
Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV




Abstract
1. Introduction
2. TB/HIV Targets
2.1. FBDD Fragment Hit Compounds against Mycobacterium Tuberculosis Targets
2.1.1. Decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase
2.1.2. KasA
2.1.3. EthR Transcriptional Repressor (EthR)
2.1.4. Antigen 85C
2.1.5. 7,8-Diaminopelargonic Acid (DAPA) Synthase (BioA)
2.1.6. Arginine Biosynthesis Pathway
2.2. Human Immunodeficiency Virus Targets
2.2.1. HIV-1 Reverse Transcriptase
2.2.2. HIV-1 Integrase
2.2.3. HIV-1 Protease
2.2.4. Gp120
2.2.5. Gp41
2.2.6. CCR5 Co-Receptor
2.2.7. TAR–tat Interaction
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Toossi, Z.; Johnson, J.L.; Kanost, R.A.; Wu, M.; Luzze, H.; Peters, P.; Okwera, A.; Joloba, M.; Mugyenyi, P.; Mugerwa, R.D.; et al. Increased Replication of HIV-1 at Sites of Mycobacterium Tuberculosis Infection: Potential Mechanisms of Viral Activation. JAIDS J. Acquir. Immune Defic. Syndr. 2001, 28, 1–8. [Google Scholar] [CrossRef]
- Sonnenberg, P.; Murray, J.; Glynn, J.R.; Shearer, S.; Kambashi, B.; Godfrey-Faussett, P. HIV-1 and Recurrence, Relapse, and Reinfection of Tuberculosis after Cure: A Cohort Study in South African Mineworkers. Lancet 2001, 358, 1687–1693. [Google Scholar] [CrossRef]
- Narayanan, S.; Swaminathan, S.; Supply, P.; Shanmugam, S.; Narendran, G.; Hari, L.; Ramachandran, R.; Locht, C.; Jawahar, M.S.; Narayanan, P.R. Impact of HIV Infection on the Recurrence of Tuberculosis in South India. J. Infect. Dis. 2010, 201, 691–703. [Google Scholar] [CrossRef]
- World Health Organization; Global Tuberculosis Programme. Meeting Report of the WHO Expert Consultation on the Definition of Extensively Drug-Resistant Tuberculosis; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Gandhi, N.R.; Shah, N.S.; Andrews, J.R.; Vella, V.; Moll, A.P.; Scott, M.; Weissman, D.; Marra, C.; Lalloo, U.G.; Friedland, G.H. HIV Coinfection in Multidrug- and Extensively Drug-Resistant Tuberculosis Results in High Early Mortality. Am. J. Respir. Crit. Care Med. 2010, 181, 80–86. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty Years on: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Togre, N.S.; Vargas, A.M.; Bhargavi, G.; Mallakuntla, M.K.; Tiwari, S. Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges. Int. J. Mol. Sci. 2022, 23, 10669. [Google Scholar] [CrossRef]
- Manina, G.; Pasca, R.M.; Buroni, S.; De Rossi, E.; Riccardi, G. Decaprenylphosphoryl-β-D-Ribose 2-Epimerase from Mycobacterium Tuberculosis Is a Magic Drug Target. Curr. Med. Chem. 2010, 17, 3099–3108. [Google Scholar] [CrossRef]
- Borthwick, J.A.; Alemparte, C.; Wall, I.; Whitehurst, B.C.; Argyrou, A.; Burley, G.; de Dios-Anton, P.; Guijarro, L.; Monteiro, M.C.; Ortega, F.; et al. Mycobacterium Tuberculosis Decaprenylphosphoryl-β-d-Ribose Oxidase Inhibitors: Expeditious Reconstruction of Suboptimal Hits into a Series with Potent in Vivo Activity. J. Med. Chem. 2020, 63, 2557–2576. [Google Scholar] [CrossRef]
- Carroll, P.; Faray-Kele, M.-C.; Parish, T. Identifying Vulnerable Pathways in Mycobacterium Tuberculosis by Using a Knockdown Approach. Appl. Environ. Microbiol. 2011, 77, 5040–5043. [Google Scholar] [CrossRef]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the Essentiality of the Decaprenyl-Phospho-D-Arabinofuranose Pathway in M Ycobacterium Tuberculosis Using Conditional Mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; KR, P.; et al. 1,4-Azaindole, a Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium Tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef]
- Stanley, S.A.; Grant, S.S.; Kawate, T.; Iwase, N.; Shimizu, M.; Wivagg, C.; Silvis, M.; Kazyanskaya, E.; Aquadro, J.; Golas, A.; et al. Identification of Novel Inhibitors of M. Tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 2012, 7, 1377–1384. [Google Scholar] [CrossRef]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a Small Molecule with Activity against Drug-Resistant and Persistent Tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, 1110. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a New Combination Therapy for Tuberculosis with next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Crellin, P.K.; Brammananth, R.; Coppel, R.L. Decaprenylphosphoryl-β-D-Ribose 2′-Epimerase, the Target of Benzothiazinones and Dinitrobenzamides, Is an Essential Enzyme in Mycobacterium Smegmatis. PLoS ONE 2011, 6, e16869. [Google Scholar] [CrossRef]
- Gao, C.; Ye, T.-H.; Wang, N.-Y.; Zeng, X.-X.; Zhang, L.-D.; Xiong, Y.; You, X.-Y.; Xia, Y.; Xu, Y.; Peng, C.-T.; et al. Synthesis and Structure–Activity Relationships Evaluation of Benzothiazinone Derivatives as Potential Anti-Tubercular Agents. Bioorg. Med. Chem. Lett. 2013, 23, 4919–4922. [Google Scholar] [CrossRef]
- Landge, S.; Mullick, A.B.; Nagalapur, K.; Neres, J.; Subbulakshmi, V.; Murugan, K.; Ghosh, A.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; et al. Discovery of Benzothiazoles as Antimycobacterial Agents: Synthesis, Structure–Activity Relationships and Binding Studies with Mycobacterium Tuberculosis Decaprenylphosphoryl-β-d-Ribose 2′-Oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. [Google Scholar] [CrossRef]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Kumar, J.L.; Koushik, K.; et al. 4-Aminoquinolone Piperidine Amides: Noncovalent Inhibitors of DprE1 with Long Residence Time and Potent Antimycobacterial Activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines Kill Mycobacterium Tuberculosis through Noncovalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Park, Y.; Engelhart, C.A.; Wallach, J.B.; Schnappinger, D.; Arora, K.; Manikkam, M.; Gac, B.; Wang, H.; Murgolo, N.; et al. Discovery and Structure–Activity-Relationship Study of N -Alkyl-5-Hydroxypyrimidinone Carboxamides as Novel Antitubercular Agents Targeting Decaprenylphosphoryl-β-D-Ribose 2′-Oxidase. J. Med. Chem. 2018, 61, 9952–9965. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of Pyrazolopyridones as a Novel Class of Noncovalent DprE1 Inhibitor with Potent Anti-Mycobacterial Activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef] [PubMed]
- Rogacki, M.K.; Pitta, E.; Balabon, O.; Huss, S.; Lopez-Roman, E.M.; Argyrou, A.; Blanco-Ruano, D.; Cacho, M.; Vande Velde, C.M.L.; Augustyns, K.; et al. Identification and Profiling of Hydantoins—A Novel Class of Potent Antimycobacterial DprE1 Inhibitors. J. Med. Chem. 2018, 61, 11221–11249. [Google Scholar] [CrossRef] [PubMed]
- Price, A.C.; Choi, K.-H.; Heath, R.J.; Li, Z.; White, S.W.; Rock, C.O. Inhibition of β-Ketoacyl-Acyl Carrier Protein Synthases by Thiolactomycin and Cerulenin. J. Biol. Chem. 2001, 276, 6551–6559. [Google Scholar] [CrossRef]
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; et al. Platensimycin Is a Selective FabF Inhibitor with Potent Antibiotic Properties. Nature 2006, 441, 358–361. [Google Scholar] [CrossRef]
- Kapilashrami, K.; Bommineni, G.; Machutta, C.; Kim, P.; Lai, C.-T.; Simmerling, C.; Picart, F.; Tonge, P. Thiolactomycin-Based β-Ketoacyl-AcpM Synthase A (KasA) Inhibitors. J. Biol. Chem. 2013, 288, 6045–6052. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Using Fragment-Based Approaches to Discover New Antibiotics. SLAS Discov. 2018, 23, 495–510. [Google Scholar] [CrossRef]
- Wang, F.; Langley, R.; Gulten, G.; Dover, L.G.; Besra, G.S.; Jacobs, W.R.; Sacchettini, J.C. Mechanism of Thioamide Drug Action against Tuberculosis and Leprosy. J. Exp. Med. 2007, 204, 73–78. [Google Scholar] [CrossRef]
- Baulard, A.R.; Betts, J.C.; Engohang-Ndong, J.; Quan, S.; McAdam, R.A.; Brennan, P.J.; Locht, C.; Besra, G.S. Activation of the Pro-Drug Ethionamide Is Regulated in Mycobacteria. J. Biol. Chem. 2000, 275, 28326–28331. [Google Scholar] [CrossRef] [PubMed]
- Willand, N.; Dirié, B.; Carette, X.; Bifani, P.; Singhal, A.; Desroses, M.; Leroux, F.; Willery, E.; Mathys, V.; Déprez-Poulain, R.; et al. Synthetic EthR Inhibitors Boost Antituberculous Activity of Ethionamide. Nat. Med. 2009, 15, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, B.; Flipo, M.; Blondiaux, N.; Crauste, C.; Malaquin, S.; Leroux, F.; Piveteau, C.; Villeret, V.; Brodin, P.; Villoutreix, B.O.; et al. Ligand Efficiency Driven Design of New Inhibitors of Mycobacterium Tuberculosis Transcriptional Repressor EthR Using Fragment Growing, Merging, and Linking Approaches. J. Med. Chem. 2014, 57, 4876–4888. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, P.O.; Surade, S.; Blaszczyk, M.; Delorme, V.; Brodin, P.; Baulard, A.R.; Blundell, T.L.; Abell, C. A Fragment Merging Approach towards the Development of Small Molecule Inhibitors of Mycobacterium Tuberculosis EthR for Use as Ethionamide Boosters. Org. Biomol. Chem. 2016, 14, 2318–2326. [Google Scholar] [CrossRef]
- Nikiforov, P.O.; Blaszczyk, M.; Surade, S.; Boshoff, H.I.; Sajid, A.; Delorme, V.; Deboosere, N.; Brodin, P.; Baulard, A.R.; Barry, C.E.; et al. Fragment-Sized EthR Inhibitors Exhibit Exceptionally Strong Ethionamide Boosting Effect in Whole-Cell Mycobacterium Tuberculosis Assays. ACS Chem. Biol. 2017, 12, 1390–1396. [Google Scholar] [CrossRef]
- Flipo, M.; Willand, N.; Lecat-Guillet, N.; Hounsou, C.; Desroses, M.; Leroux, F.; Lens, Z.; Villeret, V.; Wohlkönig, A.; Wintjens, R.; et al. Discovery of Novel N -Phenylphenoxyacetamide Derivatives as EthR Inhibitors and Ethionamide Boosters by Combining High-Throughput Screening and Synthesis. J. Med. Chem. 2012, 55, 6391–6402. [Google Scholar] [CrossRef]
- Belisle, J.T.; Vissa, V.D.; Sievert, T.; Takayama, K.; Brennan, P.J.; Besra, G.S. Role of the Major Antigen of Mycobacterium Tuberculosis in Cell Wall Biogenesis. Science 1997, 276, 1420–1422. [Google Scholar] [CrossRef]
- Mendes, V.; Blundell, T.L. Targeting Tuberculosis Using Structure-Guided Fragment-Based Drug Design. Drug Discov. Today 2017, 22, 546–554. [Google Scholar] [CrossRef]
- Ronning, D.R.; Klabunde, T.; Besra, G.S.; Vissa, V.D.; Belisle, J.T.; Sacchettini, J.C. Crystal Structure of the Secreted Form of Antigen 85C Reveals Potential Targets for Mycobacterial Drugs and Vaccines. Nat. Struct. Biol. 2000, 7, 141–146. [Google Scholar] [CrossRef]
- Scheich, C.; Puetter, V.; Schade, M. Novel Small Molecule Inhibitors of MDR Mycobacterium Tuberculosis by NMR Fragment Screening of Antigen 85C. J. Med. Chem. 2010, 53, 8362–8367. [Google Scholar] [CrossRef]
- Marquet, A.; Tse Sum Bui, B.; Florentin, D. Biosynthesis of Biotin and Lipoic Acid. Vitam. Horm. 2001, 61, 51–101. [Google Scholar] [PubMed]
- Eisenberg, M.A.; Hsiung, S.C. Mode of Action of the Biotin Antimetabolites Actithiazic Acid and Alpha-Methyldethiobiotin. Antimicrob. Agents Chemother. 1982, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Okami, Y.; Kitahara, T.; Hamada, M.; Naganawa, H.; Kondo, S.; Maeda, K.; Takeuchi, T.; Umezawa, H. Studies On A New Amino Acid Antibiotic, Amiclenomycin. J. Antibiot. 1974, 27, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, T.; Hotta, K.; Yoshida, M.; Okami, Y. Biological Studies on Amiclenomycin. J. Antibiot. 1975, 28, 215–221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hotta, K.; Kitahara, T.; Okami, Y. Studies on the Mode of Action of Amiclenomycin. J. Antibiot. 1975, 28, 222–228. [Google Scholar] [CrossRef][Green Version]
- Poetsch, M.; Zähner, H.; Werner, R.G.; Kern, A.; Jung, G. Metabolic Products from Microorganisms. 230. Amiclenomycin-Peptides, New Antimetabolites of Biotin. Taxonomy, Fermentation and Biological Properties. J. Antibiot. 1985, 38, 312–320. [Google Scholar] [CrossRef]
- Kern, A.; Kabatek, U.; Jung, G.; Werner, R.G.; Poetsch, M.; Zähner, H. Amiclenomycin Peptides—Isolation and Structure Elucidation of New Biotin Antimetabolites. Liebigs Ann. Der Chem. 1985, 1985, 877–892. [Google Scholar] [CrossRef]
- Shi, C.; Aldrich, C.C. Design and Synthesis of Potential Mechanism-Based Inhibitors of the Aminotransferase BioA Involved in Biotin Biosynthesis. J. Org. Chem. 2012, 77, 6051–6058. [Google Scholar] [CrossRef][Green Version]
- Woong Park, S.; Klotzsche, M.; Wilson, D.J.; Boshoff, H.I.; Eoh, H.; Manjunatha, U.; Blumenthal, A.; Rhee, K.; Barry, C.E.; Aldrich, C.C.; et al. Evaluating the Sensitivity of Mycobacterium Tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PLoS Pathog. 2011, 7, e1002264. [Google Scholar] [CrossRef]
- Keer, J.; Smeulders, M.J.; Gray, K.M.; Williams, H.D. Mutants of Mycobacterium Smegmatis Impaired in Stationary-Phase Survival The GenBank Accession Numbers for the Sequences Determined in This Work Are: AJ277088 (Mutant 272A), AJ277089 (Mutant 272E), AJ27790 (Mutant 317C), AJ277152 (Mutant 492A) and AJ27688. Microbiology 2000, 146, 2209–2217. [Google Scholar] [CrossRef][Green Version]
- Dai, R.; Wilson, D.J.; Geders, T.W.; Aldrich, C.C.; Finzel, B.C. Inhibition of Mycobacterium Tuberculosis Transaminase BioA by Aryl Hydrazines and Hydrazides. ChemBioChem 2014, 15, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chan, D.S.H.; Coyne, A.G.; Abell, C. Fragment-Based Approaches to TB Drugs. Parasitology 2018, 145, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Berney, M.; Berney-Meyer, L.; Wong, K.-W.; Chen, B.; Chen, M.; Kim, J.; Wang, J.; Harris, D.; Parkhill, J.; Chan, J.; et al. Essential Roles of Methionine and S -Adenosylmethionine in the Autarkic Lifestyle of Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10008–10013. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; van Tonder, A.J.; Vilchèze, C.; Mendes, V.; Thomas, S.E.; Malek, A.; Chen, B.; Chen, M.; Kim, J.; Blundell, T.L.; et al. Arginine-Deprivation–Induced Oxidative Damage Sterilizes Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Thomas, S.E.; Zaidan, S.A.; Pasillas, M.A.; Cory-Wright, J.; Sebastián-Pérez, V.; Burgess, A.; Cattermole, E.; Meghir, C.; Abell, C.; et al. A Fragment-Based Approach to Assess the Ligandability of ArgB, ArgC, ArgD and ArgF in the L-Arginine Biosynthetic Pathway of Mycobacterium Tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3491–3506. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Dubar, F.; Blaise, M.; Bernut, A.; Pawlik, A.; Bouchier, C.; Brosch, R.; Guérardel, Y.; Lelièvre, J.; et al. A New Piperidinol Derivative Targeting Mycolic Acid Transport in Mycobacterium Abscessus. Mol. Microbiol. 2016, 101, 515–529. [Google Scholar] [CrossRef]
- Trapero, A.; Pacitto, A.; Singh, V.; Sabbah, M.; Coyne, A.G.; Mizrahi, V.; Blundell, T.L.; Ascher, D.B.; Abell, C. Fragment-Based Approach to Targeting Inosine-5′-Monophosphate Dehydrogenase (IMPDH) from Mycobacterium Tuberculosis. J. Med. Chem. 2018, 61, 2806–2822. [Google Scholar] [CrossRef]
- Jaegle, M.; Wong, E.L.; Tauber, C.; Nawrotzky, E.; Arkona, C.; Rademann, J. Protein-Templated Fragment Ligations-From Molecular Recognition to Drug Discovery. Angew. Chem. Int. Ed. 2017, 56, 7358–7378. [Google Scholar] [CrossRef]
- Tran, A.T.; West, N.P.; Britton, W.J.; Payne, R.J. Elucidation of Mycobacterium Tuberculosis Type II Dehydroquinase Inhibitors Using a Fragment Elaboration Strategy. ChemMedChem 2012, 7, 1031–1043. [Google Scholar] [CrossRef]
- Kumar, A.; Imran Siddiqi, M.; Miertus, S. New Molecular Scaffolds for the Design of Mycobacterium Tuberculosis Type II Dehydroquinase Inhibitors Identified Using Ligand and Receptor Based Virtual Screening. J. Mol. Model. 2010, 16, 693–712. [Google Scholar] [CrossRef]
- Heikal, A.; Nakatani, Y.; Jiao, W.; Wilson, C.; Rennison, D.; Weimar, M.R.; Parker, E.J.; Brimble, M.A.; Cook, G.M. ‘Tethering’ Fragment-Based Drug Discovery to Identify Inhibitors of the Essential Respiratory Membrane Protein Type II NADH Dehydrogenase. Bioorg. Med. Chem. Lett. 2018, 28, 2239–2243. [Google Scholar] [CrossRef] [PubMed]
- Batt, S.M.; Jabeen, T.; Bhowruth, V.; Quill, L.; Lund, P.A.; Eggeling, L.; Alderwick, L.J.; Fütterer, K.; Besra, G.S. Structural Basis of Inhibition of Mycobacterium Tuberculosis DprE1 by Benzothiazinone Inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 11354–11359. [Google Scholar] [CrossRef] [PubMed]
- Noto, T.; Miyakawa, S.; Oishi, H.; Endo, H.; Okazaki, H. Thiolactomycin, a New Antibiotic. III. In Vitro Antibacterial Activity. J. Antibiot. 1982, 35, 401–410. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium Tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.T.; Ramey, M.E.; Massoudi, L.M.; Carter, C.L.; Zimmerman, M.; Kaya, F.; Graham, B.G.; Gruppo, V.; Hastings, C.; Woolhiser, L.K.; et al. Comparative Analysis of Pharmacodynamics in the C3HeB/FeJ Mouse Tuberculosis Model for DprE1 Inhibitors TBA-7371, PBTZ169, and OPC-167832. Antimicrob. Agents Chemother. 2021, 65, e0058321. [Google Scholar] [CrossRef] [PubMed]
- Willand, N.; Desroses, M.; Toto, P.; Dirié, B.; Lens, Z.; Villeret, V.; Rucktooa, P.; Locht, C.; Baulard, A.; Deprez, B. Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chem. Biol. 2010, 5, 1007–1013. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The Structural Biology of HIV-1: Mechanistic and Therapeutic Insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and Function of HIV-1 Reverse Transcriptase: Molecular Mechanisms of Polymerization and Inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef]
- Goff, S.P. Retroviral Reverse Transcriptase: Synthesis, Structure, and Function. J. Acquir. Immune Defic. Syndr. 1990, 3, 817–831. [Google Scholar]
- Ding, J.; Das, K.; Tantillo, C.; Zhang, W.; Clark, A., Jr.; Jessen, S.; Lu, X.; Hsiou, Y.; Jacobo-Molina, A.; Andries, K.; et al. Structure of HIV-1 Reverse Transcriptase in a Complex with the Non-Nucleoside Inhibitor α-APA R 95845 at 2.8 å Resolution. Structure 1995, 3, 365–379. [Google Scholar] [CrossRef]
- Sluis-Cremer, N. The Emerging Profile of Cross-Resistance among the Nonnucleoside HIV-1 Reverse Transcriptase Inhibitors. Viruses 2014, 6, 2960–2973. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Erman, B.; Jernigan, R.L.; Atilgan, A.R.; Covell, D.G. Collective Motions in HIV-1 Reverse Transcriptase: Examination of Flexibility and Enzyme Function. J. Mol. Biol. 1999, 285, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Geitmann, M.; Elinder, M.; Seeger, C.; Brandt, P.; de Esch, I.J.P.; Danielson, U.H. Identification of a Novel Scaffold for Allosteric Inhibition of Wild Type and Drug Resistant HIV-1 Reverse Transcriptase by Fragment Library Screening. J. Med. Chem. 2011, 54, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.D.; Patel, D.; Arnold, E. Fragment Screening and HIV Therapeutics. Fragment-Based Drug Discovery and X-ray Crystallography; Davies, T.G., Hyvönen, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 181–200. [Google Scholar]
- Curreli, F.; Kwon, Y.D.; Zhang, H.; Scacalossi, D.; Belov, D.S.; Tikhonov, A.A.; Andreev, I.A.; Altieri, A.; Kurkin, A.V.; Kwong, P.D.; et al. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. [Google Scholar] [CrossRef]
- Iusupov, I.R.; Curreli, F.; Spiridonov, E.A.; Markov, P.O.; Ahmed, S.; Belov, D.S.; Manasova, E.V.; Altieri, A.; Kurkin, A.V.; Debnath, A.K. Design of Gp120 HIV-1 Entry Inhibitors by Scaffold Hopping via Isosteric Replacements. Eur. J. Med. Chem. 2021, 224, 113681. [Google Scholar] [CrossRef]
- La, J.; Latham, C.F.; Tinetti, R.N.; Johnson, A.; Tyssen, D.; Huber, K.D.; Sluis-Cremer, N.; Simpson, J.S.; Headey, S.J.; Chalmers, D.K.; et al. Identification of Mechanistically Distinct Inhibitors of HIV-1 Reverse Transcriptase through Fragment Screening. Proc. Natl. Acad. Sci. USA 2015, 112, 6979–6984. [Google Scholar] [CrossRef]
- Brown, P.O. Integration of Retroviral DNA. Retroviruses: Strategies of Replication; Swanstrom, R., Vogt, P.K., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 19–48. [Google Scholar]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an Evolutionarily Conserved Domain in Human Lens Epithelium-Derived Growth Factor/Transcriptional Co-Activator P75 (LEDGF/P75) That Binds HIV-1 Integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef]
- Cherepanov, P.; Ambrosio, A.L.B.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural Basis for the Recognition between HIV-1 Integrase and Transcriptional Coactivator P75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef]
- McKee, C.J.; Kessl, J.J.; Shkriabai, N.; Dar, M.J.; Engelman, A.; Kvaratskhelia, M. Dynamic Modulation of HIV-1 Integrase Structure and Function by Cellular Lens Epithelium-Derived Growth Factor (LEDGF) Protein. J. Biol. Chem. 2008, 283, 31802–31812. [Google Scholar] [CrossRef]
- Kessl, J.J.; Li, M.; Ignatov, M.; Shkriabai, N.; Eidahl, J.O.; Feng, L.; Musier-Forsyth, K.; Craigie, R.; Kvaratskhelia, M. FRET Analysis Reveals Distinct Conformations of IN Tetramers in the Presence of Viral DNA or LEDGF/P75. Nucleic Acids Res. 2011, 39, 9009–9022. [Google Scholar] [CrossRef]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical Profile of BI 224436, a Novel HIV-1 Non-Catalytic-Site Integrase Inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.; Balakrishnan, M.; Brizgys, G.; Cai, R.; Lansdon, E.; Mulato, A.; Osier, M.; Wang, J.; Yu, H.; Sakowicz, R. Novel Non-Catalytic Site Integrase Inhibitor with Improved Resistance Profile. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, WA, USA, 13–16 February 2017. [Google Scholar]
- Roberts, R.A.; Campbell, R.A.; Sikakana, P.; Sadler, C.; Osier, M.; Xu, Y.; Feng, J.Y.; Mitchell, M.; Sakowicz, R.; Chester, A.; et al. Species-Specific Urothelial Toxicity With an Anti-HIV Noncatalytic Site Integrase Inhibitor (NCINI) Is Related to Unusual PH-Dependent Physicochemical Changes. Toxicol. Sci. 2021, 183, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Kessl, J.J.; Kvaratskhelia, M. Allosteric Inhibition of HIV-1 Integrase Activity. Curr. Opin. Chem. Biol. 2013, 17, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Engelman, A. Multimodal Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. Expert Rev. Mol. Med. 2013, 15, e14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Métifiot, M.; Marchand, C.; Pommier, Y. HIV Integrase Inhibitors. 20-Year Landmark and Challenges. Adv. Pharmacol. 2013, 67, 75–105. [Google Scholar]
- Johnson, B.C.; Métifiot, M.; Ferris, A.; Pommier, Y.; Hughes, S.H. A Homology Model of HIV-1 Integrase and Analysis of Mutations Designed to Test the Model. J. Mol. Biol. 2013, 425, 2133–2146. [Google Scholar] [CrossRef]
- Krishnan, L.; Li, X.; Naraharisetty, H.L.; Hare, S.; Cherepanov, P.; Engelman, A. Structure-Based Modeling of the Functional HIV-1 Intasome and Its Inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 15910–15915. [Google Scholar] [CrossRef]
- Wielens, J.; Headey, S.J.; Deadman, J.J.; Rhodes, D.I.; Parker, M.W.; Chalmers, D.K.; Scanlon, M.J. Fragment-Based Design of Ligands Targeting a Novel Site on the Integrase Enzyme of Human Immunodeficiency Virus 1. ChemMedChem 2011, 6, 258–261. [Google Scholar] [CrossRef]
- Rhodes, D.I.; Peat, T.S.; Vandegraaff, N.; Jeevarajah, D.; Le, G.; Jones, E.D.; Smith, J.A.; Coates, J.A.; Winfeld, L.J.; Thienthong, N.; et al. Structural Basis for a New Mechanism of Inhibition of H I V-1 Integrase Identified by Fragment Screening and Structure-Based Design. Antivir. Chem. Chemother. 2011, 21, 155–168. [Google Scholar] [CrossRef]
- Tiefenbrunn, T.; Stout, C.D. Towards Novel Therapeutics for HIV through Fragment-Based Screening and Drug Design. Prog. Biophys. Mol. Biol. 2014, 116, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawsawi, L.Q.; Christ, F.; Dayam, R.; Debyser, Z.; Neamati, N. Inhibitory Profile of a LEDGF/P75 Peptide against HIV-1 Integrase: Insight into Integrase-DNA Complex Formation and Catalysis. FEBS Lett. 2008, 582, 1425–1430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peat, T.S.; Rhodes, D.I.; Vandegraaff, N.; Le, G.; Smith, J.A.; Clark, L.J.; Jones, E.D.; Coates, J.A.V.; Thienthong, N.; Newman, J.; et al. Small Molecule Inhibitors of the LEDGF Site of Human Immunodeficiency Virus Integrase Identified by Fragment Screening and Structure Based Design. PLoS ONE 2012, 7, e40147. [Google Scholar] [CrossRef] [PubMed]
- Wielens, J.; Headey, S.J.; Jeevarajah, D.; Rhodes, D.I.; Deadman, J.; Chalmers, D.K.; Scanlon, M.J.; Parker, M.W. Crystal Structure of the HIV-1 Integrase Core Domain in Complex with Sucrose Reveals Details of an Allosteric Inhibitory Binding Site. FEBS Lett. 2010, 584, 1455–1462. [Google Scholar] [CrossRef]
- Shen, L.; Peterson, S.; Sedaghat, A.R.; McMahon, M.A.; Callender, M.; Zhang, H.; Zhou, Y.; Pitt, E.; Anderson, K.S.; Acosta, E.P.; et al. Dose-Response Curve Slope Sets Class-Specific Limits on Inhibitory Potential of Anti-HIV Drugs. Nat. Med. 2008, 14, 762–766. [Google Scholar] [CrossRef]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.L.; Nalivaika, E.A.; Özen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef]
- Wensing, A.M.J.; van Maarseveen, N.M.; Nijhuis, M. Fifteen Years of HIV Protease Inhibitors: Raising the Barrier to Resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
- Perryman, A.L.; Zhang, Q.; Soutter, H.H.; Rosenfeld, R.; McRee, D.E.; Olson, A.J.; Elder, J.E.; David Stout, C. Fragment-Based Screen against HIV Protease. Chem. Biol. Drug Des. 2010, 75, 257–268. [Google Scholar] [CrossRef]
- Perryman, A.L.; Lin, J.-H.; McCammon, J.A. HIV-1 Protease Molecular Dynamics of a Wild-Type and of the V82F/I84V Mutant: Possible Contributions to Drug Resistance and a Potential New Target Site for Drugs. Protein Sci. 2004, 13, 1108–1123. [Google Scholar] [CrossRef]
- Tiefenbrunn, T.; Forli, S.; Happer, M.; Gonzalez, A.; Tsai, Y.; Soltis, M.; Elder, J.H.; Olson, A.J.; Stout, C.D. Crystallographic Fragment-Based Drug Discovery: Use of a Brominated Fragment Library Targeting HIV Protease. Chem. Biol. Drug Des. 2014, 83, 141–148. [Google Scholar] [CrossRef]
- Blaney, J.; Nienaber, V.; Burley, S.K. Fragment-Based Lead Discovery and Optimization Using X-Ray Crystallography, Computational Chemistry, and High-throughput Organic Synthesis. In Fragment-Based Approaches in Drug Discovery; Jahnke, W., Erlanson, D.A., Eds.; Wiley: Hoboken, NJ, USA, 2006; pp. 215–248. [Google Scholar]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-Phenyl-N′-(2,2,6,6-Tetramethyl-Piperidin-4-Yl)-Oxalamides as a New Class of HIV-1 Entry Inhibitors That Prevent Gp120 Binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Haque, K.; Xie, L.; Qiu, Q.; Xu, J.; Yong, W.; Tong, X.; Debnath, A.K. Synthesis, Antiviral Activity and Resistance of a Novel Small Molecule HIV-1 Entry Inhibitor. Bioorg. Med. Chem. 2015, 23, 7618–7628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Curreli, F.; Kwon, Y.D.; Zhang, H.; Yang, Y.; Scacalossi, D.; Kwong, P.D.; Debnath, A.K. Binding Mode Characterization of NBD Series CD4-Mimetic HIV-1 Entry Inhibitors by X-Ray Structure and Resistance Study. Antimicrob. Agents Chemother. 2014, 58, 5478–5491. [Google Scholar] [CrossRef]
- Belov, D.S.; Curreli, F.; Kurkin, A.V.; Altieri, A.; Debnath, A.K. Guanidine-Containing Phenyl-Pyrrole Compounds as Probes for Generating HIV Entry Inhibitors Targeted to Gp120. ChemistrySelect 2018, 3, 6450–6453. [Google Scholar] [CrossRef]
- Korazim, O.; Sackett, K.; Shai, Y. Functional and Structural Characterization of HIV-1 Gp41 Ectodomain Regions in Phospholipid Membranes Suggests That the Fusion-Active Conformation Is Extended. J. Mol. Biol. 2006, 364, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Sackett, K.; Shai, Y. The HIV-1 Gp41 N-Terminal Heptad Repeat Plays an Essential Role in Membrane Fusion. Biochemistry 2002, 41, 4678–4685. [Google Scholar] [CrossRef]
- Haqqani, A.A.; Tilton, J.C. Entry Inhibitors and Their Use in the Treatment of HIV-1 Infection. Antivir. Res. 2013, 98, 158–170. [Google Scholar] [CrossRef]
- Navratilova, I.; Besnard, J.; Hopkins, A.L. Screening for GPCR Ligands Using Surface Plasmon Resonance. ACS Med. Chem. Lett. 2011, 2, 549–554. [Google Scholar] [CrossRef]
- Davidson, A.; Begley, D.W.; Lau, C.; Varani, G. A Small-Molecule Probe Induces a Conformation in HIV TAR RNA Capable of Binding Drug-Like Fragments. J. Mol. Biol. 2011, 410, 984–996. [Google Scholar] [CrossRef]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X. Crystal Structures of HIV-1 Reverse Transcriptase with Etravirine (TMC125) and Rilpivirine (TMC278): Implications for Drug Design. J. Med. Chem. 2010, 53, 4295–4299. [Google Scholar] [CrossRef]
- Kwon, Y.D.; LaLonde, J.M.; Yang, Y.; Elban, M.A.; Sugawara, A.; Courter, J.R.; Jones, D.M.; Smith, A.B.; Debnath, A.K.; Kwong, P.D. Crystal Structures of HIV-1 Gp120 Envelope Glycoprotein in Complex with NBD Analogues That Target the CD4-Binding Site. PLoS ONE 2014, 9, e85940. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Structure | Fragment Hit | Reference |
|---|---|---|
 |  | [12,21,66] |
 |  | [32,67] |
 |  | [55,56] |
 |  | [39] |
 |  | [44] |
| S.No. | Target | Fragment Molecule | Potency Parameters | References |
|---|---|---|---|---|
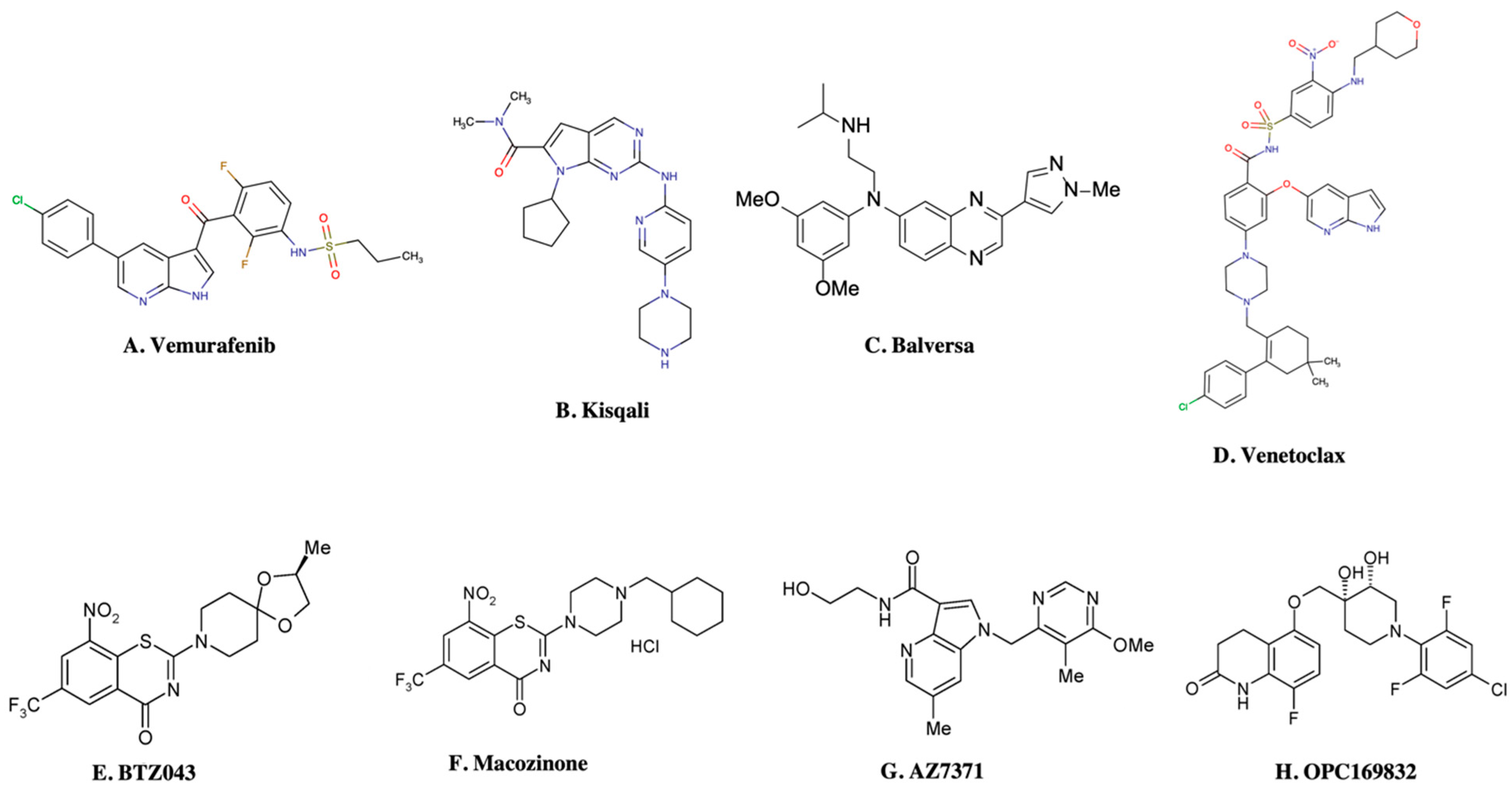
| 1. | DprE1 |  BTZ043 | MIC99 1 ng mL−1; 2.3 nM (TD50 of 5 μg mL−1) | [12,21,66,68] |
 PBTZ169 | MIC99 ≤ 0.19 ng mL−1; 2.3 nM (TD50 of 58 μg mL−1) | [12,21,66] | ||
 AZ7371 | 0.64 μg mL−1 | [18] | ||
 OPC167832 | 0.24 to 2 ng mL−1 | [69] | ||
 Piperidinylpyrimidines derivatives 3 | MIC90 (μM): 35.6 | [12] | ||
 Piperidinylpyrimidines derivatives 4 | MIC90 (μM): 15.6 | [12] | ||
| 2. | KasA |  Thiolactomycin (TLM) | IC50 n.d. | [32] |
 Pantetheine analog, PK940 | IC50 n.d. | [32] | ||
| 3. | EthR |  4-Iodo-N-prop-2-ynyl benzene sulfonamide (Compound 3) | SPR IC50 160 µM | [37] |
 Compound 18 (4-Iodo-N-prop-2-ynylbenzenesulfonamide derivative) | IC50 5 µM EC50 6 µM LE 0.34 | [37] | ||
 Compound 21 (4-Iodo-N-prop-2-ynylbenzenesulfonamide derivative) | IC50280 µM LE 0.36 | [33,37] | ||
 Compound 22 (4-Iodo-N-prop-2-ynylbenzenesulfonamide derivative) | IC50 1 µM LE 0.36 | [37] | ||
 Compound 23 (4-Iodo-N-prop-2-ynylbenzenesulfonamide derivative) | IC50 n.d. EC50 0.04 µM | [37] | ||
 Compound 20 (4-Iodo-N-prop-2-ynylbenzenesulfonamide derivative) 4-Iodo-N-(1-{2-oxo-2-[4-(3-thiophen-2-yl-1,2,4-oxadiazol-5-yl)-piperidin-1-yl]-ethyl}-1H-1,2,3-triazol-4-ylmethyl)-benzenesulfonamide | IC50 = 580 µM | [70] | ||
 Compound 1 (N-phenyl-149 phenoxy acetamide derivative | IC50 = 2.9 µM | [40] | ||
 Compound 4 (N-(4-benzothiazol-2-ylphenyl)-2-(3-methoxyphenoxy)acetamide) | EC50 0.21 µM | [40] | ||
 Compound 5 (N-(4-benzothiazol-2-ylphenyl)-2-(3-methoxyphenoxy)acetamide) | EC50 0.34 µM | [40] | ||
| 4. | 7,8-Diaminopelargonic acid (DAPA) synthase (BioA) |  2-(aminomethyl)-benzothiazole 23 | IC50 n.d. | [56] |
 2-(aminomethyl)-benzothiazole 23 hydrazine derivative | IC50 n.d. | [56] |
| Target | Analogue | Mode of Action | Reference |
|---|---|---|---|
| HIV-1 Reverse Transcriptase | NBD compound 17 | Antiviral activity Cytotoxicity | [73] |
| RT drug resistant mutants | p-Hydroxyaniline 8 | Inhibit Rnase H activity | [74] |
| HIV-1 Integrase | N-benzyl indoline 15 and 19, and Y3 compounds | Inhibit HIV-1 IN activity In vitro and In vivo cell culture | [97,100] |
| KM-SB3 | Inhibit HIV-1 IN activity | [101] | |
| HIV-1 Protease | IF1 and 4D9 | Inhibit HIV-1 PR | [105,107] |
| HIV-1 gp120 | NBD-556 and its amine and acid form (NBD-10111) | Antiviral activity Cytotoxicity | [80] |
| Mutation of HIV-1 gp120 (Arg59 and Asp368) with CD4 receptor | N (2-(2-guanidinoacetamido) -1-(4-(hydroxymethyl) thiazol- 2-yl-ethyl) 54(trifluoromethyl)phenyl)1H-pyrrole-2-carboxamide (Compound 10) | High antiviral activity and cytotoxicity | [112] |
| Compound 882376 | Antiviral activity prevents the interaction of gp120 and CD4 Prevents cell to cell fusion | [110] | |
| NBD-14010 NBD-14171 NBD-14136 NBD-14270 (Thiazole ring NBD compounds) | Moderate antiviral activity | [112] |
| Structure | Fragment Hit | Reference |
|---|---|---|
 |  | [80,81,118] |
 |  | [101] |
 |  | [105] |
 |  | [119] |
| S.No. | Target | Fragment Molecule | Potency | References |
|---|---|---|---|---|
| 1. | HIV-1 Reverse transcriptase |  (4-bromo-1-indanone) | IC50 < 25 μM | [77] |
 amine piperidine ring fragment 17 NBD compound | 1.7 ± 0.2 | [80] | ||
 4, 5 (oxime), and 8 (P-hydroxyaniline) | μM range | [81] | ||
| 2. | HIV-1 Integrase |  Compound Y3a | IC50-259 μM | [97,98] |
 Compound Y3b | IC50 5 μM | [97,98] | ||
 Compound 19 (N-benzyl indolinone analog) | EC50 29 μM | [100] | ||
 KM-SB3 | IC50 170 μM | [101] | ||
| 3. | HIV-1 Protease |  IF1 (derivative of indole-6- propionic acid) | >1 mM | [98] |
| 4. | Gp120 |  NBD-556 and its amine and acid form (NBD-10111) | IC50 n.d. | [80] |
| Mutation of HIV-1 gp120 (Arg59 and Asp368) with CD4 receptor |  N (2-(2-guanidinoacetamido) -1-(4-(hydroxymethyl) thiazol- 2-yl-ethyl) 54(trifluoromethyl)phenyl)1H-pyrrole-2-carboxamide (Compound 10) | IC50 n.d. | [112] | |
 Compound 882376 | IC50 n.d. | [110] | ||
 NBD-14010 | IC50 n.d. | [112] | ||
 NBD-14171 | IC50 n.d. | [112] | ||
 NBD-14136 NBD-14270 (Thiazole ring NBD compounds) | IC50 n.d. | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallakuntla, M.K.; Togre, N.S.; Santos, D.B.; Tiwari, S. Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV. Pharmaceuticals 2022, 15, 1415. https://doi.org/10.3390/ph15111415
Mallakuntla MK, Togre NS, Santos DB, Tiwari S. Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV. Pharmaceuticals. 2022; 15(11):1415. https://doi.org/10.3390/ph15111415
Chicago/Turabian StyleMallakuntla, Mohan Krishna, Namdev S. Togre, Destiny B. Santos, and Sangeeta Tiwari. 2022. "Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV" Pharmaceuticals 15, no. 11: 1415. https://doi.org/10.3390/ph15111415
APA StyleMallakuntla, M. K., Togre, N. S., Santos, D. B., & Tiwari, S. (2022). Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV. Pharmaceuticals, 15(11), 1415. https://doi.org/10.3390/ph15111415