (d, J = 8.1 Hz, 1H, 9-Hquinazolinyl), 8.22 (dd, J = 7.9, 1.0 Hz, 1H, 6-Hquinazolinyl), 8.75 (dd, J = 4.5, 1.4 Hz, 2H, 2-Hpyridyl/6-Hpyridyl), 13.21 (s, 1H, NH). 13C NMR (151 MHz, DMSO-d6) δ (in ppm) = 114.7 (1C, C-9quinazolinyl), 117.2 (1C, C-5aquinazolinyl), 120.2 (2C, C-3pyridyl/C-5pyridyl), 126.5 (1C, C-7quinazolinyl), 128.4 (1C, C-6quinazolinyl), 135.4 (1C, C-8quinazolinyl), 135.6 (1C, C-9aquinazolinyl), 137.3 (2C, C-4pyridyl), 149.2 (1C, C-3aquinazolinyl), 150.5 (2C, C-2pyridyl/C-6pyridyl), 158.5 (1C, C-2quinazolinyl), 159.7 (1C, C-5quinazolinyl, C=O). IR (neat): ṽ [cm−1] = 2978, 2920, 2662, 1682, 1609, 1485, 1427, 1381, 1119, 1011, 837, 748, 698, 679. HRMS (APCI): m/z = 264.0880 calculated for [M + H]+, found: 264.0902. HPLC: tR = 11.8 min, purity: 98.9%.
1-{5-[(4-methoxybenzyl)amino]-3-(pyridin-3-yl)-1H-pyrazol-1-yl}-2,2-dimethylpropan-1-one (24b). According to general procedure D, aminopyrazole 8b (100 mg, 357 µmol) was acylated using pivaloyl chloride (55 µL, 428 µmol, 1.20 eq.) in dry pyridine/THF (4.0 mL/2.0 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24b as a yellowish solid (56.7 mg, 44%). M.p.: 130 °C. TLC: Rf = 0.52 (CH/EtOAc = 1/1). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.01 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.59 (dd, J = 4.8/1.6 Hz, 1H, 6-Hpyridyl); 8.16 (ddd, J = 7.9/2.3/1.7 Hz, 1H, 4-Hpyridyl); 7.73 (t, J = 6.1 Hz, 1H, NH); 7.48 (dd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.38–7.33 (m, 2H, 2/6-Hmethoxyphenyl); 6.94–6.86 (m, 2H, 3/5-Hmethoxyphenyl); 6.03 (s, 1H, 4-Hpyrazolyl); 4.31 (d, J = 6.1 Hz, 2H, CH2); 3.72 (s, 3H, CH3); 1.48 (s, 9H, C(CH3)3). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.7 (1C, C=O); 158.4 (1C, C-4methoxyphenyl); 153.8 (1C, C-5pyrazolyl); 150.2 (1C, C-3pyrazolyl); 149.9 (1C, C-6pyridyl); 146.9 (1C, C-2pyridyl); 133.0 (1C, C-4pyridyl); 130.5 (1C, C-1methoxyphenyl); 128.8 (2C, C-2/6methoxyphenyl); 127.9 (1C, C-3pyridyl), 123.8 (1C, C-5pyridyl), 113.8 (2C, C-3/5methoxyphenyl); 82.6 (1C, C-4pyrazolyl); 54.9 (1C, CH3); 47.2 (1C, CH2); 41.7 (1C, C(CH3)3). IR (neat): ṽ [cm−1] = 3395, 3105, 1686, 1514, 1422, 1364, 1319, 1256, 1188, 1065, 1028, 943, 797, 772, 706. HRMS (APCI): m/z = 365.2107, calculated for C21H25N4O2+ [M + H]+ 365.1972. HPLC: tR = 20.1 min, purity: 99.6%.
2,2-dimethyl-1-(5-{[(naphthalen-1-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)propan-1-one (24c). According to general procedure D, aminopyrazole 8c (150 mg, 499 µmol) was acylated using pivaloyl chloride (75 µL, 599 µmol, 1.20 eq.) in dry pyridine/THF (6.0 mL/3.0 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24c as a beige solid (76.8 mg, 40%). M.p.: 130–131 °C. TLC: Rf = 0.20 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.02 (dd, J = 2.2/0.9 Hz, 1H, 2-Hpyridyl); 8.59 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 8.21–8.16 (m, 2H, 4-Hpyridyl, 9-Hnaphthyl); 7.99–7.96 (m, 1H, Hnaphthyl); 7.89–7.85 (m, 1H, Hnaphthyl); 7.80 (t, J = 6.0 Hz, NH); 7.65–7.54 (m, 3H, Hnaphthyl); 7.51–7.45 (m, 2H, Hnaphthyl, 5-Hpyridyl); 6.17 (s, 1H, 4-Hpyrazolyl); 4.89 (d, J = 5.9 Hz, 2H, CH2), 1.49 (s, 9H, C(CH3)3). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.9 (1C, C=O); 154.0 (1C, C-5pyrazolyl); 150.3 (1C, C-3pyrazolyl); 149.8 (1C, C-6pyridyl); 147.0 (1C, C-2pyridyl); 133.4 (1C, Cnaphthyl); 133.4 (1C, Cnaphthyl); 133.0 (1C, C-4pyridyl); 130.8 (1C, Cnaphthyl); 128.5 (1C, Cnaphthyl); 127.8 (1C, C-3pyridyl); 127.7 (1C, Cnaphthyl); 126.3 (1C, Cnaphthyl); 125.8 (1C, Cnaphthyl); 125.4 (1C, Cnaphthyl); 125.1 (1C, Cnaphthyl); 123.8 (1C, Cnaphthyl); 123.4 (1C, C-9naphthyl); 82.7 (1C, C-4pyrazolyl); 46.0 (1C, CH2); 41.8 (1C, C(CH3)3); 27.1 (3C, C(CH3)3). IR (neat): ṽ [cm−1] = 1738, 1682, 1572, 1422, 1366, 1317, 1217, 1190, 959, 795, 766, 708. HRMS (APCI): m/z = 385.2031, calculated for C24H25N4O+ [M + H]+ 385.2023. HPLC: tR = 21.8 min, purity: 99.6%.
1-(5-{[(furan-2-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (24d). According to general procedure D, aminopyrazole 8d (130 mg, 541 µmol) was acylated using pivaloyl chloride (80 µL, 649 µmol, 1.20 eq.) in dry pyridine/THF (6.0 mL/3.0 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24d as a brownish solid (99.1 mg, 56%). M.p.: 114–115 °C. TLC: Rf = 0.17 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.03 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.60 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 8.18 (ddd, J = 7.9/2.3/1.7 Hz, 1H, 4-Hpyridyl); 7.65 (t, J = 6.1 Hz, 1H, NH); 7.61 (dd, J = 1.9/0.9 Hz, 1H, 5-Hfuranyl); 7.49 (ddd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl); 6.48–6.45 (m, 1H, 3-Hfuranyl); 6.40 (dd, J = 3.2/1.8 Hz, 1H, 4-Hfuranyl); 6.15 (s, 1H, 4-Hpyrazolyl); 4.40 (d, J = 6.0 Hz, 2H, CH2); 1.48 (s, 9H, C(CH3)3). IR (neat): ṽ [cm−1] = 3383, 1676, 1597, 1520, 1418, 1335, 1229, 1053, 980, 949, 814, 748, 706. HRMS (APCI): m/z = 325.1671, calculated for C18H21N4O2+ [M + H]+ 325.1659. HPLC: tR = 19.0 min, purity: 99.3%.
1-(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (24e). According to general procedure D, aminopyrazole 8e (150 mg, 516 µmol) was acylated using pivaloyl chloride (75 µL, 619 µmol, 1.20 eq.) in dry pyridine/THF (6.0 mL/3.0 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24e as a yellowish solid (101 mg, 52%). M.p.: 96–97 °C. TLC: Rf = 0.13 (CH/EtOAc = 80/20). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.02 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl); 8.60 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl); 8.17 (ddd, J = 7.9/2.2/1.7 Hz, 1H, 4-Hpyridyl); 7.88 (m, 1H, NH); 7.49 (ddd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl); 7.06 (dd, J = 3.7/0.9 Hz, 1H, 3-Hchlorothiophenyl); 6.97 (d, J = 3.7 Hz, 1H, 4-Hchlorothiophenyl); 6.14 (s, 1H, 4-Hpyrazolyl); 4.51 (dd, J = 6.4/1.0 Hz, 2H, CH2); 1.48 (s, 9H, C(CH3)3). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.5 (1C, C=O); 153.2 (1C, C-5pyrazolyl); 150.1 (1C, C-3pyrazolyl); 149.9 (1C, C-6pyridyl); 147.0 (1C, C-2pyridyl); 141.5 (1C, C-2chlorothiophenyl); 132.9 (1C, C-4pyridyl); 127.8 (1C, C-3pyridyl); 126.9 (1C, C-5chlorothiophenyl); 126.2 (1C, C-4chlorothiophenyl); 126.1 (1C, C-3chlorothiophenyl); 123.9 (1C, C-5pyridyl); 82.9 (1C, C-4pyrazolyl); 43.2 (1C, CH2); 41.7 (1C, C(CH3)3); 27.1 (3C, C(CH3)3). IR (neat): ṽ [cm−1] = 3374, 1692, 1574, 1454, 1422, 1364, 1317, 1217, 1005, 795. HRMS (APCI): m/z = 375.1177, calculated for C18H20ClN4OS+ [M + H]+ 375.1041. HPLC: tR = 21.0 min, purity: 99.2%.
1-(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-phenyl-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (24g). According to general procedure D, aminopyrazole 8g (60 mg, 207 µmol) was acylated using pivaloyl chloride (30 µL, 248 µmol, 1.20 eq.) in dry pyridine/THF (2.0 mL/1.2 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24g as a yellowish solid (34.2 mg, 44%). TLC: Rf = 0.51 (CH/EtOAc = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 9.04 (dd, J = 2.2/0.9 Hz, 1H, Hphenyl); 8.61 (dd, J = 4.8/1.6 Hz, 1H, Hphenyl); 8.20 (ddd, J = 7.9/1.9 Hz, 1H, Hphenyl); 7.65 (t, J = 6.1 Hz, 1H, NH); 7.60 (ddd, J = 1.9/0.9/0.3 Hz, 1H, Hphenyl); 7.51 (ddd, J = 7.9/4.8/0.9 Hz, 1H, Hphenyl); 6.46 (d, J = 3.2/0.8 Hz, 1H, 3-Hchlorothiophenyl); 6.40 (dd, J = 3.2/1.9 Hz, 1H, 4-Hchlorothiophenyl); 6.15 (s, 1H, 4-Hpyrazolyl); 4.40 (d, J = 6.0 Hz, 2H, CH2); 1.48 (s, 9H, C(CH3)3). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.7 (1C, C=O); 153.6 (1C, C-5pyrazolyl); 151.5 (1C, C-2chlorothiophenyl); 150.1 (1C, C-3pyrazolyl); 149.7 (1C, Cphenyl); 146.8 (1C, Cphenyl); 142.5 (1C, Cphenyl); 133.1 (1C, Cphenyl); 127.9 (1C, C-5chlorothiophenyl); 123.9 (1C, Cphenyl); 110.4 (1C, C-4chlorothiophenyl); 107.8 (1C, C-3chlorothiophenyl); 82.7 (1C, C-4pyrazolyl); 41.7 (1C, C(CH3)3); 41.0 (1C, CH2); 27.1 (3C, C(CH3)3). IR (neat): ṽ [cm−1] = 3387, 2928, 1682, 1578, 1522, 1504, 1481, 1450, 1395, 1358, 1327, 1234, 1200, 1061, 1003, 945, 193, 731. HRMS (APCI): m/z = 374.1091, calculated for C19H21ClN3OS+ [M + H]+ 374.1088. HPLC: tR = 26.5 min, purity: 98.1%.
1-[5-(benzylamino)-3-cyclohexyl-1H-pyrazol-1-yl]-2,2-dimethylpropan-1-one (24h). According to general procedure D, aminopyrazole 8h (100 mg, 392 µmol) was acylated using pivaloyl chloride (60 µL, 470 µmol, 1.20 eq.) in dry pyridine/THF (4.0 mL/2.0 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24h as a colorless solid (70.1 mg, 53%). M.p.: 47 °C. TLC: Rf = 0.68 (CH/EtOAc = 95/5). 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.56 (t, J = 6.1 Hz, 1H, NH); 7.39–7.30 (m, 4H, 2/4/5/6-Hphenyl); 7.25 (t, J = 6.7 Hz, 1H, 4-Hphenyl); 5.21 (s, 1H, 4-Hpyrazolyl); 4.28 (d, J = 5.9 Hz, 2H, CH2); 2.45–2.32 (m, 1H, Hcyclohexyl); 1.89–1.79 (m, 2H, Hcyclohexyl); 1.74–1.66 (m, 2H, Hcyclohexyl); 1.65–1.58 (m, 1H, Hcyclohexyl); 1.41 (s, 9H, C(CH3)3); 1.37–1.24 (m, 4H, Hcyclohexyl); 1.25–1.13 (m, 1H, Hcyclohexyl). 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 179.5 (1C, C=O); 160.0 (1C, C-3pyrazolyl); 152.9 (1C, C-5pyrazolyl); 138.8 (1C, C-1phenyl); 128.2 (2C, C-2/6phenyl); 127.2 (1C, C-3/5phenyl); 126.9 (1C, C-4phenyl); 82.9 (1C, C-4pyrazolyl); 47.9 (1C, CH2); 41.5 (1C, C(CH3)3); 36.9 (1C, Ccyclohexyl); 31.3 (2C, Ccyclohexyl); 27.0 (3C, C(CH3)3); 25.6 (1C, Ccyclohexyl); 25.4 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3406, 2918, 2851, 1682, 1584, 1518, 1481, 1391, 1337, 1227, 1157, 1119, 993, 976, 947, 814, 737. HRMS (APCI): m/z = 340.2431, calculated for C21H30N3O+ [M + H]+ 340.2383. HPLC: tR = 28.0 min, purity: 98.0%.
1-(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-cyclohexyl-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (24i). According to general procedure D, aminopyrazole 8i (50 mg, 169 µmol) was acylated using pivaloyl chloride (25 µL, 203 µmol, 1.20 eq.) in dry pyridine/THF (2.0 mL/1.2 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 24i as a yellowish solid (19.3 mg, 30%). M.p.: 92–93 °C. TLC: Rf = 0.63 (CH/EtOAc = 95/5). 1H-NMR (600 MHz, DMSO-d6): δ (ppm) = 7.61 (t, J = 6.2 Hz, 1H, NH); 6.98–6.94 (m, 2H, 3/4-Hchlorothiophenyl); 5.35 (s, 1H, 4-Hpyrazolyl); 4.40 (d, J = 6.0 Hz, 2H, CH2); 2.47–2.38 (m, 1H, Hcyclohexyl); 1.92–1.81 (m, 2H, Hcyclohexyl); 1.76–1.68 (m, 2H, Hcyclohexyl); 1.66–1.58 (m, 1H, Hcyclohexyl); 1.40 (s, 9H, C(CH3)3); 1.37–1.27 (m, 4H, Hcyclohexyl); 1.25–1.14 (m, 1H, Hcyclohexyl). 13C-NMR (151 MHz, DMSO-d6): δ (ppm) = 179.8 (1C, C=O); 160.5 (1C, C-3pyrazolyl); 152.8 (1C, C-5pyrazolyl); 142.4 (1C, C-2chlorothiophenyl); 127.2 (1C, C-5chlorothiophenyl); 126.8 (1C, C-4chlorothiophenyl); 126.2 (1C, C-3chlorothiophenyl); 84.0 (1C, C-4pyrazolyl); 43.7 (1C, CH2); 42.0 (1C, C(CH3)3); 37.4 (1C, Ccyclohexyl); 31.9 (2C, Ccyclohexyl); 27.6 (3C, C(CH3)3); 26.1 (1C, Ccyclohexyl); 25.9 (2C, Ccyclohexyl). IR (neat): ṽ [cm−1] = 3397, 2922, 2851, 1682, 1585, 1518, 1452, 1391, 1364, 1335, 1261, 1227, 1117, 1003, 976, 941, 814, 797. HRMS (APCI): m/z = 380.1563, calculated for C19H27ClN3OS+ [M + H]+ 380.1558. HPLC: tR = 28.5 min, purity: 96.6%.
(5-{[(5-chlorothiophen-2-yl)methyl]amino}-3-(pyridin-3-yl)-1H-pyrazol-1-yl)(phenyl)methanone (25). According to general procedure D, aminopyrazole 8e (200 mg, 688 µmol) was acylated using benzoyl chloride (79.9 µL, 688 µmol, 1.00 eq.) in dry pyridine/THF (6.4 mL/3.2 mL). Flash column chromatography (CH/EtOAc = 1/0 → 0/1) yielded 25 as a colorless solid (152 mg, 56%). M.p.: 127–128 °C. TLC: Rf = 0.21 (CH/EtOAc = 70/30). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 4.59 (d, J = 6.3 Hz, 2H, CH2), 6.28 (s, 1H, 4-Hpyrazolyl), 6.98 (d, J = 3.8 Hz, 1H, 4-Hthiophenyl), 7.10–7.19 (m, 1H, 3-Hthiophenyl), 7.46 (ddd, J = 7.9/4.7/0.9 Hz, 1H, 5-Hpyridyl), 7.54–7.58 (m, 2H, 3/5-Hbenzoyl), 7.64–7.67 (m, 1H, 4-Hbenzoyl), 7.96 (t, J = 6.3 Hz, 1H, NH), 8.06–8.09 (m, 2H, 2/6-Hbenzoyl), 8.10–8.13 (m, 1H, 4-Hpyridyl), 8.59 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl), 8.99 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 43.4 (1C, CH2), 84.1 (1C, C-4pyrazolyl), 123.9 (1C, C-5pyridyl), 126.2 (1C, C-3thiophenyl), 126.3 (1C, C-4thiophenyl), 127.0 (1C, C-5thiophenyl), 127.7 (1C, C-3pyridyl), 127.9 (2C, C-3/5benzoyl), 130.9 (2C, C-2/6benzoyl), 132.5 (1C, C-4benzoyl), 132.8 (1C, C-1benzoyl), 133.2 (1C, C-4pyridyl), 141.5 (1C, C-2thiophenyl), 147.2 (1C, C-2pyridyl), 150.1 (1C, C-6pyridyl), 151.6 (1C, C-3pyrazolyl), 153.3 (1C, C-5pyrazolyl), 169.3 (1C, CON). IR (neat): ṽ [cm−1] = 3302, 3059, 1685, 1585, 1573, 1512, 1454, 1446, 1419, 1346, 1327, 1226, 1199, 1056, 999, 914, 794, 736, 705, 690, 659. HRMS (APCI): m/z = 395.0728 calculated for [M + H]+, found: 395.0733. HPLC: tR = 20.2 min, purity: 93.2%.
N-[3-(pyridin-3-yl)-1H-pyrazol-5-yl]benzamide (26a). According to general procedure D, aminopyrazole 7a (56.0 mg, 350 µmol) was acylated using BzCl (40.6 µL, 350 µmol, 1.00 eq.) in dry pyridine/THF (3.00 mL/1.50 mL) and stirred first for 1 h at 0 °C and then for 2 h at rt. Flash column chromatography (CH2Cl2/CH3OH = 95/5) yielded 26a as a colorless solid (63 mg, 238 µmol, 68%). M.p.: 270 °C (decomp.). TLC: Rf = 0.15 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 7.19 (bs, 0.2H, 4-Hpyrazolyl*), 7.19 (bs, 0.8H, 4-Hpyrazolyl), 7.43–7.65 (m, 4H, 3/4/5-Hbenzoyl, 5-Hpyridyl), 7.99–8.08 (m, 2H, 2/6-Hbenzoyl), 8.13–8.19 (m, 1H, 4-Hpyridyl), 8.50–8.59 (m, 1H, 6-Hpyridyl), 8.98–9.05 (m, 1H, 2-Hpyridyl), 10.91 (bs, 0.8H, CONH), 10.91 (bs, 0.2H, CONH*), 12.86 (bs, 0.2H, NH*), 13.13 (bs, 0.8H, NH). The ratio of tautomers is 8:2, the minor tautomer is marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 95.5 (1C, C-4pyrazolyl), 124.0 (1C, C-5pyridyl), 125.4 (1C, C-3pyridyl), 127.8 (2C, C-2/6benzoyl), 128.3 (2C, C-3/5benzoyl), 131.6 (1C, C-4benzoyl), 132.3 (1C, C-4pyridyl), 134.0 (1C, C-1benzoyl), 138.9 (1C, C-3pyrazolyl), 146.1 (1C, C-2pyridyl), 148.6 (1C, C-5pyrazolyl), 149.0 (1C, C-6pyridyl), 164.6 (1C, CONH). The signals of the major tautomer are given. IR (neat): ṽ [cm−1] = 3275, 1651, 1577, 1562, 1543, 1311, 1002, 960, 894, 790, 705, 690, 655. HRMS (APCI): m/z = 265.1084 calculated for [M + H]+, found: 265.1058. HPLC: tR = 12.4 min, purity: 99.4%.
N-(3-phenyl-1H-pyrazol-5-yl)benzamide (26b). According to general procedure D, aminopyrazole 7b (50.0 mg, 314 µmol) was acylated using BzCl (36.5 µL, 314 µmol, 1.00 eq.) in dry pyridine/THF (3.00 mL/1.50 mL) and stirred first for 1 h at 0 °C and then for 2 h at rt. The solid was filtered off and washed with H2O (2×) to yield 26b as a colorless solid (64 mg, 77%). M.p.: 194–195 °C. TLC: Rf = 0.13 (CH/EtOAc = 60/40). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 6.55 (bs, 0.1H, 4-Hpyrazolyl*), 7.07 (bs, 0.9H, 4-Hpyrazolyl), 7.32–7.39 (m, 1H, 4-Hphenyl), 7.44–7.49 (m, 2H, 3/5-Hphenyl), 7.49–7.53 (m, 2H, 3/5-Hbenzoyl), 7.55–7.60 (m, 1H, 4-Hbenzoyl), 7.73–7.83 (m, 2H, 2/6-Hphenyl), 8.00–8.07 (m, 2H, 2/6-Hbenzoyl), 10.85 (bs, 0.9H, CONH), 11.07 (bs, 0.1H, CONH*), 12.66 (bs, 0.1H, NH*), 12.95 (bs, 0.9H, NH). The ratio of tautomers is 9:1, the minor tautomer is marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 94.8 (1C, C-4pyrazolyl), 125.0 (2C, C-2/6phenyl), 127.8 (2C, C-2/6benzoyl), 128.1 (1C, C-4phenyl), 128.3 (2C, C-3/5benzoyl), 129.0 (2C, C-3/5phenyl), 129.4 (1C, C-1phenyl), 131.6 (1C, C-4benzoyl), 134.1 (1C, C-1benzoyl), 141.8 (1C, C-3pyrazolyl), 148.3 (1C, C-5pyrazolyl), 164.6 (1C, CONH). The signals of the major tautomer are given. IR (neat): ṽ [cm−1] = 3282, 3059, 1654, 1570, 1543, 1489, 1307, 1280, 1002, 894, 794, 759, 675. HRMS (APCI): m/z = 264.1131 calculated for [M + H]+, found: 264.1107. HPLC: tR = 17.5 min, purity: 99.6%.
3-oxo-3-phenylpropanenitrile (28b). Under N2, at ™78 °C, n-BuLi (4.58 mL, 7.32 mmol, 1.10 eq, 1.6m in n-hexane) was added dropwise over 20 min to a solution of CH3CN (417 µL, 7.99 mmol, 1.20 eq.) in dry THF (12.0 mL) and the reaction mixture was stirred for 1 h. A solution of ethyl benzoate (952 µL, 6.66 mmol, 1.00 eq.) in dry THF (3.00 mL) was added dropwise to the reaction mixture at ™78 °C and the mixture was allowed to warm up to rt over 16 h. The suspension was quenched with aqueous HCl (4M), the aqueous phase was extracted with EtOAc (3×), the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (CH/EtOAc = 1/0 → 50/50) to yield 28b as a yellow solid (566 mg, 3.90 mmol, 59%). M.p.: 79–80 °C. TLC: Rf = 0.37 (CH/EtOAc = 60/40). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 4.76 (s, 2H, CH2), 7.53–7.61 (m, 2H, 3/5-Hphenyl), 7.68–7.73 (m, 1H, 4-Hphenyl), 7.90–7.97 (m, 2H, 2/6-Hphenyl). 13C NMR (101 MHz, DMSO-d6): δ (ppm) = 30.0 (1C, CH2), 115.9 (1C, CN), 128.4 (2C, C-2/6phenyl), 128.9 (2C, C-3/5phenyl), 134.2 (1C, C-4phenyl), 134.6 (1C, C-1phenyl), 189.7 (1C, CO). IR (neat): ṽ [cm−1] = 3360, 3070, 2978, 2954, 2924, 2256, 1685, 1597, 1581, 1450, 1392, 1327, 1215, 999, 925, 752, 682. HRMS (APCI): m/z = 146.0600 calculated for [M + H]+, found: 146.0569. HPLC: tR = 14.4 min, purity: 94.6%.
[5-amino-3-(pyridin-3-yl)-1H-pyrazol-1-yl](phenyl)methanone (29a). According to general procedure B, β-ketonitrile sodium enolate 6a (200 mg, 1.19 mmol, 1.00 eq.) was dissolved in aqueous HCl (5 mL, 1M), followed by work-up as described above. The orange oily residue of the keto-form of β-ketonitrile (28a) and benzhydrazide (113 mg, 833 µmol, 0.70 eq.) were reacted in a solution of methanesulfonic acid (7.7 µL, 103 µmmol, 0.1 eq.) in dry EtOH (2.00 mL). Flash column chromatography (CH2Cl2/CH3OH = 1/0 → 90/10) yielded 29a as a yellow solid (124 mg, 56%, calculated for 0.70 eq benzhydrazide). TLC: Rf = 0.25 (CH2Cl2/CH3OH = 95/5). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 5.99 (s, 1H, 4-Hpyrazolyl), 6.93 (bs, 2H, NH2), 7.45 (ddd, J = 7.9/4.8/0.9 Hz, 1H, 5-Hpyridyl), 7.54–7.58 (m, 2H, 3/5-Hbenzoyl), 7.64–7.68 (m, 1H, 4-Hbenzoyl), 8.06–8.09 (m, 2H, 2/6-Hbenzoyl), 8.11 (ddd, J = 7.9/2.3/1.7 Hz, 1H, 4-Hpyridyl), 8.58 (dd, J = 4.8/1.7 Hz, 1H, 6-Hpyridyl), 8.96 (dd, J = 2.3/0.9 Hz, 1H, 2-Hpyridyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 84.9 (1C, C-4pyrazolyl), 123.8 (1C, C-5pyridyl), 127.88 (2C, C-3/5benzoyl), 127.91 (1C, C-3pyridyl), 130.9 (2C, C-2/6benzoyl), 132.4 (1C, C-4benzoyl), 133.0 (1C, C-1benzoyl), 133.2 (1C, C-4pyridyl), 147.2 (1C, C-2pyridyl), 149.9 (1C, C-6pyridyl), 151.5 (1C, C-3pyrazolyl), 153.3 (1C, C-5pyrazolyl), 169.5 (1C, CON). IR (neat): ṽ [cm−1] = 3452, 3275, 3197, 3066, 1681, 1627, 1597, 1581, 1450, 1365, 1334, 1315, 941, 921, 740, 721, 698. HRMS (APCI): m/z = 265.1084 calculated for [M + H]+, found: 265.1068. HPLC: tR = 14.2 min, purity: 98.3%.
(5-amino-3-phenyl-1H-pyrazol-1-yl)(phenyl)methanone (29b). According to general procedure B, β-ketonitrile keto-form 28b (150 mg, 1.03 mmol, 1.00 eq.) and benzhydrazide (141 mg, 1.03 mmol, 1.00 eq.) were reacted in a solution of methanesulfonic acid (6.7 µL, 103 µmmol, 0.1 eq.) in dry EtOH (2.00 mL). Flash column chromatography (CH/EtOAc = 1/0 → 50/50) yielded 29b as a yellow solid (264 mg, 97%). TLC: Rf = 0.56 (CH/EtOAc = 60/40). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 5.90 (s, 1H, 4-Hpyrazolyl), 6.85 (bs, 2H, NH2), 7.36–7.44 (m, 3H, 3/4/5-Hphenyl), 7.54–7.58 (m, 2H, 3/5-Hbenzoyl), 7.63–7.67 (m, 1H, 4-Hbenzoyl), 7.74–7.77 (m, 2H, 2/6-Hphenyl), 8.05–8.08 (m, 2H, 2/6-Hbenzoyl). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 84.9 (1C, C-4pyrazolyl), 126.0 (2C, C-2/6phenyl), 127.8 (2C, C-3/5benzoyl), 128.6 (2C, C-3/5phenyl), 129.0 (1C, C-4phenyl), 130.8 (2C, C-2/6benzoyl), 132.1 (1C, C-1phenyl), 132.3 (1C, C-4benzoyl), 133.2 (1C, C-1benzoyl), 153.2 (1C, C-5pyrazolyl), 153.9 (1C, C-3pyrazolyl), 169.5 (1C, CON). IR (neat): ṽ [cm−1] = 3406, 3294, 3209, 3116, 1670, 1612, 1577, 1473, 1446, 1361, 1311, 1222, 1188, 1134, 921, 752, 678, 651. HRMS (APCI): m/z = 264.1131 calculated for [M + H]+, found: 264.1150. HPLC: tR = 21.1 min, purity: 98.3%.
2-fluoro-3-oxo-3-phenylpropanenitrile (
31). Synthesis was performed as previously reported [
29]. Fluoroacetonitrile (300 mg, 5.1 mmol, 1.00 eq.) and Ph
2P(O)Cl (1.20 g, 5.1 mmol, 1.00 eq.) were dissolved in dry THF (45 mL) in an inert atmosphere. At ™78 °C, LiHMDS (10 mL, 10.2 mmol, 1 M in THF, 2.00 eq.) was slowly added, leading to a red solution. After stirring for 15 min at ™78 °C, benzoyl chloride (786 mg, 5.6 mmol, 1.10 eq.) was added dropwise and the temperature was allowed to rise to room temperature. After 1 h, saturated brine (30 mL) was added to the reaction mixture, and the aqueous layer was extracted with diethyl ether (3 × 30 mL). The combined organic layers were dried with Na
2SO
4. After filtration and evaporation of the solvents in vacuo, the crude product was purified by flash chromatography (hexane/EtOAc 7:3,
Rf = 0.30) to yield product
31 as a red oil (144 mg, 0.9 mmol, 17%).
1H NMR (400 MHz, CDCl
3)
δ (in ppm) = 6.15 (dt,
J = 46.7, 1.9 Hz, 1H, CHF), 7.57 (t,
J = 8.0 Hz, 2H, 3-H
phenyl/5-H
phenyl), 7.72 (t,
J = 7.5 Hz, 4-H
phenyl), 8.00 (d,
J = 7.5 Hz, 2H, 2-H
phenyl/6-H
phenyl).
1H NMR spectral data of
31 were in accordance with those reported in the literature [
29].
3-amino-4-fluoro-5-phenyl-1H-pyrazole (
32). Synthesis was performed as previously reported [
29]. In a 50 mL flask,
31 (100 mg, 613.0 μmol, 1.00 eq.) of was dissolved in isopropanol (2.2 mL). To this solution, hydrazine hydrate (31 mg, 98% in water, 613.0 μmol, 1.00 eq.) was added and the mixture was refluxed for 3 h. After completion of the reaction, the solvents were evaporated in vacuo and the crude oil was purified by flash chromatography (EtOAc,
Rf = 0.33) to yield product
31 as an orange solid (60 mg, 389.0 μmol, 55% yield).
1H NMR (400 MHz, CDCl
3):
δ (in ppm) = 3.67 (br s, 2H, NH
2), 7.34–7.38 (m, 1H, 4-H
phenyl), 7.42–7.47 (m, 2H, 3-H
phenyl/5-H
phenyl), 7.58–7.60 (m, 2H, 2-H
phenyl/6-H
phenyl), NH invisible.
19F NMR (376 MHz, CDCl
3):
δ (in ppm) = –186.2 (1F, s, CF).
13C NMR (101 MHz, CDCl
3):
δ (in ppm) = 125.3 (d,
J = 3.8 Hz, 2C, C-2
phenyl/C-6
phenyl), 127.5 (1C, C-1
phenyl), 127.9 (d,
J = 4.3 Hz, 1C, C-5
pyrazolyl), 128.7 (1C, C-4
phenyl), 129.2 (2C, C-3
phenyl/C-5
phenyl), 135.7 (d,
JC-F = 244.1 Hz, 1C, C-4
pyrazolyl), C-3
pyrazolyl invisible.
1H and
13C NMR spectral data of
31 were in accordance with those reported in the literature [
29]. HRMS (APCI):
m/
z = 178.0775 calculated for [M + H]
+, found: 178.0784.
N-((5-chlorothiophen-2-yl)methyl)-4-fluoro-5-phenyl-1H-pyrazol-3-amine (33). Under N2, 32 (54 mg, 305.0 μmol, 1.00 eq.) was dissolved in dry EtOH (3 mL). To this solution, glacial acetic acid (18 mg, 305.0 μmol, 1.00 eq.) and molecular sieves (3 Å) were added. After cooling the mixture to 0 °C, 5-chlorothiophene-2-carbaldehyde (67 mg, 457.0 μmol, 1.50 eq.) was added dropwise over a period of 10 min, and the solution was stirred at 0 °C for 1 h and for another 23 h, while gradually warming to room temperature. After cooling the solution again to 0 °C, NaBH4 (69 mg, 1.8 mmol, 6.00 eq.) was added, and the mixture was stirred for another 24 h, while gradually warming to room temperature. The suspension was cooled to 0 °C and diluted with H2O (3 mL) and HCl (1 M, 3 mL, aq.), filtered over Celite® and was washed with H2O (10 mL), CH3OH (10 mL) and EtOAc (10 mL). The organic layer was washed with HCl (1 M, 3x10 mL, aq.) and subsequent neutralization of the combined aqueous layers were carried out with NaOH (1 M, 30 mL, aq.). After re-extraction of the combined aqueous layers with EtOAc (3x50 mL), the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure, and the residue was purified by flash column chromatography (CH/EtOAc 1:0 → 0:1) to yield product 33 as a colorless solid (72 mg, 234.0 μmol, 77%). M.p.: 121–122 °C. 1H NMR (600 MHz, CDCl3): δ (in ppm) = 3.92 (br s, 1H, NHCH2); 4.56 (s, 2H, CH2); 6.75 (d, J = 3.7 Hz, 1H, 3-Hchlorothiophenyl); 6.80 (d, J = 3.8 Hz, 1H, 4-Hchlorothiophenyl); 7.37 (t, J = 7.5 Hz, 1H, 4-Hphenyl); 7.45 (t, J = 7.7 Hz, 2H, 3-Hphenyl/5-Hphenyl); 7.56 (t, J = 7.0 Hz, 2H, 2-Hphenyl/6-Hphenyl); 8.86 (br s, 1H, NH). 19F NMR (376 MHz, CDCl3): δ (in ppm) = –186.0 (1F, s, CF). 13C NMR (151 MHz, CDCl3): δ (in ppm) = 43.6 (1C, CHz); 124.8 (1C, C-3chlorothiophenyl); 125.3 (d, JC-F = 3.5 Hz, 2C, C-2phenyl/C-6phenyl); 125.8 (1C, C-4chlorothiophenyl); 127.6 (1C, C-1phenyl), 128.8 (1C, C-4phenyl); 129.2 (1C, C-2chlorothiophenyl); 129.3 (2C, C-3phenyl/C-5phenyl); 133.5 (d, JC-F = 256.7 Hz, 1C, C-4pyrazolyl); 141.9 (1C, C-5chlorothiophenyl); 144.6 (1C, C-3pyrazolyl); C-5pyrazolyl invisible. IR (neat): ṽ [cm–1] = 3381, 3177, 2901, 2857, 1560, 1518, 1456, 1443, 1420, 1354, 1236, 1177, 1065, 1003, 959, 800, 762, 719, 685, 662. HRMS (APCI): m/z = 308.0419 calculated for [M + H]+, found: 308.0413.
1-(5-(((5-chlorothiophen-2-yl)methyl)amino)-4-fluoro-3-phenyl-1H-pyrazol-1-yl)-2,2-dimethylpropan-1-one (34a). According to general procedure D, aminopyrazole 33 (81 mg, 262.0 μmol, 1.00 eq.) was acylated using pivaloyl chloride (38 mg, 314.0 μmol, 1.20 eq.) in dry pyridine/THF (3.0 mL/1.5 mL). Flash column chromatography (CH/EtOAc = 9/1) yielded 34a as a colorless oil (22 mg, 21%). 1H NMR (600 MHz, CDCl3): δ (in ppm) = 1.53 (s, 9H, t-Bu); 4.63 (ddd, J = 6.7, 1.7, 0.9 Hz, 2H, NHCH2); 6.76 (d, J = 3.8 Hz, 1H, 3-Hchlorothiophenyl); 6.79 (d, J = 3.8 Hz, 1H, 4-Hchlorothiophenyl); 7.14 (t, J = 6.7 Hz, 1H, NH); 7.40 (t, J = 7.3 Hz, 1H, 4-Hphenyl); 7.45 (t, J = 7.3 Hz, 2H, 3-Hphenyl/5-Hphenyl); 7.91 (d, J = 7.0 Hz, 2H, 2-Hphenyl/6-Hphenyl). 19F NMR (376 MHz, CDCl3): δ (in ppm) = –184.9 (1F, s, CF). 13C NMR (151 MHz, CDCl3): δ (in ppm) = 27.9 (3C, C(CH3)3); 42.6 (1C, C(CH3)3); 43.0 (d, J = 6.3 Hz, 1C, CH2), 125.0 (1C, C-3chlorothiophenyl); 126.0 (1C, C-4chlorothiophenyl); 126.9 (d, J = 4.1 Hz, 2C, C-2phenyl/C-6phenyl); 128.8 (2C, C-3phenyl/C-5phenyl); 129.4 (1C, C-4phenyl); 129.5 (1C, C-2chlorothiophenyl); 130.5 (d, J = 4.0 Hz, 1C, C-1phenyl); 132.2 (d, JC-F = 242.8 Hz, 1C, C-4pyrazolyl); 137.0 (d, JC-F = 17.3 Hz, 1C, C-3pyrazolyl); 141.1 (d, JC-F = 6.9 Hz, 1C, C-5chlorothiophenyl), 141.5 (d, JC-F = 6.9 Hz, 1C, C-5pyrazolyl), 181.7 (1C, CO). IR (neat): ṽ [cm–1] = 3366, 2957, 2930, 2870, 1694, 1632, 1518, 1416, 1395, 1360, 1323, 1234, 1001, 943, 795, 770, 692. HRMS (APCI): m/z = 392.0994 calculated for [M + H]+, found: 392.1027. HPLC: tR = 27.3 min, purity: 99.1%.
(5-(((5-chlorothiophen-2-yl)methyl)amino)-4-fluoro-3-phenyl-1H-pyrazol-1-yl)(phenyl)methanone (34b). According to general procedure D, aminopyrazole 33 (79 mg, 255.0 μmol, 1.00 eq.) was acylated using benzoyl chloride (43 mg, 306.0 μmol, 1.20 eq.) in dry pyridine/THF (3.0 mL/1.5 mL). Flash column chromatography (CH/EtOAc = 9/1) yielded 34b as a yellowish oil (36 mg, 34%). 1H NMR (600 MHz, CDCl3): δ (in ppm) = 4.71 (d, J = 6.8 Hz, 2H, NHCH2); 6.78 (d, J = 3.7 Hz, 1H, 3-Hchlorothiophenyl); 6.84 (d, J = 3.8 Hz, 1H, 4-Hchlorothiophenyl); 7.16 (t, J = 6.7 Hz, 1H, NH); 7.45–7.38 (m, 3H, 3-Hphenyl/4-Hphenyl/5-Hphenyl); 7.52–7.47 (m, 2H, 3-Hbenzoyl/5-Hbenzoyl); 7.60 (ddt, J = 8.8, 7.0, 1.3 Hz, 1H, 4-Hbenzoyl); 7.87 (ddt, J = 6.9, 2.3, 1.1 Hz, 2H, 2-Hphenyl/6-Hphenyl); 8.11 (dd, J = 8.4, 1.4 Hz, 2H, 2-Hbenzoyl/6-Hbenzoyl). 19F NMR (376 MHz, CDCl3): δ (in ppm) = –182.9 (1F, s, CF). 13C NMR (151 MHz, CDCl3): δ (in ppm) = 43.1 (d, J = 5.8 Hz, 1C, CH2); 125.1 (1C, C-3chlorothiophenyl); 126.1 (1C, C-4chlorothiophenyl); 127.1 (d, J = 3.9 Hz, 2C, C-2phenyl/C-6phenyl); 128.0 (2C, C-3benzoyl/C-5benzoyl); 128.8 (2C, C-3phenyl/C-5phenyl); 129.6 (1C, C-4phenyl); 129.7 (1C, C-2chlorothiophenyl); 130.1 (d, JC-F = 3.9 Hz, 1C, C-1phenyl); 131.7 (2C, C-2benzoyl/C-6benzoyl); 132.3 (1C, C-1benzoyl); 132.8 (d, JC-F = 244.5 Hz, 1C, C-4pyrazolyl); 132.9 (1C, C-4benzoyl); 137.32 (d, JC-F = 17.9 Hz, 1C, C-3pyrazolyl); 141.0 (1C, C-5chlorothiophenyl); 143.3 (d, JC-F = 7.0 Hz, 1C, C-5pyrazolyl); 171.0 (1C, CO). IR (neat): ṽ [cm–1] = 3368, 3061, 2930, 2857, 1682, 1630, 1580, 1518, 1462, 1414, 1348, 1202, 1001, 920, 795, 772, 710, 691. HRMS (APCI): m/z = 412.0681 calculated for [M + H]+, found: 412.0715. HPLC: tR = 25.7 min, purity: 98.3%.
X-ray diffraction. Data sets for compounds
10a and
19 were collected with a Bruker D8 Venture Photon III Diffractometer. Programs used: data collection:
APEX4 Version 2021.4-0 [
39]; cell refinement:
SAINT Version 8.40B [
39]; data reduction:
SAINT Version 8.40B [
39]; absorption correction,
SADABS Version 2016/2 [
39]; structure solution
SHELXT-Version 2018-3 [
40]; structure refinement
SHELXL-Version 2018-3 [
41] and graphics,
XP [
42]. R-values are given for observed reflections, and wR
2 values are given for all reflections. CCDC-2211140 (
10a) and CCDC-2211141 (
19) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif, accessed on 14 October 2022. X-ray crystal structure analysis of
10a and
19 is given in the
Supporting Information.
Serine protease inhibition assays. The inhibitory activity of synthesized compounds towards thrombin, FXIIa, FXa, FXIa, plasmin, plasma kallikrein, chymotrypsin, and trypsin was measured by quantifying the hydrolysis rate of the fluorogenic substrate as reported previously [
14,
17]. Briefly, the activity was tested in buffer (10 mM Tris-Cl, 150 mM NaCl, 10 mM MgCl
2·6H
2O, 1 mM CaCl
2·2H
2O, 0.1%
w/
v BSA, 0.01%
v/
v Triton-X100, pH = 7.4) utilizing clear flat-bottom, black polystyrene 96 well-plates. The enzymes (human β-FXIIa, HFXIIAB, >95% purity; Molecular Innovations, 2.5 nM–final concentration; human α-thrombin (active) protein, ABIN2127880, >95% purity; antibodies-online, 0.25 nM–final concentration; human Factor Xa, HFXA, >95% purity; Molecular Innovations, 2.5 nM–final concentration; human Factor XIa, HFXIA, >95% purity; Molecular Innovations, 0.5 nM–final concentration; human plasmin, >95% purity; Innovative Research, 10 nM–final concentration; human plasma kallikrein, >94% purity; Innovative Research, 0.25 nM–final concentration; porcine trypsin; Merck, 3.5 nM–final concentration; human chymotrypsin; >95% purity; Innovative Research, 0.5 nM–final concentration) and the fluorogenic substrates for thrombin: Boc-Val-Pro-Arg-AMC (Pepta Nova, 25 µM–final concentration,
Km = 18 µM); for FXIIa: Boc-Gln-Gly-Arg-AMC (Pepta Nova, 25 µM–final concentration,
Km = 167 µM); for FXa: Boc-Ile-Glu-Gly-Arg-AMC (Pepta Nova, 25 µM–final concentration); for FXIa: Boc-Glu(OBzl)-Ala-Arg-AMC (Pepta Nova, 25 µM–final concentration); for plasmin: Boc-Val-Leu-Lys-AMC (Pepta Nova, 25 µM–final concentration); for plasma kallikrein: Z-Phe-Lys-AMC (Pepta Nova, 25 µM–final concentration); for chymotrypsin: Suc-Ala-Ala-Pro-Phe-AMC (Sigma-Aldrich, 25 µM–final concentration); for trypsin: Z-Gly-Gly-Arg-AMC (Sigma-Aldrich, 25 µM–final concentration). Dilutions of test-compounds ranging from 2 nM to 32 µM (final concentrations) in DMSO were prepared. The fluorogenic substrate solution was added into the wells followed by the addition of 2 µL of test-compounds solution, and the reaction was triggered by addition of the enzyme solution (final testing volume–152 µL). In case of blank (substrate + buffer) and control (substrate + enzyme) wells, 2 µL of DMSO was added instead of the test-compounds’ solution. Fluorescence intensity was measured with Microplate Reader Mithras LB 940 (Berthold Technologies, excitation at 355 nm, emission at 460 nm) for a period of 1 h (thrombin, FXIIa, FXa, FXIa, and trypsin), 30 min (plasma kallikrein, chymotrypsin), or 15 min (plasmin) with a read every minute. The reactions were performed at 25 °C. To derive IC
50 values, endpoint RFU (single fluorescence reading) was used. Sigmoidal curves were prepared in GraphPad Prism software and IC
50 values were derived from the fitted curves [
14,
17].
In vitro plasma coagulation assays (aPTT and PT). All measurements were performed using commercially available citrated (3.8%) human pooled plasma (Dunn Labortechnik GmbH, Germany) on a semi-automated coagulometer (Thrombotimer-2, Behnk Elektronik, Germany) according to the manufacturer instructions as previously reported [
14,
17]. For aPTT measurement, plasma (100 μL) was placed into the incubation cuvettes of the instrument and incubated for 2 min at 37 °C. Then, test compound solution (10 μL) or solvent (DMSO, 10 μL) was added with a pipette. After 1 min of incubation, 100 μL of prewarmed (37 °C) aPTT reagent (Convergent Technologies, Germany) was added and incubated for additional 2 min. The cuvettes were transferred to a measuring position, the coagulation was initiated by addition of 100 μL of CaCl
2 solution (25 mM, prewarmed at 37 °C, Convergent Technologies, Germany) and the clotting time was recorded. For PT assays, plasma (100 μL) was incubated for 2 min at 37 °C. Then, test compound solution (10 μL) or solvent (DMSO, 10 μL) was added with a pipette. After 3 min of incubation, the cuvettes were transferred to a measuring position, the coagulation was initiated by addition of 100 μL of PT assay reagent already containing CaCl
2 (prewarmed at 37 °C, Biolabo, France) and the clotting time was recorded [
14,
17].
Analysis of the covalent thrombin-inhibitor complex by LC/ESI-MS. The analysis of covalent thrombin-inhibitor complex was performed utilizing LC/ESI-MS as reported previously [
14,
17] with variations. Briefly, 2 µL of the stock solution (128 µM) of human α-thrombin (active) protein (ABIN2127880, >95% purity; antibodies-online) were diluted with 68 µL of purified water. An aliquot of 35 µL was mixed with 3 µL of inhibitor
24e solution (1 mM in DMSO) to prepare the enzyme-inhibitor solution, which was then analyzed via LC/ESI-MS. Reversed phase liquid chromatography was performed using a Discovery BIO Wide Pore C5 column (100 × 2.1 mm, 3 µm particle size, 300 Å pore size) from Supelco (Bellefonte, PA, USA). For chromatographic analysis of the sample solutions, 0.1% formic acid in purified water (A) and 0.1% formic acid in acetonitrile (B) were used as mobile phase. The following gradient of 15 min in total was applied: starting with 5% B, B is increased up to 40% within 3 min. After holding 40% B for 1.5 min, B is decreased to 5% within 0.5 min and kept at 5% for 1 min. B is then increased to 95% within 4 min and kept at this value for 1 min. After decreasing B to 5% within 1 min, this ratio is kept for 3 min. The flow rate was 0.4 mL/min, the column oven temperature was 30 °C and the injection volume was set to 5 μL. Mass spectrometric detection was carried out using a timsTOF time-of-flight mass spectrometer from Bruker Daltonics (Bremen, Germany) equipped with an electrospray ionization interface. The enzyme-inhibitor complex was analyzed in the positive ion mode using the following ESI-MS parameters: The mass range was set to
m/
z 1000–4000, nebulizer gas (nitrogen) was 1.6 bar, dry gas (nitrogen) was 9.0 L/min, dry gas temperature was 200 °C, Funnel 1 RF was 450 V, pre pulse storage time was 30 µs and transfer time was 100 µs. Deconvoluted mass spectra were obtained by averaging frames from 4.0 min to 4.6 min retention time and subsequent deconvolution using multiple charge states of the respective protein species.
Molecular Modeling. To perform molecular docking studies, the crystal structure of human thrombin (PDB ID: 6CYM [
16]) was used as a protein model. The protein was prepared using the LigX module of MOE (version 2019.01; Chemical Computing Group, Montreal, Canada) according to the following procedure. All solvent particles were removed, hydrogen atoms were automatically added, and tautomeric forms and protonation states of the amino acid residues were automatically assigned, using the “Protonate 3D” module at pH 7.0, and model was subjected to restrained energy minimization as reported before [
43]. Energy minimized structures of the ligands were generated in MOE modeling software using the MMFF94x force field within the RMS gradient of 0.1 kcal/mol/Å. Covalent docking with pharmacophore-based constrains was carried out using the MOE Covalent Docking procedure (“Rigid Receptor”) with a protocol defined by a chemical reaction in which the hydroxy group of a Ser residue undergoes a nucleophilic addition to a carbonyl carbon atom. The active serine residues (Ser219 in structure 6CYM [
16]) was selected as an attachment point.

 ,
, 
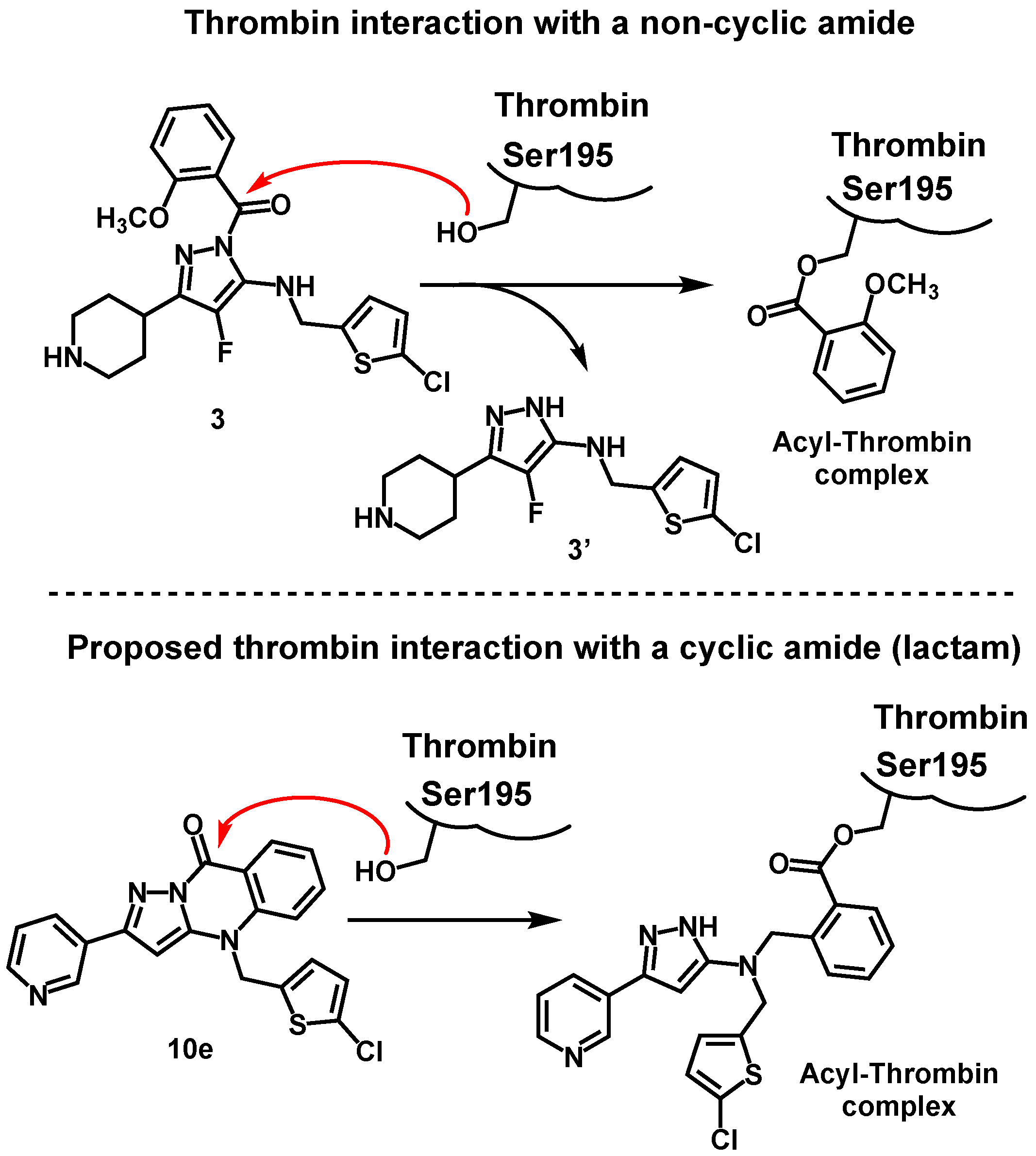











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























