
Structural Basis of Parasitic HSP90 ATPase Inhibition by Small Molecules




Abstract
:1. Introduction
2. Protozoan Parasite HSPs and HSP90
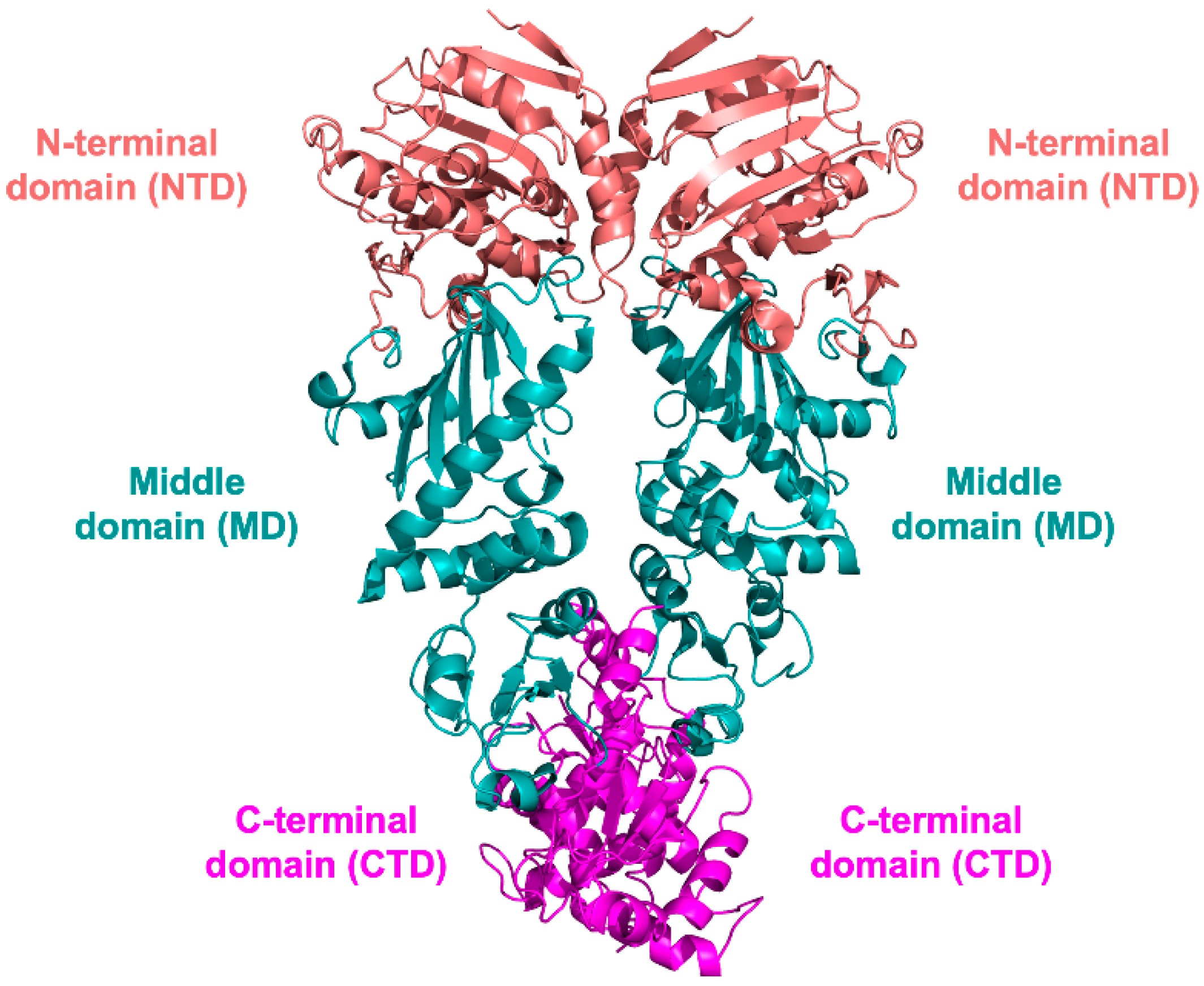
3. HSP90 Structure
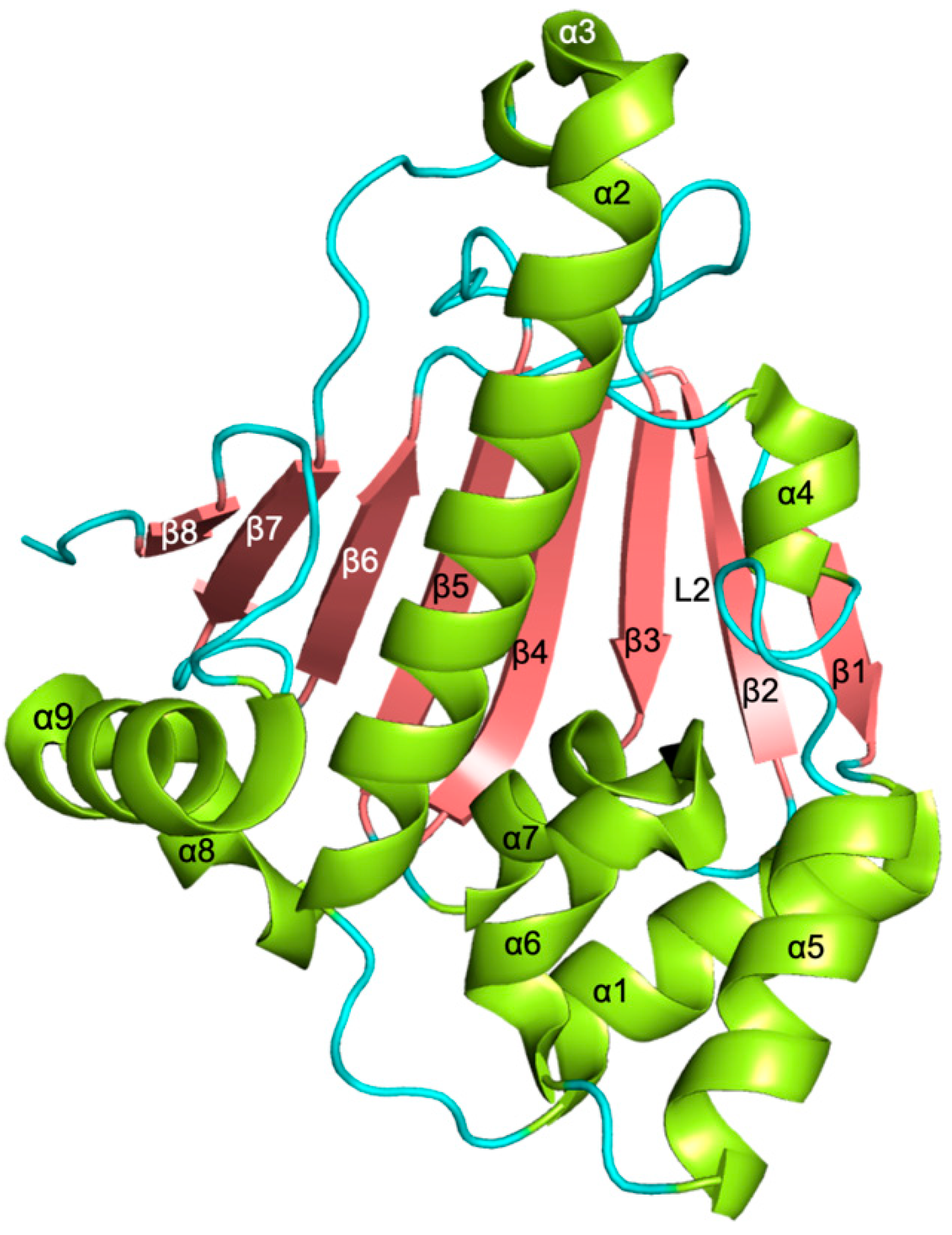
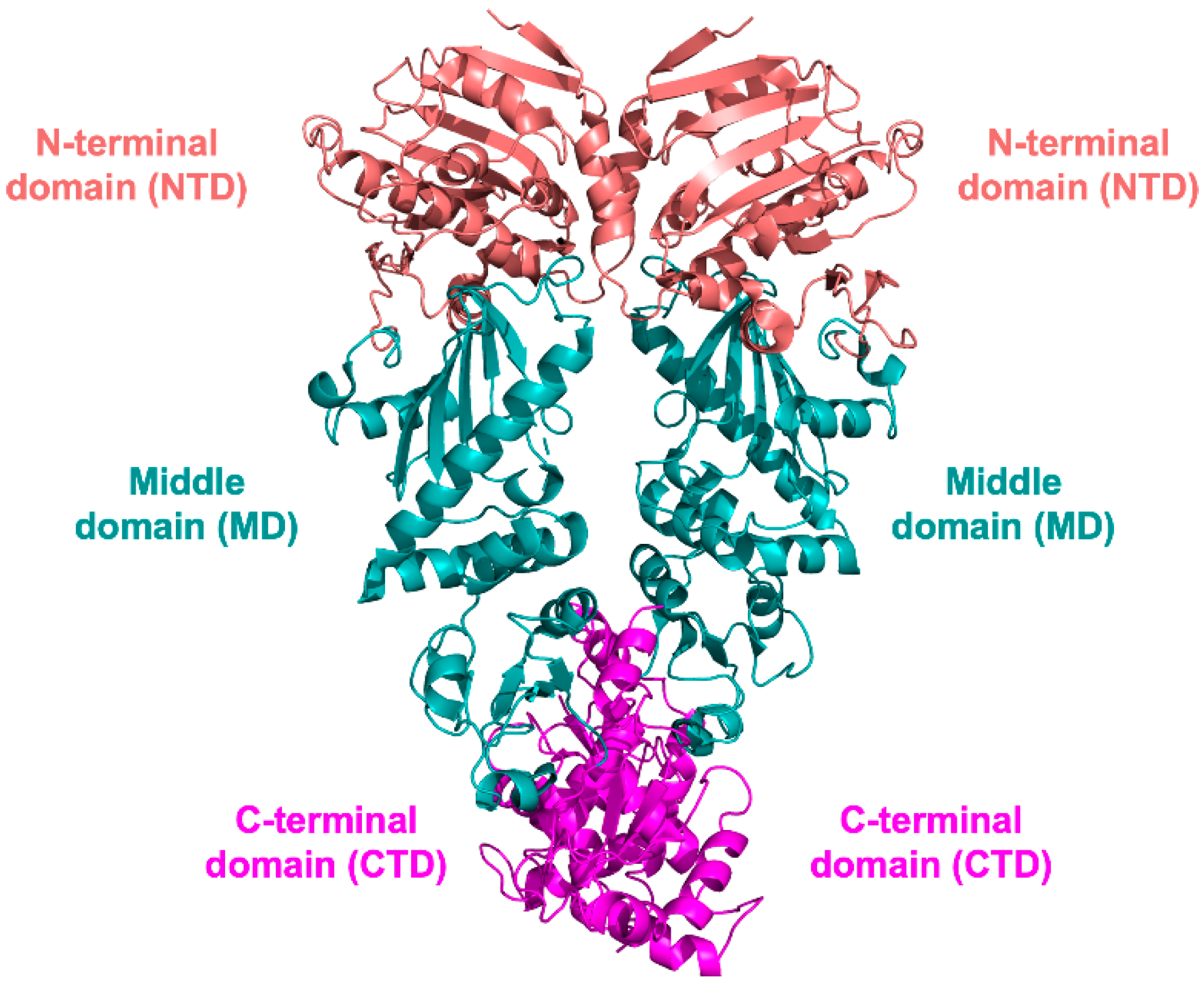
3.1. NTD Structure
3.2. MD and CTD Structure
4. The HSP90 Machinery: Co-Chaperones, Clients, and Mechanism of Action
4.1. Co-Chaperones
4.2. Client Proteins
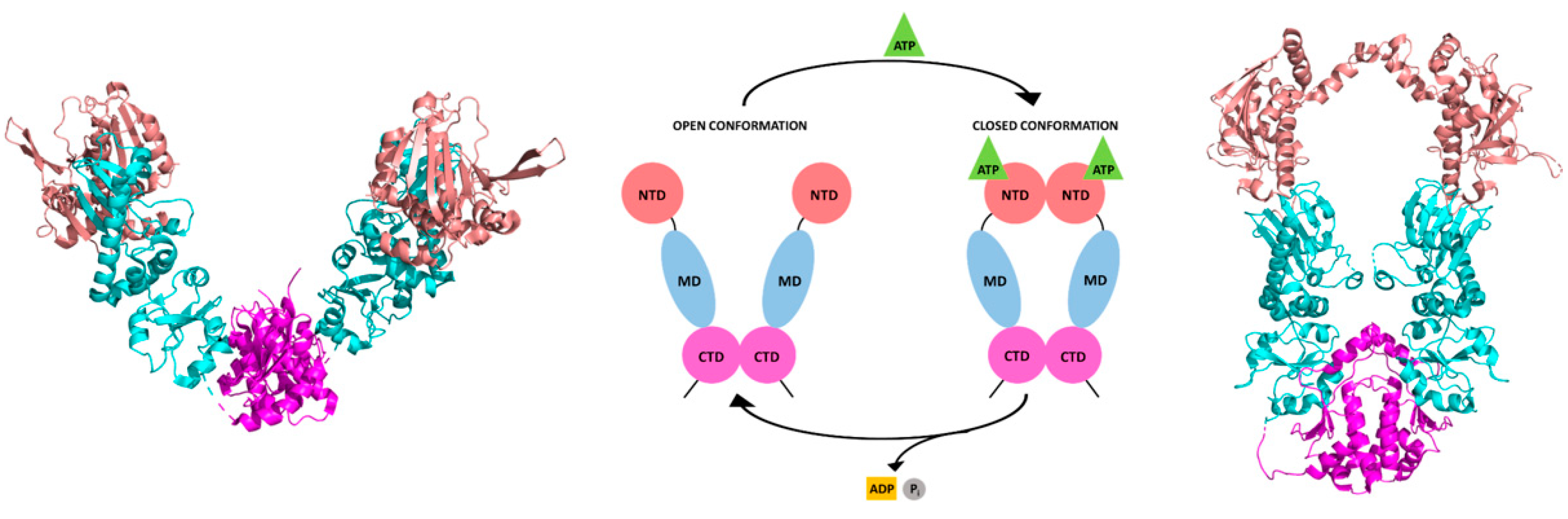
4.3. Mechanistic Insight on Parasite HSP90
5. HSP90 of Protozoan Parasites as Drug Target
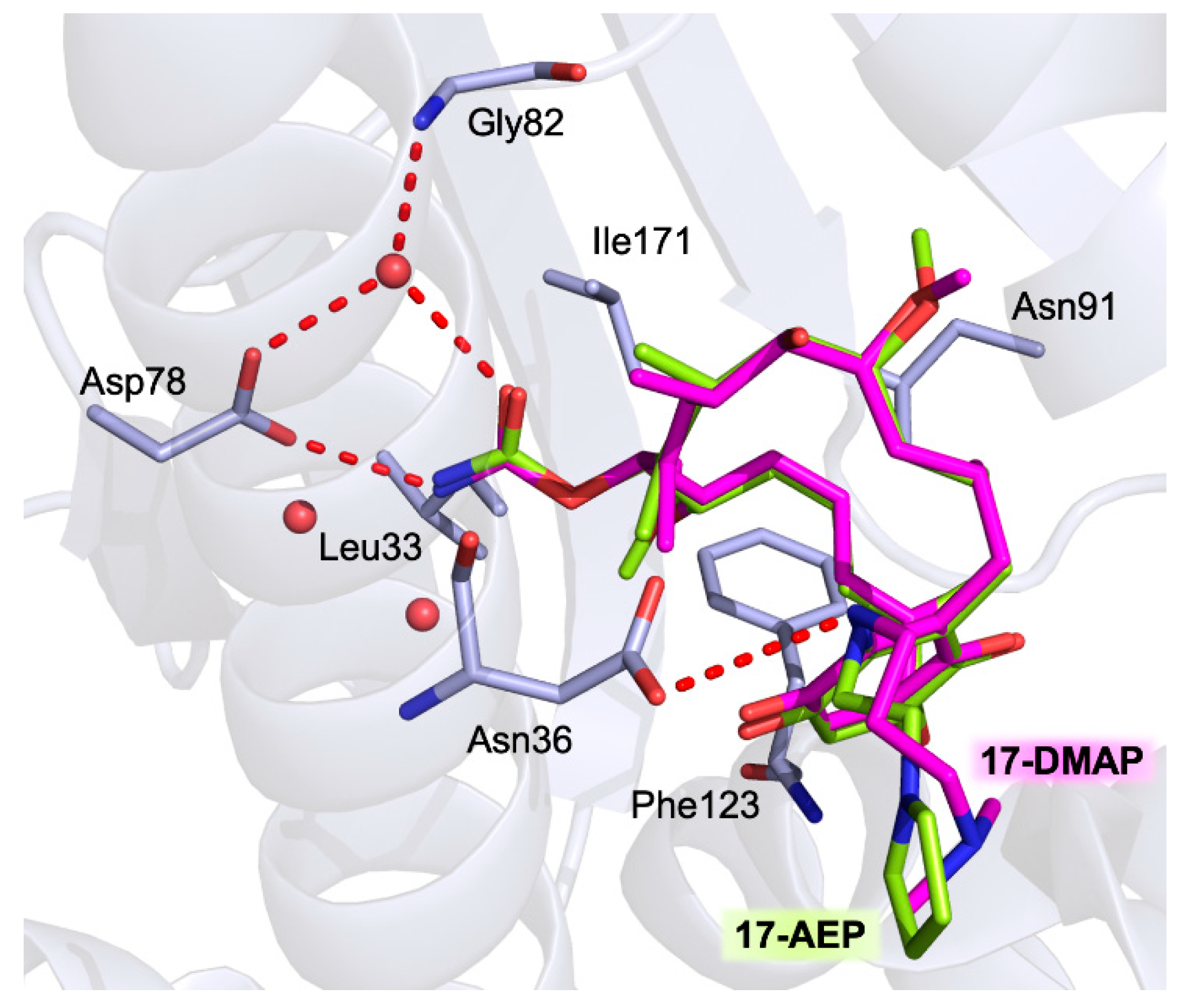
5.1. Natural Macrocyclic Compounds and Their Derivatives
5.2. Other Natural Products and Their Derivatives Acting as Parasite HSP90 Inhibitors
5.2.1. Harmine and Its Derivatives
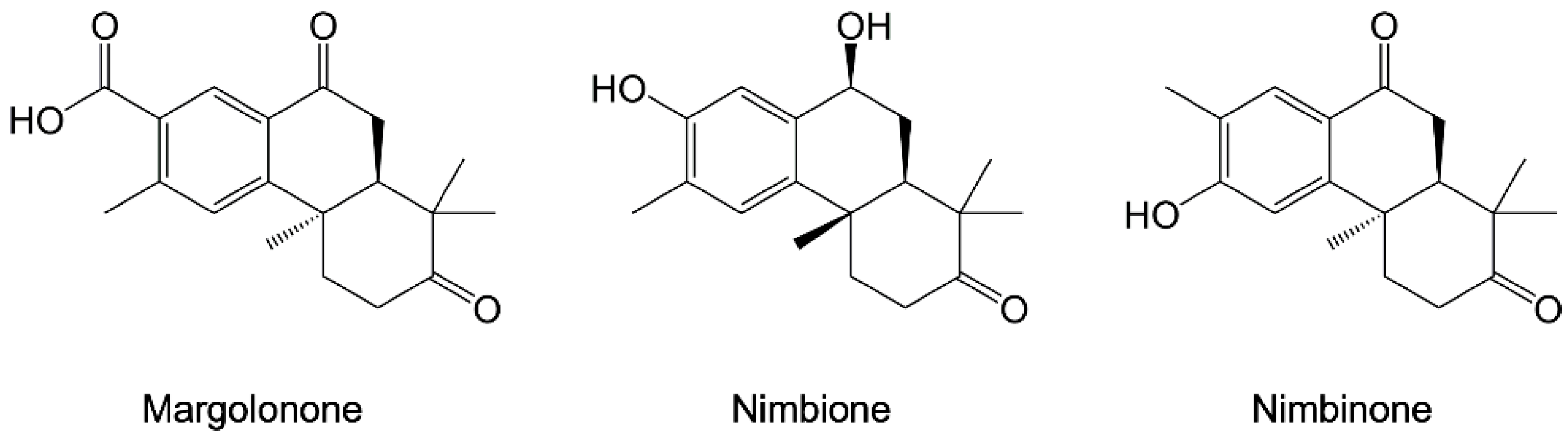
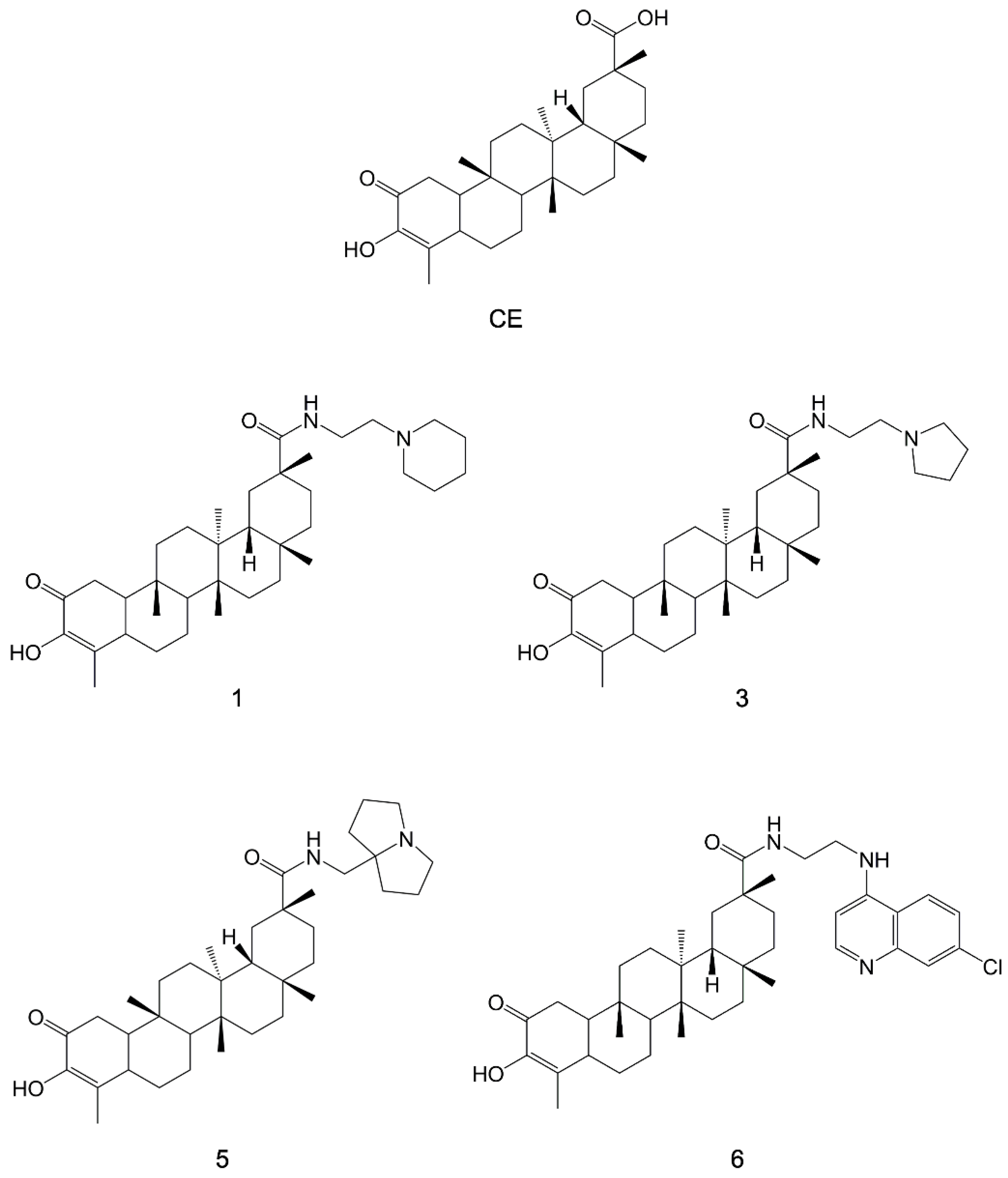
5.2.2. Antiparasite Phytochemicals and Their Synthetic Derivatives
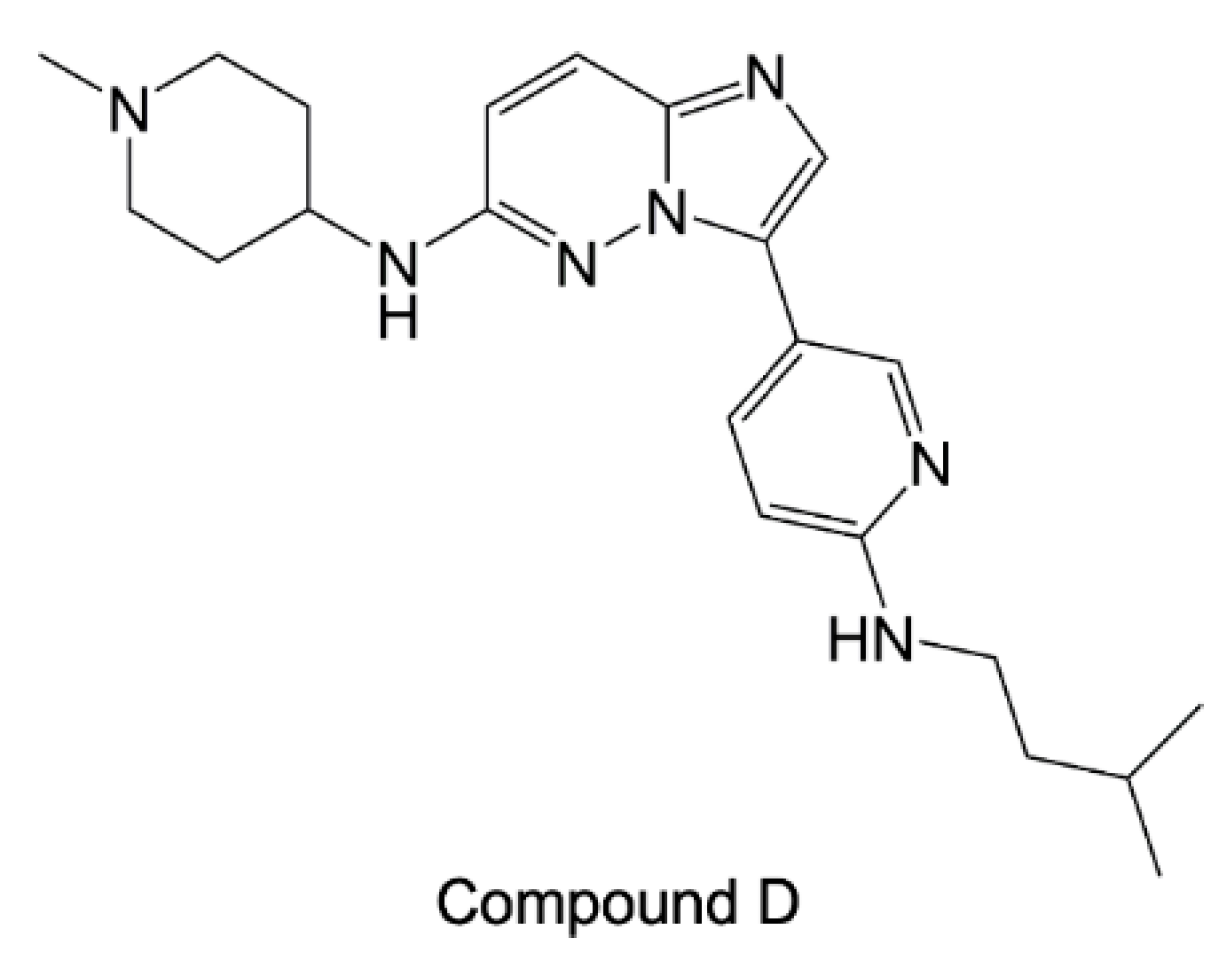
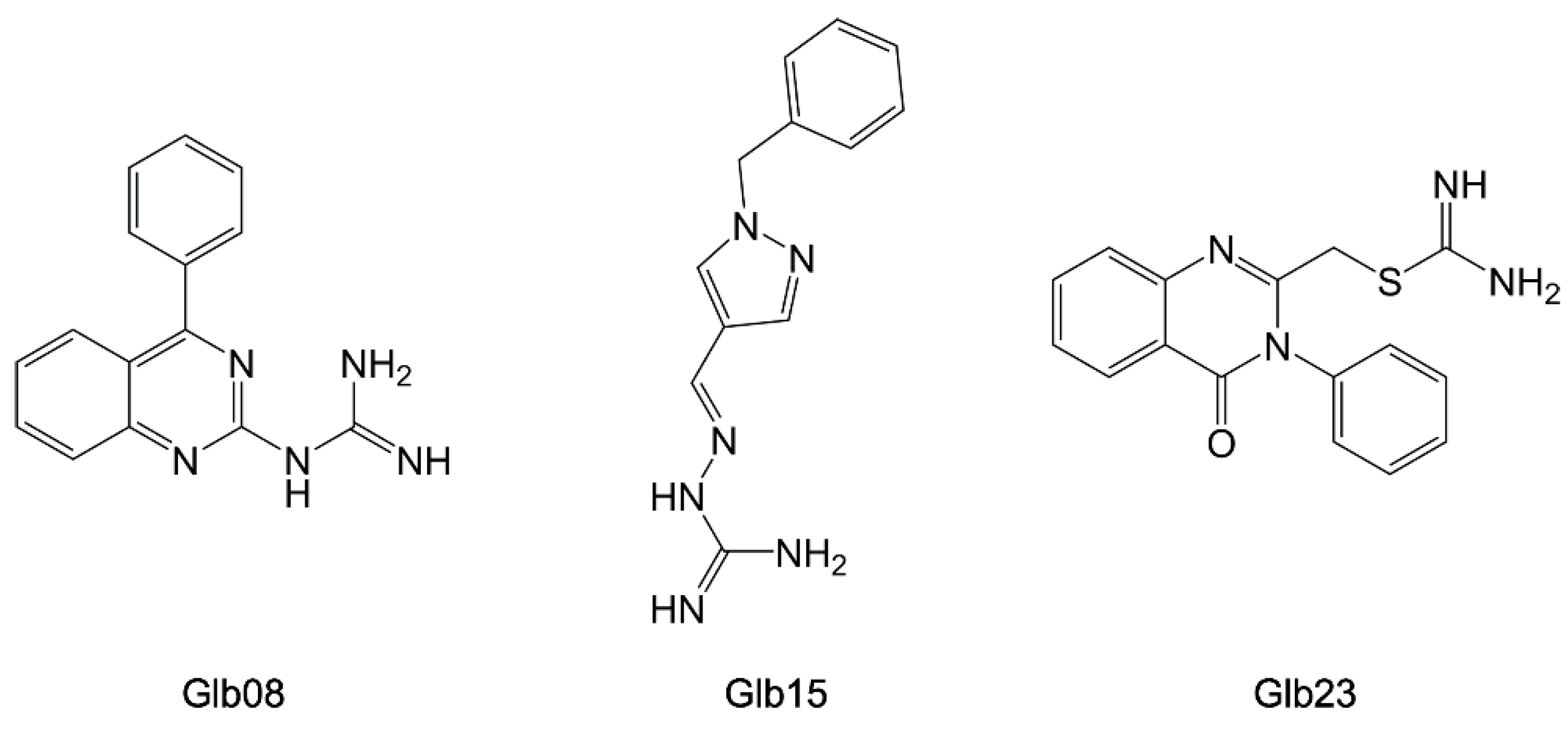
5.3. Synthetic Parasite HSP90 Inhibitors
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Available online: Http://Www.Who.Int (accessed on 1 September 2022).
- Mackey, T.K.; Liang, B.A.; Cuomo, R.; Hafen, R.; Brouwer, K.C.; Lee, D.E. Emerging and Reemerging Neglected Tropical Diseases: A Review of Key Characteristics, Risk Factors, and the Policy and Innovation Environment. Clin. Microbiol. Rev. 2014, 27, 949–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related Protozoan Pathogens, Different Diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Zekar, L.; Sharman, T. Plasmodium Falciparum Malaria. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Weiss, L.M.; Dubey, J.P. Toxoplasmosis: A History of Clinical Observations. Int. J. Parasitol. 2009, 39, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Nii-Trebi, N.I. Emerging and Neglected Infectious Diseases: Insights, Advances, and Challenges. BioMed Res. Int. 2017, 2017, 5245021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, E.E.; Caminade, C.; Thomas, B.N. Climate Change Contribution to the Emergence or Re-Emergence of Parasitic Diseases. Infect. Dis. 2017, 10, 1178633617732296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steverding, D. The Spreading of Parasites by Human Migratory Activities. Virulence 2020, 11, 1177–1191. [Google Scholar] [CrossRef]
- Barrett, M.P.; Croft, S.L. Management of Trypanosomiasis and Leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alday, P.H.; Doggett, J.S. Drugs in Development for Toxoplasmosis: Advances, Challenges, and Current Status. Drug Des. Dev. Ther. 2017, 11, 273–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, E.A.; Phyo, A.P. Drugs in Development for Malaria. Drugs 2018, 78, 861–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirol, E.; Schrumpf, D.; Amici Heradi, J.; Riedel, A.; de Patoul, C.; Quere, M.; Chappuis, F. Nifurtimox-Eflornithine Combination Therapy for Second-Stage Gambiense Human African Trypanosomiasis: Médecins Sans Frontières Experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2013, 56, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Maxmen, A. Sleeping Sickness Can Now Be Cured with Pills. Nature 2017, 550, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole--a New Oral Nitroimidazole Drug Candidate Entering Clinical Development for the Treatment of Sleeping Sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theel, E.S.; Pritt, B.S. Parasites. Microbiol. Spectr. 2016, 4, 39. [Google Scholar] [CrossRef]
- Billker, O.; Lindo, V.; Panico, M.; Etienne, A.E.; Paxton, T.; Dell, A.; Rogers, M.; Sinden, R.E.; Morris, H.R. Identification of Xanthurenic Acid as the Putative Inducer of Malaria Development in the Mosquito. Nature 1998, 392, 289–292. [Google Scholar] [CrossRef]
- Fang, J.; McCutchan, T.F. Thermoregulation in a Parasite’s Life Cycle. Nature 2002, 418, 742. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.; Marchetti, R.; Kirk, K.; Matthews, K.R. A Surface Transporter Family Conveys the Trypanosome Differentiation Signal. Nature 2009, 459, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, S.; Morris, J.C.; Morris, M.T. Environmentally Regulated Glycosome Protein Composition in the African Trypanosome. Eukaryot. Cell 2013, 12, 1072–1079. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Clos, J. No Stress—Hsp90 and Signal Transduction in Leishmania. Parasitology 2014, 141, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Graefe, S.E.B.; Wiesgigl, M.; Gaworski, I.; Macdonald, A.; Clos, J. Inhibition of HSP90 in Trypanosoma Cruzi Induces a Stress Response but No Stage Differentiation. Eukaryot. Cell 2002, 1, 936–943. [Google Scholar] [CrossRef] [Green Version]
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular Protozoan Parasites of Humans: The Role of Molecular Chaperones in Development and Pathogenesis. Protein Pept. Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, R.; Roy, N.; Nageshan, R.K.; Talukdar, P.; Pavithra, S.R.; Reddy, R.; Venketesh, S.; Kumar, R.; Gupta, A.K.; Singh, R.K.; et al. Heat Shock Protein 90 as a Drug Target against Protozoan Infections: Biochemical Characterization of HSP90 from Plasmodium Falciparum and Trypanosoma Evansi and Evaluation of Its Inhibitor as a Candidate Drug. J. Biol. Chem. 2010, 285, 37964–37975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinas, D.; Pillai, D.R. Role of Hsp90 in Plasmodium Falciparum Malaria. Adv. Exp. Med. Biol. 2021, 1340, 125–139. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.C.D.; Shiburah, M.E.; Paiva, S.C.; Vieira, M.R.; Morea, E.G.O.; da Silva, M.S.; de Alves, C.S.; Segatto, M.; Gutierrez-Rodrigues, F.; Borges, J.C.; et al. Possible Involvement of Hsp90 in the Regulation of Telomere Length and Telomerase Activity During the Leishmania Amazonensis Developmental Cycle and Population Proliferation. Front. Cell Dev. Biol. 2021, 9, 713415. [Google Scholar] [CrossRef] [PubMed]
- Kalesh, K.; Sundriyal, S.; Perera, H.; Cobb, S.L.; Denny, P.W. Quantitative Proteomics Reveals That Hsp90 Inhibition Dynamically Regulates Global Protein Synthesis in Leishmania Mexicana. mSystems 2021, 6, e00089-21. [Google Scholar] [CrossRef] [PubMed]
- Angel, S.O.; Matrajt, M.; Echeverria, P.C. A Review of Recent Patents on the Protozoan Parasite HSP90 as a Drug Target. Recent Pat. Biotechnol. 2013, 7, 2–8. [Google Scholar] [CrossRef]
- Sõti, C.; Nagy, E.; Giricz, Z.; Vígh, L.; Csermely, P.; Ferdinandy, P. Heat Shock Proteins as Emerging Therapeutic Targets. Br. J. Pharmacol. 2005, 146, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.V.; Workman, P. Inhibitors of the Heat Shock Response: Biology and Pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [Green Version]
- Giannini, G.; Battistuzzi, G. Exploring in Vitro and in Vivo Hsp90 Inhibitors Activity against Human Protozoan Parasites. Bioorg. Med. Chem. Lett. 2015, 25, 462–465. [Google Scholar] [CrossRef]
- Sidera, K.; Patsavoudi, E. HSP90 Inhibitors: Current Development and Potential in Cancer Therapy. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Zininga, T.; Shonhai, A. Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites. Int. J. Mol. Sci. 2019, 20, 5930. [Google Scholar] [CrossRef]
- Li, Z.; Srivastava, P.; Li, Z.; Srivastava, P. Heat-Shock Proteins. In Current Protocols in Immunology, Current Protocols in Immunology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. A.1T.1–A.1T.6. ISBN 978-0-471-14273-7. [Google Scholar]
- Pozzi, C.; Tassone, G.; Mangani, S. Chapter Five—X-Ray Crystallography Contributions to Drug Discovery Against Parasite. In Annual Reports in Medicinal Chemistry; Botta, M., Ed.; Neglected Diseases: Extensive Space for Modern Drug Discovery; Academic Press: Cambridge, MA, USA, 2018; Volume 51, pp. 175–230. [Google Scholar]
- Pérez-Morales, D.; Espinoza, B. The Role of Small Heat Shock Proteins in Parasites. Cell Stress Chaperones 2015, 20, 767–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narberhaus, F. Alpha-Crystallin-Type Heat Shock Proteins: Socializing Minichaperones in the Context of a Multichaperone Network. Microbiol. Mol. Biol. Rev. 2002, 66, 64–93, table of contents. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M. SHsps and Their Role in the Chaperone Network. Cell Mol. Life Sci. 2002, 59, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- MacRae, T.H. Structure and Function of Small Heat Shock/Alpha-Crystallin Proteins: Established Concepts and Emerging Ideas. Cell. Mol. Life Sci. 2000, 57, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Viitanen, P.V.; Lubben, T.H.; Reed, J.; Goloubinoff, P.; O’Keefe, D.P.; Lorimer, G.H. Chaperonin-Facilitated Refolding of Ribulosebisphosphate Carboxylase and ATP Hydrolysis by Chaperonin 60 (GroEL) Are K+ Dependent. Biochemistry 1990, 29, 5665–5671. [Google Scholar] [CrossRef]
- McCallum, C.D.; Do, H.; Johnson, A.E.; Frydman, J. The Interaction of the Chaperonin Tailless Complex Polypeptide 1 (TCP1) Ring Complex (TRiC) with Ribosome-Bound Nascent Chains Examined Using Photo-Cross-Linking. J. Cell Biol. 2000, 149, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Abdeen, S.; Salim, N.; Mammadova, N.; Summers, C.M.; Goldsmith-Pestana, K.; McMahon-Pratt, D.; Schultz, P.G.; Horwich, A.L.; Chapman, E.; Johnson, S.M. Targeting the HSP60/10 Chaperonin Systems of Trypanosoma Brucei as a Strategy for Treating African Sleeping Sickness. Bioorg. Med. Chem. Lett. 2016, 26, 5247–5253. [Google Scholar] [CrossRef] [Green Version]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of Regulation of Hsp70 Chaperones by DnaJ Cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, S.; Schneider-Mergener, J.; Bukau, B. Its Substrate Specificity Characterizes the DnaJ Co-Chaperone as a Scanning Factor for the DnaK Chaperone. EMBO J. 2001, 20, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Cheetham, M.E.; Caplan, A.J. Structure, Function and Evolution of DnaJ: Conservation and Adaptation of Chaperone Function. Cell Stress Chaperones 1998, 3, 28–36. [Google Scholar] [CrossRef]
- Schilke, B.A.; Ciesielski, S.J.; Ziegelhoffer, T.; Kamiya, E.; Tonelli, M.; Lee, W.; Cornilescu, G.; Hines, J.K.; Markley, J.L.; Craig, E.A. Broadening the Functionality of a J-Protein/Hsp70 Molecular Chaperone System. PLoS Genet. 2017, 13, e1007084. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.-B.; Shao, Y.-M.; Miao, S.; Wang, L. The Diversity of the DnaJ/Hsp40 Family, the Crucial Partners for Hsp70 Chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-Y.; Lee, S.; Cyr, D.M. Mechanisms for Regulation of Hsp70 Function by Hsp40. Cell Stress Chaperones 2003, 8, 309–316. [Google Scholar] [CrossRef]
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein Disaggregation Mediated by Heat-Shock Protein Hsp104. Nature 1994, 372, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 Targets Hsp100 Chaperones to Substrates for Protein Disaggregation and Prion Fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogk, A.; Kummer, E.; Bukau, B. Cooperation of Hsp70 and Hsp100 Chaperone Machines in Protein Disaggregation. Front. Mol. Biosci. 2015, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Nageshan, R.K.; Ranade, S.; Tatu, U. Heat Shock Protein 90 from Neglected Protozoan Parasites. Biochim. Biophys. Acta 2012, 1823, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Dutta, T.; Singh, H.; Edkins, A.L.; Blatch, G.L. Hsp90 and Associated Co-Chaperones of the Malaria Parasite. Biomolecules 2022, 12, 1018. [Google Scholar] [CrossRef]
- Sun, H.; Zhuo, X.; Zhao, X.; Yang, Y.; Chen, X.; Yao, C.; Du, A. The Heat Shock Protein 90 of Toxoplasma Gondii Is Essential for Invasion of Host Cells and Tachyzoite Growth. Parasite 2017, 24, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Buchner, J. Structure, Function and Regulation of the Hsp90 Machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal Structure of an Hsp90–Nucleotide–P23/Sba1 Closed Chaperone Complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B. ATP Binding and Hydrolysis Are Essential to the Function of the Hsp90 Molecular Chaperone Invivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H. Review: The HSP90 Molecular Chaperone-an Enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90: Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.-T.; Scroggins, B.T.; Xu, W.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 Charged-Linker Truncation Reverses the Functional Consequences of Weakened Hydrophobic Contacts in the N Domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar] [CrossRef]
- Hainzl, O.; Lapina, M.C.; Buchner, J.; Richter, K. The Charged Linker Region Is an Important Regulator of Hsp90 Function. J. Biol. Chem. 2009, 284, 22559–22567. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, T.; Ohara-Nemoto, Y.; Ota, M.; Takagi, T.; Yokoyama, K. Mechanism of Dimer Formation of the 90-KDa Heat-Shock Protein. Eur. J. Biochem. 1995, 233, 1–8. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic Structure of Hsp90-Cdc37-Cdk4 Reveals That Hsp90 Traps and Stabilizes an Unfolded Kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, K.D.; Berger, J.M. Structure of the ATP-Binding Domain of Plasmodium Falciparum Hsp90. Proteins 2010, 78, 2738–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, J.C.; Hills, T.; Senisterra, G.; Wernimont, A.K.; Mackenzie, C.; Norcross, N.R.; Ferguson, M.A.J.; Wyatt, P.G.; Gilbert, I.H.; Hui, R. Exploring the Trypanosoma Brucei Hsp83 Potential as a Target for Structure Guided Drug Design. PLoS Negl. Trop. Dis. 2013, 7, e2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO Server for Macromolecular Structure Model Optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sun, L.; Xu, C.; Yu, F.; Zhou, H.; Zhao, Y.; Zhang, J.; Cai, J.; Mao, C.; Tang, L.; et al. Structure Insights into Mechanisms of ATP Hydrolysis and the Activation of Human Heat-Shock Protein 90. Acta Biochim. Biophys. Sin. 2012, 44, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 Chaperone Machinery: Conformational Dynamics and Regulation by Co-Chaperones. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuehlke, A.; Johnson, J.L. Hsp90 and Co-Chaperones Twist the Functions of Diverse Client Proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Zinn, N.; Faelth-Savitski, M.; Poeckel, D.; Gade, S.; Becher, I.; Muelbaier, M.; Wagner, A.J.; Strohmer, K.; Werner, T.; et al. Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 2018, 173, 260–274.e25. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Mimnaugh, E.; Schulte, T.W. Hsp90 as an Anti-Cancer Target. Drug Resist. Updates 1999, 2, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal Structure of an Hsp90–Geldanamycin Complex: Targeting of a Protein Chaperone by an Antitumor Agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational Dynamics of the Molecular Chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The Charged Linker of the Molecular Chaperone Hsp90 Modulates Domain Contacts and Biological Function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef] [Green Version]
- Morán Luengo, T.; Mayer, M.P.; Rüdiger, S.G.D. The Hsp70–Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 2019, 29, 164–177. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. Coli Hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 Inhibitor, Ameliorates Polyglutamine-Mediated Motor Neuron Degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin Activates a Heat Shock Response and Inhibits Huntingtin Aggregation in a Cell Culture Model of Huntington’s Disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagatell, R.; Whitesell, L. Altered Hsp90 Function in Cancer: A Unique Therapeutic Opportunity. Mol. Cancer Ther. 2004, 3, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for Cancer Therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat Shock Protein 90 in Neurodegenerative Diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, N.-N.; You, Q.-D.; Xu, X.-L. An Updated Patent Review of Anticancer Hsp90 Inhibitors (2013-Present). Expert. Opin. Ther. Pat. 2021, 31, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Debnath, A.; Shahinas, D.; Bryant, C.; Hirata, K.; Miyamoto, Y.; Hwang, G.; Gut, J.; Renslo, A.R.; Pillai, D.R.; Eckmann, L.; et al. Hsp90 Inhibitors as New Leads to Target Parasitic Diarrheal Diseases. Antimicrob. Agents Chemother. 2014, 58, 4138–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faya, N.; Penkler, D.L.; Tastan Bishop, Ö. Human, Vector and Parasite Hsp90 Proteins: A Comparative Bioinformatics Analysis. FEBS Open Biol. 2015, 5, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banumathy, G.; Singh, V.; Pavithra, S.R.; Tatu, U. Heat Shock Protein 90 Function Is Essential for Plasmodium Falciparum Growth in Human Erythrocytes. J. Biol. Chem. 2003, 278, 18336–18345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Shiomi, K.; Kamei, K.; Sugoh-Hagino, M.; Enomoto, Y.; Fang, F.; Yamaguchi, Y.; Masuma, R.; Zhang, C.G.; Zhang, X.W.; et al. Antimalarial Activity of Radicicol, Heptelidic Acid and Other Fungal Metabolites. J. Antibiot. 1998, 51, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi-Ostad-Kalayeh, S.; Stahl, F.; Scheper, T.; Kock, K.; Herrmann, C.; Heleno Batista, F.A.; Borges, J.C.; Sasse, F.; Eichner, S.; Ongouta, J.; et al. Heat Shock Proteins Revisited: Using a Mutasynthetically Generated Reblastatin Library to Compare the Inhibition of Human and Leishmania Hsp90s. ChemBioChem 2018, 19, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, O.S.; Atolani, O.; Awakan, O.J.; Olaolu, T.D.; Nwonuma, C.O.; Alejolowo, O.; Otohinoyi, D.A.; Rotimi, D.; Owolabi, A.; Batiha, G.E.-S. In Vitro Screening to Identify Anti-Toxoplasma Compounds and In Silico Modeling for Bioactivities and Toxicity. Yale J. Biol. Med. 2019, 92, 369–383. [Google Scholar]
- Posfai, D.; Eubanks, A.L.; Keim, A.I.; Lu, K.-Y.; Wang, G.Z.; Hughes, P.F.; Kato, N.; Haystead, T.A.; Derbyshire, E.R. Identification of Hsp90 Inhibitors with Anti-Plasmodium Activity. Antimicrob. Agents Chemother. 2018, 62, e01799-17. [Google Scholar] [CrossRef] [Green Version]
- Batista, F.A.H.; Ramos, S.L.; Tassone, G.; Leitão, A.; Montanari, C.A.; Botta, M.; Mori, M.; Borges, J.C. Discovery of Small Molecule Inhibitors of Leishmania Braziliensis Hsp90 Chaperone. J. Enzyme Inhib. Med. Chem. 2020, 35, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, L.C.; Ferreira, L.F.G.R.; de Petersen, A.L.O.A.; Dias, B.R.S.; de Menezes, J.P.B.; de Moreira, D.R.M.; Hernandes, M.Z.; Veras, P.S.T. A Docking-Based Structural Analysis of Geldanamycin-Derived Inhibitor Binding to Human or Leishmania Hsp90. Sci. Rep. 2019, 9, 14756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesgigl, M.; Clos, J. Heat Shock Protein 90 Homeostasis Controls Stage Differentiation in Leishmania Donovani. Mol. Biol. Cell 2001, 12, 3307–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiosis, G. Targeting Chaperones in Transformed Systems—A Focus on Hsp90 and Cancer. Expert. Opin. Ther. Targets 2006, 10, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Immormino, R.M.; Metzger, L.E.; Reardon, P.N.; Dollins, D.E.; Blagg, B.S.J.; Gewirth, D.T. Different Poses for Ligand and Chaperone in Inhibitor-Bound Hsp90 and GRP94: Implications for Paralog-Specific Drug Design. J. Mol. Biol. 2009, 388, 1033–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinas, D.; Folefoc, A.; Pillai, D.R. Targeting Plasmodium Falciparum Hsp90: Towards Reversing Antimalarial Resistance. Pathogens 2013, 2, 33–54. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.J.; Shapiro, T.A. Potent Antitrypanosomal Activities of Heat Shock Protein 90 Inhibitors in Vitro and in Vivo. J. Infect. Dis. 2013, 208, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-Solano, C.; Dong, C.; Sanchez, C.G.; Pizarro, J.C. Identification and Characterization of the Antiplasmodial Activity of Hsp90 Inhibitors. Malar. J. 2017, 16, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, A.L.; de, O.A.; Guedes, C.E.S.; Versoza, C.L.; Lima, J.G.B.; de Freitas, L.A.R.; Borges, V.M.; Veras, P.S.T. 17-AAG Kills Intracellular Leishmania Amazonensis While Reducing Inflammatory Responses in Infected Macrophages. PLoS ONE 2012, 7, e49496. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Macmullin, G.; Benedict, C.; Crandall, I.; Pillai, D.R. Harmine Is a Potent Antimalarial Targeting Hsp90 and Synergizes with Chloroquine and Artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinas, D.; Liang, M.; Datti, A.; Pillai, D.R. A Repurposing Strategy Identifies Novel Synergistic Inhibitors of Plasmodium Falciparum Heat Shock Protein 90. J. Med. Chem. 2010, 53, 3552–3557. [Google Scholar] [CrossRef] [PubMed]
- Bayih, A.G.; Folefoc, A.; Mohon, A.N.; Eagon, S.; Anderson, M.; Pillai, D.R. In Vitro and in Vivo Anti-Malarial Activity of Novel Harmine-Analog Heat Shock Protein 90 Inhibitors: A Possible Partner for Artemisinin. Malar. J. 2016, 15, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel Harmicines with Improved Potency against Plasmodium. Molecules 2020, 25, 4376. [Google Scholar] [CrossRef]
- Dhar, R.; Zhang, K.; Talwar, G.P.; Garg, S. Nirbhay Kumar Inhibition of the Growth and Development of Asexual and Sexual Stages of Drug-Sensitive and Resistant Strains of the Human Malaria Parasite Plasmodium Falciparum by Neem (Azadirachta Indica) Fractions. J. Ethnopharmacol. 1998, 61, 31–39. [Google Scholar] [CrossRef]
- Elumalai, P.; Arunakaran, J. Review on Molecular and Chemopreventive Potential of Nimbolide in Cancer. Genom. Inform. 2014, 12, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Daniyan, M.O.; Ojo, O.T. In Silico Identification and Evaluation of Potential Interaction of Azadirachta Indica Phytochemicals with Plasmodium Falciparum Heat Shock Protein 90. J. Mol. Graph. Model. 2019, 87, 144–164. [Google Scholar] [CrossRef]
- Bassanini, I.; Parapini, S.; Ferrandi, E.E.; Gabriele, E.; Basilico, N.; Taramelli, D.; Sparatore, A. Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90. Biomolecules 2021, 11, 56. [Google Scholar] [CrossRef]
- Wang, T.; Mäser, P.; Picard, D. Inhibition of Plasmodium Falciparum Hsp90 Contributes to the Antimalarial Activities of Aminoalcohol-Carbazoles. J. Med. Chem. 2016, 59, 6344–6352. [Google Scholar] [CrossRef] [Green Version]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical Genetics of Plasmodium Falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Moon, R.W.; Whalley, D.; Bowyer, P.W.; Wallace, C.; Rochani, A.; Nageshan, R.K.; Howell, S.A.; Grainger, M.; Jones, H.M.; et al. Imidazopyridazine Inhibitors of Plasmodium Falciparum Calcium-Dependent Protein Kinase 1 Also Target Cyclic GMP-Dependent Protein Kinase and Heat Shock Protein 90 To Kill the Parasite at Different Stages of Intracellular Development. Antimicrob. Agents Chemother. 2015, 60, 1464–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinas, D.; Folefoc, A.; Taldone, T.; Chiosis, G.; Crandall, I.; Pillai, D.R. A Purine Analog Synergizes with Chloroquine (CQ) by Targeting Plasmodium Falciparum Hsp90 (PfHsp90). PLoS ONE 2013, 8, e75446. [Google Scholar] [CrossRef] [PubMed]







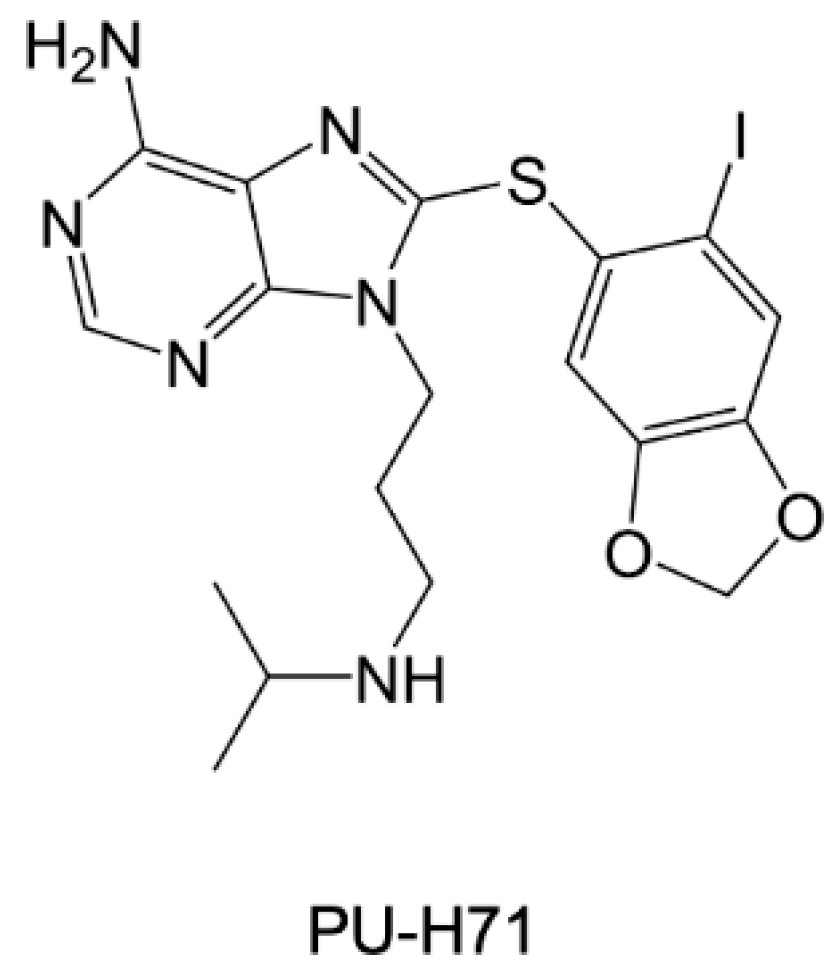
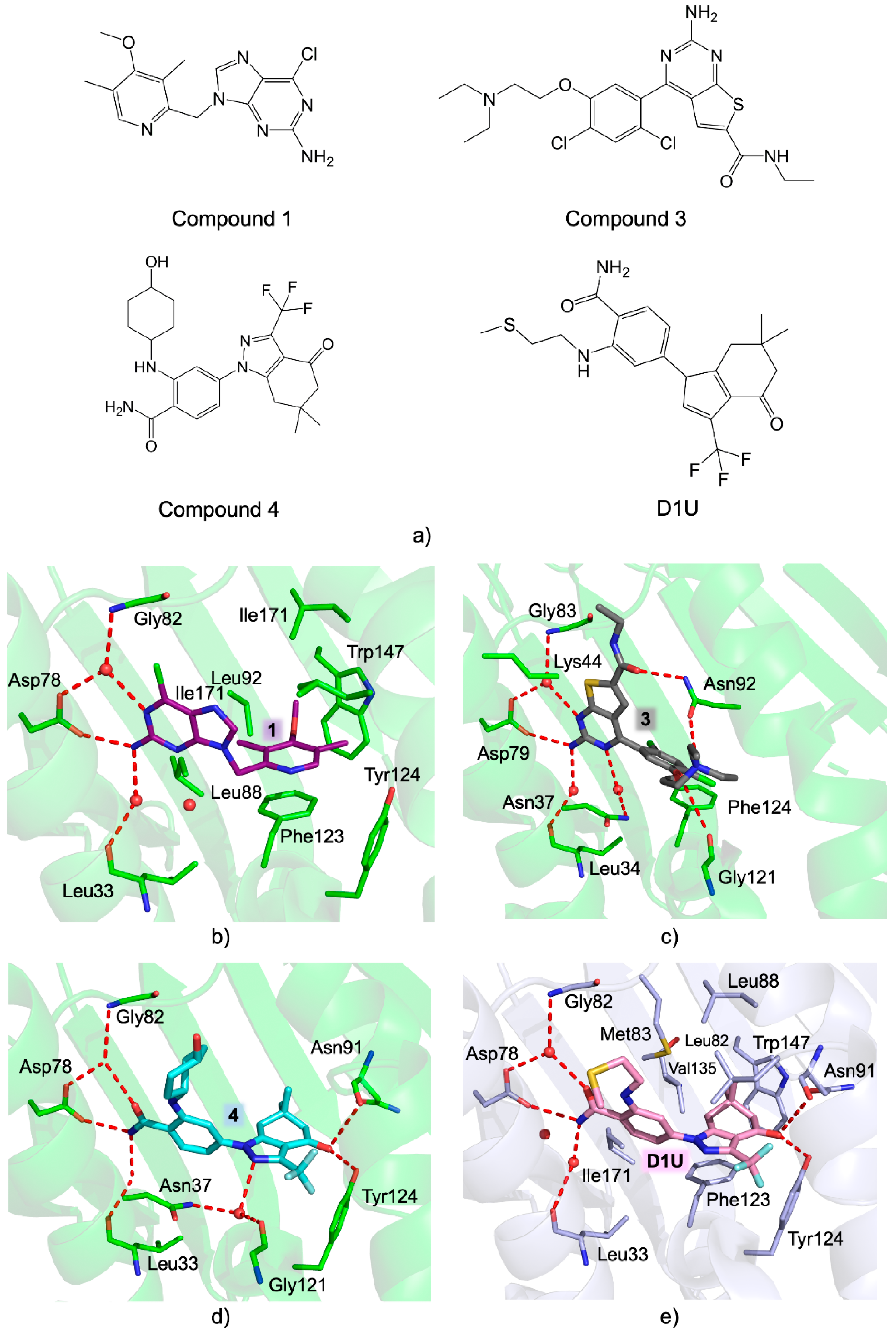

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
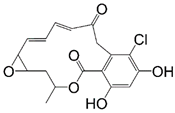 Radicicol | 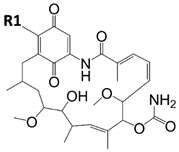 Ansamycin Scaffold | ||
|---|---|---|---|
| Compound | R1 | HSP Binding and/or Inhibition Data on Parasite HSP90 | Parasite Cytotoxicity |
| Radicicol | - | Pf-HSP90 IC50 = 0.01 ug/mL [93] Lb-HSP90 IC50 = 80 nM [94] | T. gondii IC50 = 4.9 ug/mL [95] T. brucei EC50 = 70 nM [103] |
| Geldanamycin | OCH3 | Pf-HSP90 Ki = 2.3 nM [96] Kd = 1.0 µM [23] Tb-HSP90 Kd = 0.001 nM [67] | P. falciparum IC50 = 300.0 nM [96], 214.8 nM [104], and 25 nM [23] P. berghei IC50 = 170.0 nM [96] T. brucei EC50 = 13 nM [103] L. braziliensis IC50 = 190 nM [97] L. donovani IC50 = 32 ng/mL [99] |
| Lb-HSP90 KDapp = 7.6 µM [97] Lb-HSP90 IC50 = 530 nM [94] La-HSP90 IC50 = 190 nM [98] | |||
| 17-AEP-GA |  | Tb-HSP90 Kd = 714 nM [67] | |
| 17-AAG-GA | 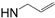 | La-HSP90 IC50 = 65 nM [105] and 110 nM [98] Lb-HSP90 IC50 = 65 nM [105] Lm-HSP90 IC50 = 80 nM [105] Pf-HSP90 IC50 = 387.5 nM [104] | P. falciparum IC50 = 160 nM [23] T. brucei EC50 = 38 nM [103] |
| 17-DMAG-GA |  | Pf-HSP90 Ki = 5.7 nM [96] La-HSP90 IC50 = 86 nM [98] | P. falciparum IC50 = 170.0 nM [96] and 118.1 nM [104] P. berghei IC50 = 580.0 nM [96] T. brucei EC50 = 3.0 nM [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, G.; Mazzorana, M.; Pozzi, C. Structural Basis of Parasitic HSP90 ATPase Inhibition by Small Molecules. Pharmaceuticals 2022, 15, 1341. https://doi.org/10.3390/ph15111341
Tassone G, Mazzorana M, Pozzi C. Structural Basis of Parasitic HSP90 ATPase Inhibition by Small Molecules. Pharmaceuticals. 2022; 15(11):1341. https://doi.org/10.3390/ph15111341
Chicago/Turabian StyleTassone, Giusy, Marco Mazzorana, and Cecilia Pozzi. 2022. "Structural Basis of Parasitic HSP90 ATPase Inhibition by Small Molecules" Pharmaceuticals 15, no. 11: 1341. https://doi.org/10.3390/ph15111341
APA StyleTassone, G., Mazzorana, M., & Pozzi, C. (2022). Structural Basis of Parasitic HSP90 ATPase Inhibition by Small Molecules. Pharmaceuticals, 15(11), 1341. https://doi.org/10.3390/ph15111341